

Summary
Drug resistance in lymphoblastic and myeloblastic leukemia cells is poorly understood, with several lines of evidence suggesting that resistance can be correlated with upregulation of human asparagine synthetase (hASNS) expression, although this hypothesis is controversial. New tools are needed to investigate this clinically important question, including potent hASNS inhibitors. In vitro experiments show an adenylated sulfoximine to be a slow-onset, tight-binding inhibitor of hASNS with nanomolar affinity. This binding affinity represents a 10-fold improvement over that reported for the only other well-characterized hASNS inhibitor. The adenylated sulfoximine has a cytostatic effect on L-asparaginase-resistant MOLT-4 cells cultured in the presence of L-asparaginase, an enzyme that depletes L-asparagine in the growth medium. These observations represent direct evidence that potent hASNS inhibitors may prove to be effective agents for the clinical treatment of acute lymphoblastic leukemia.
Introduction
The enzyme L-asparaginase (ASNase), which catalyzes the hydrolysis of L-asparagine [1], is a component of most therapeutic protocols for the treatment of acute lymphoblastic leukemia (ALL) [2–4]. The opposing reaction is catalyzed by asparagine synthetase (ASNS), which converts L-aspartic acid into L-asparagine in a transformation requiring ATP and a nitrogen source that is L-glutamine in eukaryotic cells [5]. The human enzyme (hASNS) is of clinical interest [6] because several lines of evidence suggest that the development of ASNase resistance in ALL is correlated with upregulation and expression of hASNS [7–10]. Although the molecular basis underlying the therapeutic utility of ASNase in ALL remains ill defined [6, 9], it is thought that normal and malignant lymphocytes depend on the uptake of L-asparagine from circulating plasma for growth [11], as recently reviewed elsewhere [6]. In turn, this has given rise to the hypothesis that ASNase exerts its clinical effects in ALL by depleting L-asparagine in the blood, with the subsequent efflux of cytoplasmic L-asparagine from the leukemic blasts [12]. Upregulation of hASNS activity might overcome such an effect by catalyzing production of the L-asparagine necessary for cellular growth when cells are in the presence of ASNase [7]. The clinical significance of this hypothesis has been challenged, however, by studies in which ASNS concentrations and activity were extrapolated from the levels of mRNA encoding the enzyme [13–15]. New strategies are therefore needed to probe the molecular mechanisms by which cellular hASNS contributes to ASNase-resistant ALL. In particular, cell-permeable compounds capable of specifically inhibiting the enzyme may be valuable tools in evaluating whether increased levels of intracellular asparagine biosynthesis might be the key change in cellular metabolism that underlies the appearance of drug resistance. Assays employing small-molecule libraries to identify potent ASNS inhibitors have not yet been reported, and early studies of substrate analogs failed to yield any compounds exhibiting micromolar affinity against the form of ASNS present in either murine lymphoblasts and pancreatic cells [16], or rodent neoplasm leukemia 5178Y/AR cell lines [17].
Based on the crystal structure of the glutamine-dependent ASNS from Escherichia coli (AS-B) [18], the human enzyme is likely built from two domains, each of which contains a catalytic site. The N-terminal site catalyzes the conversion of glutamine into glutamic acid and ammonia, and aspartate is reacted with ATP in the C-terminal site to yield the reactive intermediate β-aspartyl-AMP (βAspAMP) (Figure 1), the existence of which has been demonstrated by isotopic labeling experiments [19, 20]. As is the case in other glutamine-dependent amidotransferases [21], ammonia released in the N-terminal domain of ASNS travels through an intramolecular tunnel linking the active sites, and reacts with the activated acyladenylate moiety to form asparagine [6]. Recent kinetic experiments have demonstrated that βAspAMP is stabilized by the enzyme [22], and we have shown that stable analogs of this intermediate are submicromolar inhibitors of ASNS [23]. In this paper, we describe in vitro experiments showing that the adenylated sulfoximine 1 (Figure 2) [24] is a slow-onset, tight-binding inhibitor of hASNS. Perhaps more importantly, we also demonstrate that treatment of a drug-resistant MOLT-4 cell line [25] with adenylated sulfoximine 1 has a cytostatic effect. This observation is the first direct evidence that ASNS inhibitors represent interesting compounds that may prove to be effective agents for the clinical treatment of ASNase-resistant ALL, as initially discussed almost 40 years ago [12].
Figure 1. Reactions Catalyzed by ASNS.
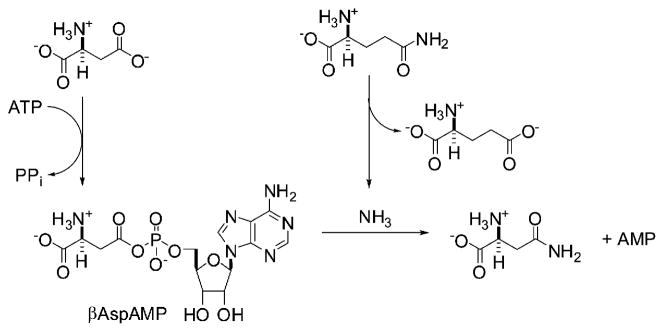
Ammonia may replace glutamine as a nitrogen source for the ASNS-catalyzed synthetase reaction.
Figure 2.

Chemical Structures of the Adenylated Sulfoximine 1 and Other Compounds Discussed in the Text
Results
The Adenylated Methylsulfoximine Moiety Is a Transition-State Analog
The adenylated sulfoximine 1, as a mixture of diastereoisomers 1a and 1b [24] (Figure 2), was first reported as an inhibitor of the ammonia-dependent variant of E. coli ASNS (AS-A), an enzyme that has been identified only in prokaryotes [26] and does not share a common ancestor with glutamine-dependent ASNS [27]. Based on the crystal structure of the complex between AS-A and 1, the adenylated sulfoximine moiety was proposed to be a stable analog of the transition state for the attack of ammonia on βAspAMP [24]. In order to verify this hypothesis, we compared the electrostatic and steric properties of a model phosphorylated sulfoximine 2 (Figure 3A) and the transition state for the addition of ammonia to the acylphosphate 3 (Figure 3B) using semi-empirical calculations [28] at the PM3 level of theory [29]. These calculations also employed a continuum solvation potential [30, 31] so as to model the effects of a polarizable medium on the charge distributions in the transition state and sulfoximine. These studies suggest that the tetrahedral sulfur atom is a good model for the rehybridized carbonyl carbon in the transition state for ammonia attack (see Tables S1 and S2 in the Supplemental Data available with this article online), at least at this relatively unsophisticated level of theory. For example, the S-CH3 bond length in the sulfoximine is 1.77 Å compared to a value of 1.64 Å for the analogous C-N bond in the model transition state, and the related O-S-CH3 and O-C-N angles in the two structures are 108.2° and 108.7°, respectively. There are also interesting steric and electronic similarities between the methyl substituent in the phosphorylated sulfoximine moiety and the ammonia molecule as it undergoes uncatalyzed reaction with the carbonyl group of the acylphosphate (Tables S2 and S3). Hence, an examination of the electrostatic potential for sulfoximine 2, computed on the electronic isodensity surface (Figure 3A), shows regions of positive potential (red) adjacent to the hydrogen substituents. These presumably result from polarization effects associated with the inductive effect of the phosphorylated sulfoximine moiety, and mimic a similar region of positive potential about the ammonia hydrogens that is seen when the electrostatic potential at the isodensity surface of the model transition state 3 is visualized (Figure 3B).
Figure 3. Computational Evaluation of the Adenylated Methylsulfoximine Moiety as a Stable Analog of the Transition State for Asparagine Formation.

(A) Structure of the model sulfoximine 2 (left), and graphical representations of the PM3-optimized molecular structure (middle) and electrostatic properties (right) of this compound.
(B) Structure of the transition state for the attack of ammonia on the model acylphosphate 3 (left), and graphical representations of the PM3-optimized molecular structure (middle) and electrostatic properties (right) of this transition state. Dotted lines represent weak interactions, and the electrostatic potential is mapped on the isodensity surface in both figures.
In Vitro Characterization of 1 as an Inhibitor of Human ASNS
We initially assayed the ability of the adenylated sulfoximine 1 (as a mixture of diastereoisomers 1a and 1b) to inhibit the ammonia-dependent activity of a C-terminally tagged form of glutamine-dependent hASNS expressed in Sf9 cells [32]. By using a coupled assay to detect inorganic pyrophosphate (PPi) [33], which is formed in a 1:1 stoichiometry with asparagine by the human enzyme [34], time-dependent inhibition was observed when reactions were initiated by the addition of hASNS to a mixture of substrates containing the transition-state analog (Figure 4A). Control experiments established that the sulfoximine did not affect the PPi detection assay (data not shown), but it was possible that the presence of 1 had decoupled asparagine and PPi formation. The Asn:PPi ratio was therefore checked by an HPLC-based end-point assay in which recombinant ASNS was incubated with substrates at various sulfoximine concentrations before reaction was terminated by the addition of trichloroacetic acid. The resulting solution was then assayed for PPi [33], and the asparagine formed was quantitated by HPLC after conversion into its 2,4-dini-trophenol derivative [22]. The results show that the product stoichiometry was not significantly affected by the inhibitor, at least within the experimental error (see Figure S1).
Figure 4. Steady-State Kinetic Behavior of hASNS in the Presence of the Adenylated Sulfoximine 1.

(A) Progress curves showing PPi formation as a function of incubation time for ammonia-dependent synthetase activity at various concentrations of 1: open circles, 0 μM; closed circles, 1 μM; open squares, 2 μM; closed squares, 4 μM; open diamonds, 6 μM; closed diamonds, 10 μM. Lines represent the fit used to obtain the kinetic parameters for inhibition.
(B) Kinetic model used in the quantitative analysis of the steady-state progress curves. E, enzyme; I, inhibitor.
(C) Reactivation of inhibited hASNS as a function of time. The line shows the exponential of best fit to the data.
(D) Progress curves showing PPi formation as a function of incubation time for glutamine-dependent synthetase activity at various concentrations of 1: open circles, 0 μM; closed circles, 1 μM; open squares, 2 μM; closed squares, 4 μM; open diamonds, 6 μM; closed diamonds, 10 μM. Lines represent the fit used to obtain the kinetic parameters for inhibition.
In previous studies of the ability of 1 to inhibit the AS-B, it was reported that the adenylated sulfoximine did not bind to the free enzyme [35]. We therefore examined whether this was the case for its interaction with hASNS by measuring the residual activity of the human enzyme after incubation with 1 for 10 min and subsequent filtration through a Sephadex G-50 spin column. These experiments showed that ASNS synthetase activity was reduced by incubation with the adenylated sulfoximine in a concentration-dependent manner (Table S1), suggesting that the inhibitor was capable of binding to the free enzyme. This contrasts with its observed kinetic behavior with AS-B under similar reaction conditions, and in the absence of a crystal structure for hASNS, it is difficult to assess the structural basis for this apparent difference in inhibition kinetics. Given the multidomain structure of the two glutamine-dependent enzymes, it is possible that differences in the conformational states of the two forms of ASNS may result in modified accessibility of the adenylated sulfoximine to its binding site. The fact that 1 could bind to free hASNS, however, permitted quantitative analysis of the progress-curve data for the ammonia-dependent reaction using a standard kinetic model for slow-onset, tight-binding inhibition (Figure 4B) [36]. Curve fitting gave values of 280 ± 43 nM and 2.5 ± 0.3 nM for KI and KI*, respectively (Table 1). This value of KI* is 100-fold smaller than that observed for the inhibition of hASNS by the N-acylsulfonamide 4 (Figure 2A), which is an analog of βAspAMP [23], and compares very favorably to the KI* value of 67 nM reported for the interaction of the diastereoisomeric mixture 1a and 1b with the bacterial, ammonia-dependent AS-A [24].
Table 1.
Steady-State Parameters for Inhibition of Human ASNS by the Adenylated Sulfoximine 1
| Nitrogen Source | KI (nM) | k5 (s−1) | k6 (s−1) | kobs/[I]tot (M−1 s−1) | K*I (nM) |
|---|---|---|---|---|---|
| NH4Cl | 285 | 2.98 × 10−3 | 2.6 × 10−5 | 436 | 2.46 |
| L-Glutamine | 985 | 2.81 × 10−3 | 7.1 × 10−5 | 219 | 24.3 |
Parameters are those obtained by fitting to a kinetic model in which the inhibitor binds to free enzyme, and assuming a Ka value for ATP of 0.2 mM. See text for full details of experimental conditions.
Of course, this analysis makes the assumption that only one of the two diastereoisomers (1a or 1b) is capable of undergoing a tight-binding interaction with hASNS. Evidence to support this assertion is provided by previous studies showing the stereochemical dependence of tight-binding inhibition by sulfoximine derivatives [37–39]. Hence, in all situations to date for which diastereoisomeric and enantiomeric mixtures of sulfoximine-based inhibitors have been separated, only one of these structures has exhibited tight binding inhibition, with the others in the mixture generally exhibiting no activity. This behavior seems reasonable given that active sites generally stabilize one of the two stereo-chemically distinct transition states for nucleophilic attack on activated carbonyl groups due to the location of the single oxyanion ‘‘hole’’ [40]. On the other hand, we did consider the situation in which one of the diastereoisomeric adenylated sulfoximines acted as a reversible inhibitor. In this situation, because the concentrations of 1a and 1b in the mixture are identical, the integrated rate expression for time-dependent product formation can be written as:
| (1) |
where k takes the following form [41]:
| (2) |
and Ki and KX are the reversible inhibition constants for the tight-binding and reversible inhibitors, respectively, and I is the total concentration of adenylated sulfoximine 1. From this equation, in situations where KX is significantly greater than Ki, the effects of the other diastereoisomer are unlikely to have a significant impact on the tight-binding inhibition constants. In this case, the use of Equation 2 with various values of KX did not improve our ability to fit the experimental progress curves.
We next evaluated the reversibility of in vitro hASNS inhibition using standard protocols [42]. Thus, after complete inactivation of the enzyme by incubating hASNS with ATP, aspartate, and ammonium chloride in the presence of the adenylated sulfoximine 1 (as judged by the cessation of PPi production), the reaction mixture was subjected to gel filtration on a Sephadex G-50 column. Fractions containing hASNS were diluted into a solution containing substrates at saturating concentration, and PPi formation was monitored spectroscopically over a period of several hours (Figure 4C). Over the time course of the experiment, the inactivated sample of human ASNS regained 56% of its activity relative to a control sample of the enzyme that had been subjected to identical treatment in the absence of sulfoximine 1, and quantitative analysis gave a value of 7.4 hr for the half-life of reactivation, which is consistent with the estimate of k6 obtained from analysis of progress-curve data (Table 1). The demonstrated reversibility of hASNS inhibition again contrasts with reported observations on the bacterial enzymes, given that both AS-A and AS-B do not regain activity after being inactivated by the adenylated sulfoximine 1 [24, 35].
Given the relatively complicated structure of the adenylated sulfoximine 1, we also examined whether hASNS was capable of catalyzing the adenylation of the structurally much simpler methylsulfoximine 5 (Figure 2), which is easily prepared as a mixture of diastereoisomers 5a and 5b from L-S-methylcysteine, and is an intermediate in the synthesis of 1 [24]. These experiments were prompted by the observation that the methionine sulfoximine 6 (Figure 2) [43] binds to the metabolically important enzyme, glutamine synthetase (GS) [44, 45], where it undergoes enzyme-catalyzed phosphorylation in the presence of ATP to yield the phosphorylated derivative 7 [46]. The latter compound is a potent GS inhibitor because it resembles the transition state formed when ammonia attacks the γ-glutamylphosphate intermediate 8 (Figure 2) [47]. Despite testing a variety of conditions, hASNS was not inhibited by the sulfoximine 5 in the presence of ATP, suggesting that the enzyme did not catalyze formation of the adenylated sulfoximine 1 (data not shown), and showing the importance of the adenylyl moiety for ASNS inhibition. On the basis of these observations, it is possible that 1 was exerting its effects by covalently modifying the enzyme. We therefore carried out a series of experiments to evaluate whether protein adenylation was indeed occurring when hASNS was incubated with 1 by using conditions that we had developed for employing electrospray time-of-flight (TOF) mass spectrometry to observe tryptic peptides from over 90% of the sequence of recombinant hASNS [32]. We incubated the enzyme with the adenylated sulfoximine 1 (10 μM) until the rate of pyrophosphate production became zero, and the protein was then subjected to in-gel digestion with trypsin under our standard conditions [32]. Mass spectrometric analysis of the peptide fragments from the sample of fully inhibited hASNS showed no fragments with an increased mass corresponding to that expected if adenylation of any protein side chains had taken place (data not shown). In this regard, we note that all peptides containing residues that are located in the synthetase active site of the enzyme can be observed using electrospray TOF mass spectrometry [32].
Given that L-glutamine is the likely physiological nitrogen source for asparagine biosynthesis in eukaryotic cells [6], we also investigated the ability of 1 to inhibit the glutamine-dependent synthetase activity of hASNS at a physiologically relevant ATP concentration (5 mM) and pH 8 (Figure 4D). Once again, slow-onset tight-binding inhibition was observed, and the quantitative analysis of the progress curves for PPi production yielded values of 1000 ± 176 nM and 24 ± 2.8 nM for KI and KI*, respectively, under these conditions (Table 1). The 10-fold decrease in the ability of 1 to inhibit the glutamine-dependent synthetase reaction may arise from conformational differences in the C-terminal domain of the human enzymes that are associated with occupancy of the N-terminal glutaminase site by substrate, L-glutamate, or the thioester intermediate [48, 49]. We therefore evaluated the effects of the adenylated sulfoximine 1 on the glutaminase activity of the enzyme, hASNS, with a continuous assay in which glutamate formed by ASNS-catalyzed glutamine hydrolysis is coupled to the production of NADH [50]. The rationale for these experiments came from previous studies showing that the presence of ATP stimulates the glutaminase activity of E. coli AS-B [51]; an effect that was used to demonstrate the functional roles of conserved residues in the N-terminal, glutaminase domain [52]. In the presence of 10 μM adenylated sulfoximine 1, kcat/Km for the glutaminase activity of hASNS was 982 M−1 s−1, which was relatively unchanged from the value of 795 M−1 s−1 determined for the reaction in the absence of the inhibitor (Figure 5). This finding was unexpected, however, on the basis of ATP-dependent stimulation of the glutaminase activity of AS-B [51], and so we assayed whether the glutaminase activity of hASNS was affected by ATP. In another interesting observation, the presence of 3 mM ATP had almost no impact on kcat/Km for the glutaminase activity of hASNS, which was determined to be 674 M−1 s−1. The lack of effect on the steady-state rate of ASNS-catalyzed glutamine hydrolysis for either ATP or the sulfoximine derivative 1 contrasts with the stimulation of glutaminase activity observed for other members of the class II glutamine-dependent amidotransferase family [53, 54], including glutamine 5′-phosphoribosyl-pyrophosphate amidotransferase [55] and glutamine fructose-6-phosphate amidotransferase [56], when nitrogen-accepting substrates are bound in the C-terminal synthetase domains of these enzymes.
Figure 5. Effect of the Adenylated Sulfoximine 1 on the Steady-State Glutaminase Activity of hASNS.

Glutamine dependence of the glutaminase activity in the presence (closed circles) and absence (open circles) of 10 μM 1. Lines represent the fit to the Michaelis-Menten equations used to obtain the steady-state kinetic parameters.
Given the ability of the adenylated sulfoximine 1 to stimulate the glutaminase activity of free hASNS, we examined whether this remained the case under conditions in which ATP and L-aspartate were present. In these experiments, we examined the Asn:Glu ratio after 20 min incubation in the presence and absence of 1 with our standard HPLC-based end-point assay [22] (Figure S2). The results showed that 10 mM 1 did increase the Asn:Glu ratio for hASNS under these conditions from 1:1.3 to 1:2.4, presumably because the enzyme continues to catalyze the hydrolysis of the glutamine amide even when the adenylated sulfoximine is bound in the synthetase active site.
Characterization of the Effects of the ASNS on the Proliferation of an Asparaginase-Resistant MOLT-4 Cell Line
After demonstrating that the adenylated sulfoximine 1 is the first ASNS inhibitor with low-nanomolar affinity for the enzyme, the effects of incubating this compound with a MOLT-4 leukemia cell line [25] were investigated. The MOLT-4(R) cell line used for these experiments is continuously maintained by us in the presence of 1 U/mg ASNase [7, 57], and has been used extensively in previous studies of molecular mechanisms mediating ASNase resistance [6, 7, 57, 58]. The MOLT-4(R) cells were transferred to medium containing the hASNS inhibitor 1 at concentrations of 0.1–1 mM. These high concentrations were chosen because we anticipated that the charged functional groups on the molecule, which are known to be important in mediating recognition [23], would negatively impact cell permeability. Incubations were performed for a total of 48 hr, and the effect of the ASNS inhibitor on cell proliferation was assayed using a dye-based (WST-1) method [59] for estimating the number of MOLT-4 cells. The adenylated sulfoximine 1 inhibited, in a concentration-dependent manner, the ability of the MOLT-4(R) cells to proliferate. Interestingly, this cytostatic effect was accentuated by the simultaneous presence of ASNase in the growth medium (Figure 6). The origin of this synergistic effect remains to be determined. One possible explanation is that the greater degree of cytostatic action in the presence of the ASNase is a consequence of the inhibitor being poorly transported into the MOLT-4(R) cells, thereby preventing complete inhibition of asparagine biosynthesis. As a result, any simultaneous depletion of pre-existing asparagine by cotreatment of the cells with ASNase is more effective than using the inhibitor alone. In light of finding that the ASNS-sulfoximine complex retains its ability to catalyze glutamine hydrolysis, alternative mechanisms in which the cellular regulation of glutamine-dependent metabolism is perturbed may also underlie the observed synergy of ASNase and the hASNS inhibitor.
Figure 6. Effect of the Adenylated Sulfoximine 1 on MOLT-4 Proliferation in the Presence and Absence of 1 U ASNase.

Note that cell proliferation is defined as the number of viable cells after 48 hr expressed as the ratio to the initial number of cells (t = 0). Error bars represent the standard deviation of triplicate experiments.
Significance
ASNS is a structurally and mechanistically complex enzyme [2] that likely contributes to cellular mechanisms mediating ASNase resistance in ALL [6]. Despite considerable research activity aimed at delineating whether upregulation of ASNS expression is a direct cause of ASNase resistance in patients [14, 60], this question remains unresolved, in part because of the absence of potent, and selective, ASNS inhibitors that prevent intracellular asparagine biosynthesis. In this paper, we describe a transition state analog that inhibits hASNS with nanomolar activity in vitro, and confirm that the adenylated sulfoximine 1 is a slow-onset, tight-binding inhibitor even when ATP is present at physiological concentration. Although the affinity of hASNS for sulfoximine 1 is dependent on the nitrogen source used by the enzyme, the reversibility of inhibition suggests that 1 forms complementary noncovalent interactions, presumably with residues located in the C-terminal synthetase active site. More importantly, the adenylated sulfoximine 1 slows the growth of ASNase-resistant MOLT-4 cells in the presence of ASNase. These experiments appear to be the first direct validation of the long-held hypothesis that hASNS inhibitors represent potential therapeutic agents for the treatment of ALL and related leukemias. On the other hand, more studies need to be performed that address (1) the nature of the molecular events in the cell underpinning the synergistic interaction of the adenylated sulfoximine 1 and ASNase, and (2) whether other leukemia cell lines and primary ALL cells will show similar behavior on treatment with 1 and/or more cell-permeable analogs. Our observations demonstrate the feasibility of employing methylsulfoximine derivatives as ‘‘lead’’ structures in future efforts to obtain potent, small-molecule hASNS inhibitors [61]. Such compounds not only have potential therapeutic value, but also represent new tools that can be used in modern profiling strategies [62, 63] for delineating the role of ASNS expression in ASNase resistance.
Experimental Procedures
Materials
All samples of the adenylated sulfoximine 1 (as a 1:1 mixture of diastereoisomers 1a and 1b) and the methylsulfoximine 5 (as a 1:1 mixture of diastereoisomers 5a and 5b) were obtained by chemical synthesis following procedures in the literature [24]. Unless otherwise stated, all other chemicals and reagents, including authentic samples of dinitrobenzene derivatives of L-asparagine, L-aspartate, and L-glutamate, were purchased from Sigma-Aldrich (St. Louis, MO), and were of the highest available purity. Protein concentrations were determined by a modified Bradford assay (Pierce, Rockford, IL) based on standard curves constructed using bovine serum albumin [64]. L-glutamine was purified by recrystallization prior to use in all assays containing this reagent. Caution: extreme care should be taken when handling solutions of 2,4-dinitrofluorobenzene (DNFB) in organic solvents, because this reagent is a potent allergen and will penetrate many types of laboratory gloves [65].
Expression and Purification of Recombinant, C-Terminally Tagged ASNS
Multimilligram amounts of wild-type, C-terminally tagged human ASNS were expressed in Sf9 cells and purified using procedures that have been reported elsewhere [32].
Steady-State Kinetic Assays and Data Analysis
Progress curves were obtained under steady-state conditions using a continuous assay in which the formation of PPi is measured by monitoring the consumption of NADH (340 nm) (Sigma Technical Bulletin BI-100). It has been shown that the asparagine and PPi are produced in a 1:1 ratio by the recombinant, C-terminally tagged form of hASNS [32]. In these experiments, purified hASNS (2 μg) was incubated at 37°C with substrates in 100 mM EPPS buffer (pH 8), containing 5 mM ATP, 10 mM MgCl2, 100 mM NH4Cl, 10 mM L-aspartic acid, and varying concentrations of the adenylated sulfoximine 1 (0–10 μM) over a period of 20 min (1 ml final volume). Reactions were initiated by addition of the enzyme. The resulting progress curves were analyzed by fitting the data to Equation 1 [36] using the Kaleidagraph v3.5 software package (Synergy, Reading, PA), where P is the PPi concentration (equivalent to the amount of asparagine) formed at time t, vo and vss are initial and steady-state velocities, respectively, and k is the apparent first-order rate constant for isomerization of EI to EI*. The values of k at different inhibitor concentrations were therefore determined by fitting, and used to find k6 from the expression:
| (3) |
The average value of k6 could then be computed, and this was subsequently used to obtain estimates of Ki and k5 by fitting to the following equation:
| (4) |
where [I] is the concentration of the adenylated sulfoximine, [ATP] = 5 mM, and Ka was taken to be 0.2 mM [23]. For experiments in which L-glutamine replaced ammonia as the nitrogen source, purified hASNS (2 μg) was incubated at 37°C in EPPS buffer (pH 8, 1 ml final volume), containing 5 mM ATP, 10 mM MgCl2, 25 mM L-glutamine, 10 mM L-aspartic acid, and varying concentrations of 1 (0–10 μM) over a period of 20 min. Control experiments using known amounts of PPi demonstrated that the assay reagent was not affected by the presence of the adenylated sulfoximine 1.
In order to check that the inhibitor 1 did not affect the 1:1 stoichiometry of Asn:PPi, an alternate HPLC-based assay [22] was employed in control experiments to measure the amount of L-aspara-gine directly. Thus, recombinant hASNS (4 μg) was incubated at 37°C with 5 mM ATP, 10 mM L-aspartic acid, 10 mM MgCl2, and 100 mM NH4Cl in 100 mM EPPS buffer (pH 8), and varying concentrations of the adenylated sulfoximine 1 (0, 2, or 10 μM) over a period of 20 min (1 ml final volume). Reactions were initiated by addition of the enzyme. After quenching with glacial AcOH, and neutralization with aq. NaOH, an aliquot of each mixture (40 μl) was diluted (200 μl final volume) with 400 mM aq. Na2CO3 (pH 9), containing 10% DMSO and 30% DNFB (as a saturated solution in EtOH). The resulting solutions were heated at 50°C for 45 min to permit reaction of DNFB with the amino acids to yield their dinitrophenyl (DNP) derivatives [66, 67]. Aliquots of each assay mixture (10 μl) were analyzed by reverse-phase HPLC (RP-HPLC) using a C18 column (Varian Inc., Palo Alto, CA). The DNP-derivatized amino acids were eluted with a step gradient of 40 mM formic acid buffer (pH 3.6), and CH3CN. In this procedure, the initial concentration of the organic phase (CH3CN) was 13.5%, which was increased to 14.5% over a period of 21 min. After this time, the amount of CH3CN was increased to 80% over a period of 30 s, and elution continued for a further 20 min. Eluted amino acid DNFB derivatives were monitored at 365 nm and identified by comparison to authentic standards. Under these conditions, DNP-asparagine exhibited a retention time of approximately 21 min, and could be quantified on the basis of its peak area. Calibration curves were constructed using solutions of pure L-asparagine derivatized in the same manner as the samples. The amount of asparagine detected in this assay was compared to PPi as determined by the coupled enzyme assay (Sigma). These experiments were also repeated for mixtures containing 0.5 mM ATP.
The glutaminase activity of hASNS was assayed by determining L-glutamate formation with glutamate dehydrogenase in the presence of NAD+ [50]. Assay mixtures (200 μl total volume) contained varying concentrations of L-glutamine (1–50 mM) and 8 mM MgCl2 in 100 mM EPPS buffer (pH 8), containing 0.5 mM DTT and 100 mM NaCl, and reaction was initiated by the addition of recombinant hASNS (2.6 μg). The resulting solution was then incubated at 37°C for 20 min before being terminated by addition of 20% trichloroacetic acid (30 μl). The mixture was added to 770 μl of the coupling reagent (300 mM glycine, 250 mM hydrazine (pH 9), containing 1 mM ADP, 1.6 mM NAD+, and 2.2 U glutamate dehydrogenase) and incubated for 30 min (1 ml final volume). The absorbance at 340 nm of the resulting solution was measured, and the amount of L-glutamate determined from a standard curve. Steady-state parameters were obtained by fitting the data to the Michaelis-Menten equation [68] using the Kaleidagraph v3.5 software package. These experiments were repeated in the presence of ATP (5 mM) or the adenylated sulfoximine 1 (10 μM).
Enzyme Binding Assay
Purified hASNS (4 μg) was incubated in the presence (5 μM or 10 μM) or absence of the adenylated sulfoximine 1 for 10 min in 100 mM EPPS (pH 8, 20 μl total volume). The resulting reaction mixtures were then loaded onto a Sephadex G-50 spin column and washed with 100 μl EPPS buffer (100 mM, pH 8). The activity of the enzyme and its concentration in each of the three solutions was determined using the coupled assay for detection of PPi and the modified Bradford assay, respectively. Experiments to monitor the synthetase activity employed saturating levels of ATP and L-aspartic acid, 10 mM MgCl2, and 100 mM NH4Cl, and PPi production was measured over a 20 min period.
Enzyme Reactivation Assay
Recombinant, wild-type hAS (at a final concentration of 0.25 μM) was added to a solution of 5 μM sulfoximine 1, 0.5 mM ATP, 10 mM MgCl2, 10 mM Asp, and 100 mM NH4Cl in 100 mM EPPS (pH 8, 1 ml total volume). The solution was incubated until synthetase activity ceased, as measured by PPi production [33]. The reaction mixture was then filtered through a Sephadex G-25 column using elution with 100 mM EPPS (pH 8), so as to remove substrates, products, and the unbound sulfoximine 1. The reactivation of AS activity was then monitored by measuring ammonia-dependent asparagine formation in aliquots (500 μl) of the fractions containing human ASNS, which were diluted 20-fold into assay mixtures containing 5 mM ATP, 10 mM MgCl2, 10 mM aspartate, and 100 mM NH4Cl in 100 mM EPPS buffer (pH 8) at 37°C.
Mass Spectrometric Analysis of Tryptic Digests
Recombinant, wild-type hASNS (at a final concentration of 0.25 μM) was added to a solution of 5 μM sulfoximine 1, 5 mM ATP, 10 mM MgCl2, 10 mM Asp, and 100 mM NH4Cl in 100 mM EPPS, (pH 8, 1 ml total volume). The solution was incubated until synthetase activity ceased, as measured by PPi production [33]. The resulting protein was isolated and a sample purified by SDS-PAGE on a 9% resolving gel at 110 V, including a lane for molecular weight markers. Staining with Coomassie brilliant blue dye revealed a protein band of 66 kDa molecular weight, which was excised and destained by soaking the gel pieces in 50% methanol (1 ml) overnight, followed by several additional washings. Cysteine residues were reduced with dithiothreitol and alkylated with iodoacetamide to give the carbamidomethylated-modified protein, which was then digested in-gel with trypsin (625 ng trypsin per gel band) on ice for 45 min and overnight at 37°C. The trypsin solution was replaced with 20 mM ammonium bicarbonate, and the reaction quenched with 5 μl glacial AcOH. After centrifugation, the sample was desalted prior to mass spectrometric analysis by elution through C18 ZipTips (Millipore, Billerica, MA) with a solution of 50% aq. CH3CN containing 1% HCO2H.
The separation of tryptic peptides produced in the protein digest was performed by capillary RP-HPLC on a 15 cm × 75 μm i.d. Pep-Map C18 column (LC Packings, San Francisco, CA) in combination with an Ultimate Capillary HPLC System (LC Packings) operating at a flow rate of 200 nl/min. Inline mass spectrometric analysis of the column eluate was accomplished with a hybrid TOF instrument (QSTAR; Applied Biosystems, Foster City, CA) equipped with a nano-electrospray source. Fragment ion data obtained on the TOF instrument were searched against the NCBI nr sequence database using the Mascot database search engine (Matrix Science, Boston, MA). Peptide masses were calculated based on the mono-isotopic peak and charge state of each ion cluster, and compared to those expected for tryptic peptides, and their corresponding adenylated derivatives, from recombinant hASNS. Probability-based MOWSE scores above the default significant value were considered for peptide identification in addition to validation by manual interpretation of tandem MS/MS data. Control experiments employed hASNS treated in an identical manner, except that the adenylated sulfoximine 1 was absent from the initial incubation mixture.
Cell-Based Assays
The effect of the adenylated sulfoximine 1 was tested on cell proliferation by first seeding MOLT-4/R-resistant cells into 96-well plates at a density of 4000 cells per well with RPMI 1640 medium with 10% FBS, and incubated in 95% air with 5% CO2 at 37°C. MOLT-4/R is a derivative of a human ALL cell line (MOLT-4) [25] that is resistant to the presence of ASNase in the medium and shows upregulated levels of ASNS expression [7, 57]. The sulfoximine 1 was diluted to give 0.1, 0.5, and 1.0 mM concentrations in culture medium, and cells were incubated with the compound in the presence or absence of 1 U ASNase. Cell viability was determined 48 hr after treatment by the WST-1 Cell Proliferation Assay (Roche Diagnostics, Indianapolis, IN). The optical density was read at 450 and 690 nm by an ELX800 Universal Microplate Reader (Bio-Tek Instruments, Inc., Winooski, VT). The mean cell titer of treated samples relative to control cells prior to treatment (time = 0) was calculated, and the data expressed as the mean ± SD of triplicate experiments.
Molecular Modeling Studies
All model structures were built using the graphical interface of the BioMedCAChe V6.5 software package (Fujitsu America Inc., Beaverton, OR), and their geometries were optimized using the PM3 semi-empirical model chemistry [29] in combination with the COSMO continuum solvation potential [30] as implemented in MOPAC V7.0 (Fujitsu America Inc.). The transition state for ammonia attack on the acetylphosphate derivative was obtained using standard methods (IRC option) [69], and all electrostatic potentials were calculated and visually displayed using algorithms implemented within BioMedCAChe V6.5 [70].
Supplementary Material
Acknowledgments
The Chiles Endowment Biomedical Research Program of the Florida Department of Health provided financial support for this work (J.A.G. and N.G.J.R). We also gratefully acknowledge the provision of funds from the National Institutes of Health (DK52064 and DK59315; M.S.K.) and a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (21COE; Kyoto University Alliance for Chemistry; J.H.).
Footnotes
Supplemental Data, including additional tables and figures, are available online at http://www.chembiol.com/cgi/content/full/13/12/1339/DC1/.
References
- 1.Aghaiypour K, Wlodawer A, Lubkowski J. Structural basis for the activity and substrate specificity of Erwinia chrysanthemi L-asparaginase. Biochemistry. 2001;40:5655–5664. doi: 10.1021/bi0029595. [DOI] [PubMed] [Google Scholar]
- 2.Barr RD, DeVeber LL, Pai KM, Andrew M, Halton J, Cairney AE, Whitton AC. Management of children with acute lymphoblastic leukemia by the Dana-Farber Cancer Institute protocol. Am J Pediatr Hematol Oncol. 1992;14:136–139. doi: 10.1097/00043426-199205000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Ertel IJ, Nesbit MG, Hammon D, Weiner J, Sather H. Effective dose of L-asparaginase for induction of remission in previously untreated children with acute lymphocytic leukemia: a report from the childrens cancer study group. Cancer Res. 1979;39:3893–3896. [PubMed] [Google Scholar]
- 4.Sutow WW, Garacia F, Starling KA, Williams TE, Lane DM, Gehan EA. L-asparaginase therapy in children with advanced leukemia. Cancer. 1971;28:819–824. doi: 10.1002/1097-0142(1971)28:4<819::aid-cncr2820280403>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 5.Richards NGJ, Schuster SM. Mechanistic issues in asparagine synthetase catalysis. Adv Enzymol Relat Areas Mol Biol. 1998;72:145–198. doi: 10.1002/9780470123188.ch5. [DOI] [PubMed] [Google Scholar]
- 6.Richards NGJ, Kilberg MS. Asparagine synthetase chemotherapy. Annu Rev Biochem. 2006;75:629–654. doi: 10.1146/annurev.biochem.75.103004.142520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aslanian AM, Kilberg MS. Asparagine synthetase expression alone is sufficient to induce L-asparaginase resistance in MOLT-4 human leukemia cells. Biochem J. 2001;357:321–328. doi: 10.1042/0264-6021:3570321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiriyama Y, Kubota M, Takimoto T, Kitoh J, Tanizawa A, Akiyama Y, Mikawa H. Biochemical characterization of U937 cells resistant to L-asparaginase: the role of asparagine synthetase. Leukemia. 1989;3:294–297. [PubMed] [Google Scholar]
- 9.Chakrabarti R, Schuster SM. L-asparaginase: perspectives on the mechanisms of action and resistance. J Pediatr Hematol Oncol. 1997;4:597–611. [Google Scholar]
- 10.Haskell CM, Canellos GP. L-asparaginase resistance in human leukemia-asparagine synthetase. Biochem Pharmacol. 1969;18:2578–2580. doi: 10.1016/0006-2952(69)90375-x. [DOI] [PubMed] [Google Scholar]
- 11.Graham ML. Pegaspargase: a review of clinical studies. Adv Drug Deliv Rev. 2003;55:1293–1302. doi: 10.1016/s0169-409x(03)00110-8. [DOI] [PubMed] [Google Scholar]
- 12.Cooney DA, Handschumacher RE. L-asparaginase and L-asparagine metabolism. Annu Rev Pharmacol. 1970;10:421–440. doi: 10.1146/annurev.pa.10.040170.002225. [DOI] [PubMed] [Google Scholar]
- 13.Appel IM, den Boer ML, Meijerink JPP, Veerman AJP, Reniers NCM, Pieters R. Up-regulation of asparagine synthetase expression is not linked to the clinical response to L-asparaginase in pediatric acute lymphoblastic leukemia. Blood. 2006;106:4244–4249. doi: 10.1182/blood-2005-06-2597. [DOI] [PubMed] [Google Scholar]
- 14.Fine BM, Kaspers GJ, Ho M, Loonen AH, Boxer LM. A genome-wide view of the in vitro response to L-asparaginase in acute lymphoblastic leukemia. Cancer Res. 2005;65:291–299. [PubMed] [Google Scholar]
- 15.Stams WAG, den Boer ML, Holleman A, Appel IM, Beverloo HB, van Wering ER, Janka-Schaub GE, Evans WE, Pieters R. Asparagine synthetase expression is linked with L-asparaginase resistance in TEL-AML1-negative but not TEL-AML1-positive pediatric acute lymphoblastic leukemia. Blood. 2005;105:4223–4225. doi: 10.1182/blood-2004-10-3892. [DOI] [PubMed] [Google Scholar]
- 16.Jayaram HN, Cooney DA. Analogs of L-aspartic acid in chemotherapy for cancer. Cancer Treat Rep. 1979;63:1095–1108. [PubMed] [Google Scholar]
- 17.Cooney DA, Driscoll JS, Milman HA, Jayaram HN, Davis RD. Inhibitors of L-asparagine synthetase in vitro. Cancer Treat Rep. 1976;60:1493–1557. [PubMed] [Google Scholar]
- 18.Larsen TM, Boehlein SK, Schuster SM, Richards NGJ, Thoden JB, Holden HM, Rayment I. Three-dimensional structure of Escherichia coli asparagine synthetase B: a short journey from substrate to product. Biochemistry. 1999;38:16146–16157. doi: 10.1021/bi9915768. [DOI] [PubMed] [Google Scholar]
- 19.Boehlein SK, Stewart JD, Walworth ES, Thirumoorthy R, Richards NGJ, Schuster SM. Kinetic mechanism of Escherichia coli asparagine synthetase B. Biochemistry. 1998;37:13230–13238. doi: 10.1021/bi981058h. [DOI] [PubMed] [Google Scholar]
- 20.Luehr CA, Schuster SM. Purification and characterization of beef pancreatic asparagine synthetase. Arch Biochem Biophys. 1985;237:335–346. doi: 10.1016/0003-9861(85)90285-1. [DOI] [PubMed] [Google Scholar]
- 21.Huang X, Holden HM, Raushel F. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem. 2001;70:149–180. doi: 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- 22.Tesson AR, Soper TS, Ciustea M, Richards NGJ. Revisiting the steady-state kinetic mechanism of glutamine-dependent asparagine synthetase from Escherichia coli. Arch Biochem Biophys. 2003;413:23–31. doi: 10.1016/s0003-9861(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 23.Koroniak L, Ciustea M, Gutierrez JA, Richards NGJ. Synthesis and characterization of an N-acylsulfonamide inhibitor of human asparagine synthetase. Org Lett. 2003;5:2033–2036. doi: 10.1021/ol034212n. [DOI] [PubMed] [Google Scholar]
- 24.Koizumi M, Hiratake J, Nakatsu T, Kato H, Oda J. A potent transition-state analogue inhibitor of Escherichia coli asparagine synthetase A. J. Am. Chem Soc. 1999;121:5799–5800. [Google Scholar]
- 25.Srivasta BI, Minowada J. Terminal deoxynucleotidyl transferase-activity in a cell line (MOLT-4) derived from peripheral blood of a patient with acute lymphoblastic leukemia. Biochem Biophys Res Commun. 1973;51:529–535. doi: 10.1016/0006-291x(73)91346-6. [DOI] [PubMed] [Google Scholar]
- 26.Cedar H, Schwartz JH. The asparagine synthetase of Escherichia coli I: biosynthetic role of the enzyme, purification and characterization of the reaction products. J Biol Chem. 1969;244:4112–4121. [PubMed] [Google Scholar]
- 27.Nakatsu T, Kato H, Oda J. Crystal structure of asparagine synthetase reveals a close evolutionary relationship to class II aminoacyl tRNA synthetase. Nat Struct Biol. 1998;5:15–19. doi: 10.1038/nsb0198-15. [DOI] [PubMed] [Google Scholar]
- 28.Cramer CJ. Essentials of Computational Chemistry. New York: Wiley; 2002. pp. 121–151. [Google Scholar]
- 29.Stewart JJP. Optimization of parameters for semiempirical methods I: method. J Comput Chem. 1989;10:209–220. [Google Scholar]
- 30.Klamt A. Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena. J Phys Chem. 1995;99:2224–2235. [Google Scholar]
- 31.Cramer CJ, Truhlar DG. Implicit solvation models: equilibria, structure, spectra, and dynamics. Chem Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 32.Ciustea M, Gutierrez JA, Abbatiello SE, Eyler JR, Richards NGJ. Efficient expression, purification and characterization of C-terminally tagged, recombinant human asparagine synthetase. Arch Biochem Biophys. 2005;440:18–27. doi: 10.1016/j.abb.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 33.O’Brien WE. A continuous spectrophotometric assay for argininosuccinate synthetase based on pyrophosphate formation. Anal Biochem. 1976;76:423–430. doi: 10.1016/0003-2697(76)90337-7. [DOI] [PubMed] [Google Scholar]
- 34.Horowitz B, Meister A. Glutamine-dependent asparagine synthetase from leukemia cells. J Biol Chem. 1972;247:6708–6719. [PubMed] [Google Scholar]
- 35.Boehlein SK, Nakatsu T, Hiratake J, Thirumoorthy R, Stewart JD, Richards NGJ, Schuster SM. Characterization of inhibitors acting at the synthetase site of Escherichia coli asparagine synthetase B. Biochemistry. 2001;40:11168–11175. doi: 10.1021/bi0155551. [DOI] [PubMed] [Google Scholar]
- 36.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 37.Tokutake N, Hiratake J, Katoh M, Irie T, Kato H, Oda J. Design, synthesis and evaluation of transition-state analogue inhibitors of Escherichia coli γ-glutamylcysteine synthetase. Bioorg Med Chem. 1998;6:1935–1953. doi: 10.1016/s0968-0896(98)00142-4. [DOI] [PubMed] [Google Scholar]
- 38.Manning JM, Moore S, Rowe WB, Meister A. Identification of L-methionine S-sulfoximine as the diastereoisomer of L-methionine SR-sulfoximine that inhibits glutamine synthetase. Biochemistry. 1969;8:2681–2685. doi: 10.1021/bi00834a066. [DOI] [PubMed] [Google Scholar]
- 39.Campbell EB, Hayward ML, Griffith OW. Analytical and preparative separation of the diastereoisomers of L-buthionine (SR)-sulfoximine, a potent inhibitor of glutathione biosynthesis. Anal Biochem. 1991;194:268–277. doi: 10.1016/0003-2697(91)90229-m. [DOI] [PubMed] [Google Scholar]
- 40.Wilmouth RC, Edman K, Neutze R, Wright PA, Clifton IJ, Schneider TR, Schofield CJ, Hadju J. X-ray snapshots of serine protease catalysis reveals a tetrahedral intermediate. Nat Struct Biol. 2001;8:689–694. doi: 10.1038/90401. [DOI] [PubMed] [Google Scholar]
- 41.Weiss PM, Cleland WW. Effect of the presence of a reversible inhibitor on the time course of slow-binding inhibition. Anal Biochem. 1987;161:438–441. doi: 10.1016/0003-2697(87)90472-6. [DOI] [PubMed] [Google Scholar]
- 42.Cha S. Tight-binding inhibitors. I: kinetics. Biochem Pharmacol. 1975;24:2177–2185. doi: 10.1016/0006-2952(75)90050-7. [DOI] [PubMed] [Google Scholar]
- 43.Logusch EW, Walker DM, McDonald JF, Franz JE, Villafranca JJ, DiIanni CL, Colanduoni JA, Li B, Schineller JB. Inhibition of Escherichia coli glutamine synthetase by α-substituted and γ-substituted phosphinothricins. Biochemistry. 1990;29:366–372. doi: 10.1021/bi00454a009. [DOI] [PubMed] [Google Scholar]
- 44.Abell LM, Villafranca JJ. Investigation of the mechanism of phosphinothricin inactivation of Escherichia coli glutamine synthetase using rapid quench techniques. Biochemistry. 1991;30:6135–6141. doi: 10.1021/bi00239a008. [DOI] [PubMed] [Google Scholar]
- 45.Liaw SH, Eisenberg D. Structural model for the reaction mechanism of glutamine synthetase, based on 5 crystal structures of enzyme-substrate complexes. Biochemistry. 1994;33:675–681. doi: 10.1021/bi00169a007. [DOI] [PubMed] [Google Scholar]
- 46.Manning JM, Moore S, Rowe WB, Meister A. Identification of L-methionine S-sulfoximine as diastereoisomer of L-methionine SR-sulfoximine that inhibits glutamine synthetase. Biochemistry. 1969;8:2681–2685. doi: 10.1021/bi00834a066. [DOI] [PubMed] [Google Scholar]
- 47.Weisbrod RE, Meister A. Studies on glutamine synthetase from Escherichia coli: formation of pyrrolidone carboxylate and inhibtion by methionine sulfoximine. J Biol Chem. 1973;248:3997–4002. [PubMed] [Google Scholar]
- 48.Schnizer HG, Boehlein SK, Stewart JD, Schuster SM, Richards NGJ. The γ-glutamyl thioester covalent intermediate in the glutaminase reaction catalyzed by Escherichia coli asparagine synthetase B. Methods Enzymol. 2002;354:260–271. doi: 10.1016/s0076-6879(02)54022-2. [DOI] [PubMed] [Google Scholar]
- 49.Schnizer HG, Boehlein SK, Stewart JD, Richards NGJ, Schuster SM. Formation and isolation of a covalent intermediate during the glutaminase reaction of a class II amidotransferase. Biochemistry. 1999;38:3677–3682. doi: 10.1021/bi981450v. [DOI] [PubMed] [Google Scholar]
- 50.Bernt E, Bergmeyer HU. L-Glutamate UV assay with glutamate dehydrogenase and NAD. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. New York: Academic Press; 1974. pp. 1704–1708. [Google Scholar]
- 51.Boehlein SK, Richards NGJ, Schuster SM. Glutamine-dependent nitrogen transfer in Escherichia coli asparagine synthetase B: searching for the catalytic triad. J Biol Chem. 1994;269:7450–7457. [PubMed] [Google Scholar]
- 52.Boehlein SK, Richards NGJ, Schuster SM. Arginine 30 and asparagine 74 have functional roles in the glutamine dependent activities of Escherichia coli asparagine synthetase B. J. Biol. Chem. 1994;269:26789–26795. [PubMed] [Google Scholar]
- 53.Zalkin H, Smith JL. Enzymes using glutamine as an amide donor. Adv Enzymol Relat Areas Mol Biol. 1998;72:87–144. doi: 10.1002/9780470123188.ch4. [DOI] [PubMed] [Google Scholar]
- 54.Zalkin H. The amidotransferases. Adv Enzymol Relat Areas Mol Biol. 1993;66:203–309. doi: 10.1002/9780470123126.ch5. [DOI] [PubMed] [Google Scholar]
- 55.Bera AK, Smith JL, Zalkin H. Dual role for the glutamine phosphoribosyl-phosphate amidotransferase ammonia channel: interdomain signaling and intermediate channeling. J Biol Chem. 2000;275:7975–7979. doi: 10.1074/jbc.275.11.7975. [DOI] [PubMed] [Google Scholar]
- 56.Badet-Denisot MA, René L, Badet B. Mechanistic investigations on glucosamine-6-phosphate synthase. Bull Soc Chim Fr. 1993;130:249–255. [Google Scholar]
- 57.Hutson RG, Kitoh T, Moraga-Amador D, Cosic S, Schuster SM, Kilberg MS. Amino acid control of asparagine synthetase: relation to asparaginase resistance in human leukemia cells. Am J Physiol. 1997;272:C1691–C1699. doi: 10.1152/ajpcell.1997.272.5.C1691. [DOI] [PubMed] [Google Scholar]
- 58.Aslanian AM, Kilberg MS. Multiple adaptive mechanisms affect asparagine synthetase substrate availability in asparaginase-resistant MOLT-4 human leukemia cells. Biochem J. 2001;357:58–67. doi: 10.1042/0264-6021:3580059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishiyama M, Tominaga H, Shiga M, Sasamoto K, Ohkura Y, Ueno K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol Pharm Bull. 1996;19:1518–1520. doi: 10.1248/bpb.19.1518. [DOI] [PubMed] [Google Scholar]
- 60.Leslie M, Case MC, Hall AG, Coulthard SA. Expression levels of asparagine synthetase in blasts from children and adults with acute lymphoblastic leukemia. Br J Haematol. 2006;132:740–742. doi: 10.1111/j.1365-2141.2005.05945.x. [DOI] [PubMed] [Google Scholar]
- 61.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 62.Butcher RA, Schreiber SL. Using genome-wide transcriptional profiling to elucidate small molecule mechanisms. Curr Opin Chem Biol. 2005;9:25–30. doi: 10.1016/j.cbpa.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 63.Smukste I, Stockwell BR. Advances in chemical genetics. Annu Rev Genomics Hum Genet. 2005;6:261–286. doi: 10.1146/annurev.genom.6.080604.162136. [DOI] [PubMed] [Google Scholar]
- 64.Bradford MM. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 65.Thompson JS, Edmonds OP. Safety aspects of handling the potent allergen FDNB. Ann Occup Hyg. 1980;23:27–33. doi: 10.1093/annhyg/23.1.27. [DOI] [PubMed] [Google Scholar]
- 66.Orth DL. HPLC determination of taurine in sports drinks. J Chem Educ. 2001;78:791–792. [Google Scholar]
- 67.Morton RC, Gerber GE. Amino acid analysis by dinitrophenylation and reverse-phase high-pressure liquid chromatography. Anal Biochem. 1988;170:220–227. doi: 10.1016/0003-2697(88)90111-x. [DOI] [PubMed] [Google Scholar]
- 68.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 69.Stewart JJP. MOPAC: a semiempirical molecular orbital program. J Comput Aided Mol Des. 1990;4:1–45. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 70.Purvis GD., III On the use of isovalued surfaces to determine molecular shape and reaction pathways. J Comput Aided Mol Des. 1991;5:55–80. doi: 10.1007/BF00173470. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.