

Abstract
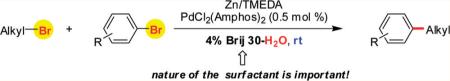
Negishi-like cross-couplings between (functionalized) alkyl and aryl bromides are described. Despite the fact that organozinc reagents are intolerant of water, their formation as well as their use in an aqueous micellar environment is discussed herein. Each component of this complex series of events leading up to C–C bond formation has an important role which has been determined insofar as the type of zinc, amine ligand, surfactant, and palladium catalyst are concerned. In particular, the nature of the surfactant has been found to be crucial in order to obtain synthetically useful results involving highly reactive, moisture-sensitive organometallics. Neither organic solvent nor heat is required for these cross-couplings to occur; just add water.
INTRODUCTION
Cross-couplings between aryl-sp2 and alkyl-sp3 centers have been intensely studied by many groups, and important advances have been achieved.1 Traditionally, this type of transformation has been performed by reacting preformed organometallic species (zinc,2,3 magnesium,2,4 boron,2,5 tin,2,6 and silicon2,7) with an appropriate aryl halide (eqs I and II, Scheme 1). More recently, reductive8 and oxidative9 couplings, as well as C–H activation10 of arenes (eq III) have been developed. A conceptually different approach that relies on simultaneous selective insertion of main-group (Mmg) and transition (Mt) metals in two different carbon–halogen bonds was presented recently by von Wangelin (Kumada-like cross couplings in THF)11 and by our group as well (Negishi-like cross couplings in water under micellar catalysis12) (eq IV; X = Br, X′ = I). Chemoselective metal insertion is based on the preferential reaction of main-group metals with sp3-halogen bonds, while transition metals tend to react more quickly with sp2-halogen centers. In order to realize good overall conversions and isolated yields, we originally used alkyl iodides rather than alkyl bromides, since the latter react more slowly, if at all, with zinc metal at room temperature. Herein we wish to report a study leading to a general solution for cross-coupling reactions focused on alkyl bromides, with aryl bromides as reaction partners, in water at room temperature using micellar nanoparticle technology (eq IV).
Scheme 1.

Aryl–Alkyl Cross-Couplings
RESULTS AND DISCUSSION
Role of the Surfactant
During development of these couplings between alkyl iodides and aryl bromides, PTS was chosen as the lead amphiphile, although the surfactant did not appear to have a major impact on the results obtained.12 However, in the absence of surfactant (i.e., “on water”), the product is formed to a very limited extent and at a much lower rate where, presumably, the rate-determining step is considered to be zinc insertion into the alkyl halide. On the other hand, in the presence of far more reactive benzylic chlorides, couplings can be performed “on water” in only a few hours (3–8 h).13 Initial attempts to utilize less reactive alkyl bromides in cross-coupling reactions with aryl bromides were unsuccessful, in contrast to reactions of alkyl iodides (Scheme 2) where significant byproduct formation was observed under micellar catalysis. Nonetheless, insertion clearly did take place at ambient temperature, while in organic solvents direct insertion of zinc into alkyl bromides occurs, without exception, only with heating.14 Thus, while after 36 h a 91% yield of product 1 is obtained starting with 1-iododecane, only a 46% yield could be isolated after 72 h using 1-bromodecane. Interestingly, under micellar conditions, competitive zinc insertion takes place with the activated aryl bromide (ethyl 4-bromobenzoate), leading to reduction and homocoupling byproducts that take their toll on the overall isolated yield (Scheme 2).
Scheme 2.
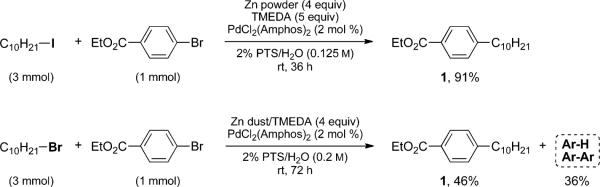
Comparison Reactions between Iodo- and Bromodecane
The effect of varying the surfactant was studied next, choosing as a model reaction the coupling between bromocyclohexane and ethyl 4-bromobenzoate in the presence of excess diamine and zinc dust (Figure 1). Surprisingly, the product was formed in moderate yield (50%, by GC) even in the absence of surfactant. Different percentages of several surfactants (2 and 4 wt %), including PTS, solutol, cremophore, and Triton X-100, all gave comparable results when used in pure water, whereas CTAC, TPGS, and Brij 35 are actually detrimental to the formation of the desired product. Using these latter surfactants, significant amounts of ethyl benzoate and biphenyl were detected. After intensive further screening, it was found that inexpensive and commercially available Brij 30, 4 wt % in water, enables the reaction to proceed with good selectivity and in high isolated yield (90%).
Figure 1.

Influence of the surfactant on the reaction profile.
The major difference between Brij 30 and the other surfactants examined is the size of its micelles. While all other surfactants have a size ranging between 10 and 25 nm, as determined by dynamic light scattering measurements (DLS), significantly larger micelles are formed in water by Brij 30 (ca. 110 nm), reflecting its shorter hydrophilic portion (Table 1). These observations include Brij 35, which, notwithstanding the fact that its lipophilic core is identical with that of Brij 30, leads to the lowest level of cross-coupling. As is the case for all surfactants examined in water to date, this larger diameter surfactant in its surface collisions with zinc may be supplying a greater “payload” of alkyl halide. Its greater diameter may also provide a more extensive buffering of the newly forming RZnX to the surrounding water, thereby increasing opportunities for this water-sensitive reagent to gain entry into the hydrophobic micellar core.
Table 1.
Diameter of Micelles Associated with Surfactants in Watera
| entry | surfactant | size of micelles (nm)a |
|---|---|---|
| 1 | Triton X-100 | 10 |
| 2 | Solutol HS 15 | 13 |
| 3 | Cremophor EL | 16 |
| 4 | TPGS | 13 |
| 5 | PTS-600 | 25 |
| 6 | Brij 35 | 15 |
| 7 | Brij 30 | 110 |
Measured by dynamic light scattering (DLS).
Optimization of Conditions
The medium of Brij 30 in water (4 wt %) was then utilized for further optimization studies with various alkyl bromides (Table 2). Noteworthy are the observations that lower catalyst loadings (0.5 vs 2 mol %) and more globally concentrated solutions (0.330 vs 0.125 M) can be employed for cross-couplings of alkyl bromides compared to the corresponding iodides.12 Additionally, the Brij 30–H2O medium is so effective that less TMEDA (activating agent) is needed to assist with zinc insertion into alkyl bromides relative to that required with the PTS–H2O system.12 Bromocyclohexane (2.5 equiv) reacted smoothly with zinc powder and TMEDA (50 mol %) to give the product 2 in 86% yield (entry 1; 2 mmol scale). With additional TMEDA no improvement was observed. Switching to zinc dust, which is somewhat more reactive than zinc powder, the yield was decreased slightly, due to faster consumption of the alkyl halide. With ethyl 4-bromobutyrate, zinc dust is required, and more TMEDA (1 equiv) is needed to achieve a 72% yield of 3 (entry 3). An excess of either TMEDA or bromo ester (3 equiv) gave lower yields of product 3. The same observations were made for ethyl 6-bromohexanoate (entry 5). The reaction with bromodecane (1.5 equiv) led to 56% of desired product 1 (entry 6) when an excess of TMEDA (3 equiv) was present. Control experiments confirm the crucial role of TMEDA (entries 1 vs 2, 3 vs 4, 6 vs 7) as well as Brij 30 (entries 1, 3, and 6). Secondary alkyl bromides react faster than do primary bromides, although primary bromides undergo insertion more rapidly when a functional group is close to the C–Br bond such that intramolecular coordination to zinc is possible. Incomplete reactions allowed for unreacted aryl bromide to be recovered from the reaction mixture, as little to no byproduct formation is common to these room-temperature cross-couplings. Excess alkyl halide is required, since some of the derived organozinc bromide is inevitably lost as the protio-quenched material. None of the starting alkyl halide is recovered from these couplings, and only trace amounts of alkyl homocoupling have been observed.
Table 2.
Optimization of Conditions

| entry | RX | n | type of zinca | TMEDA (equiv) | product | yield (%)b | ArBr recovered (%) |
|---|---|---|---|---|---|---|---|
| 1 |
|
2.5 | powder | 0.5 | 2 | 86 (45c) | 10 |
| 2 | 0 | 36 | 65 | ||||
| 3 | EtO2C–(CH2)5–Br | 2 | dust | 1 | 3 | 72 (32c) | 18 |
| 4 | 0 | 15 | 79 | ||||
| 5 | EtO2C–(CH2)5–Br | 1.5 | dust | 1 | 4 | 65 | 22 |
| 6 | n-C10H21–Br | 1.5 | dust | 3d | 1 | 56 (18c) | 35 |
| 7 | 0d | traces | 78 |
6 mmol of zinc was used in all cases.
Isolated yield.
In the absence of Brij 30.
Run over 72 h.
Unfortunately, complete conversion was not reached although much effort has to be made to remedy this situation. Changing the concentration (to between 0.125 and 0.5 M), heating the reaction (up to 40 °C), or adding catalyst over time did not improve results and, in some cases, even gave lower yields of products. Different amines (triethylamine, pyridine, hexamethylethylenetetramine, piperidine) were tested, but only traces of product were detected. Eventually, it was postulated that ZnBr2 formed during the reaction might be interfering with the course of these cross-couplings. Indeed, addition of only 0.5 equiv of ZnBr2 at the beginning of the reaction decreased yields significantly. On the other hand, we have shown earlier that salts can have a positive impact in related Pd-catalyzed cross-couplings under micellar catalysis.15 Therefore, different salts were screened, including LiCl, LiBr, KCl, KBr, NaI, and NaBr, leading to the observation that addition of sodium chloride (2 equiv) increased the yield in each of the cross-coupled products 1–3 (Scheme 3).
Scheme 3.
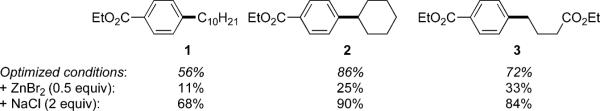
Influence of Salts
Effect of the Pd Catalyst
The catalyst PdCl2(Amphos)2 is especially important for the success of this cross-coupling reaction. It belongs to a new class of air-stable palladium catalysts synthesized in 2006 by Guram16 at Amgen. Many other catalysts (e.g., PdCl2(PPh3)2, PdCl2(PCy3)2, and PdCl2(DtBPF)2) were screened under optimized conditions, although each led to the desired product 2 in low yields (Scheme 4). Modest results were obtained with PdCl2(PR2Ar)2 analogues, although they were still far less effective than PdCl2(Amphos)2. No significant difference in reactivity was found between ligands L1 and L2, whereas substitution of tert-butyl by cyclohexyl (L3) on phosphorus affected the yield dramatically (18%). Although the higher reactivity of PdCl2(Amphos)2 relative to that of this catalyst's bis-desamino analogue (L1) might at first be attributed to electronic effects, this alone cannot account for the observed results using more electron-rich ligands; the explanation is obviously unrelated to steric effects (compare results with Amphos vs L3). Whatever the reason(s) behind the success based on the Amphos ligand, there may be a correlation between its dimethylamino group and the facility with which Et3N mediates several other metal-catalyzed couplings in aqueous surfactant media.17 Thus, the Me2N residue may be increasing the polarity of the catalyst, making it more hydrotropic and, hence, easier for it to negotiate transport into, and between, micellar arrays. Although PdCl2(Amphos)2 was routinely employed at the 0.5 mol % level (as in Scheme 4), dropping the catalyst loading to 0.1 mol % did not significantly decrease yield (80% vs 86%).
Scheme 4.
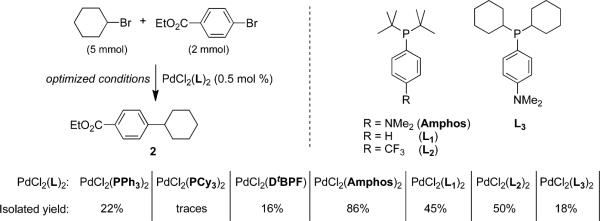
Influence of the Catalyst
Isomerization
Palladium and nickel-catalyzed coupling reactions of secondary alkyl organometallic compounds are known to oftentimes give mixtures of linear and branched products.18 The reaction has been reinvestigated recently by Molander19 and Buchwald.20 Under our micellar conditions, using four different secondary alkyl bromides (Table 3), no undesired linear product was detected by NMR of the crude reaction mixture. Surprisingly, a mixture of branched products (ca. 1:1) was obtained with 3-bromopentane as coupling partner (entry 3). The challenging case of ethyl 4-bromovalerate gave a single branched product in 82% isolated yield (entry 4). Thus, it appears that no isomerization between primary and secondary carbons occurs and that functional groups can suppress any isomerization between two secondary centers.
Table 3.
Couplings of Secondary Alkyl Bromides
| entry | RBr | product | yield (%) | branched/lineara |
|---|---|---|---|---|
| 1 |
|
5 | 69b,c | 100:0 |
| 2 |
|
6 | 78b | 100:0 |
| 3 |
|
7 | 86b,d | - |
| 4 |
|
8 | 82e,f | 100:0 |
Determined by NMR.
NMR yield of the crude reaction.
5 mmol of 2-bromopropane was used.
Obtained as a 1:1 mixture of branched products.
Isolated yield.
Obtained as only one branched product.
Scope
Table 4 illustrates representative aryl bromides that have been alkylated. As anticipated for organozinc chemistry in general, functionality present in both reaction partners is tolerated, including halides (entries 2–4), esters (entries 1, 2, and 5), nitrile (entry 6), and ketones (entries 3 and 5). The case of an especially electron-rich aryl bromide (entry 7) appeared to couple to the same extent as the more electron-deficient examples studied, suggesting a lack of sensitivity to oxidative addition as the rate-determining step. In most cases, the isolated yield is modest as shown, although this reflects the extent of conversion and not the cleanliness of these reactions. It is likely that the use of salts in the medium, in particular NaCl as noted previously (Scheme 3), would afford improved results.
Table 4.
Scope of the Cross-Coupling Reaction
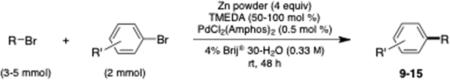
Isolated yield.
3 equiv of TMEDA were used (72 h).
As for limitations noted to date, several substrates were found to be unresponsive under these micellar conditions (Scheme 5). Thus, bromocyclopropane as well as α- and β-bromo ester derivatives are quantitatively protio-quenched, with the aryl bromide being fully recoverable. A nitrile residue on an alkyl bromide is not well tolerated and gives a low yield of cross-coupled product. All cases examined where an aryl bromide contains a relatively acidic hydrogen comparable in pKa to that of water also fail to afford the desired product in yields >10%. These observations are independent of the surfactant involved.
Scheme 5.
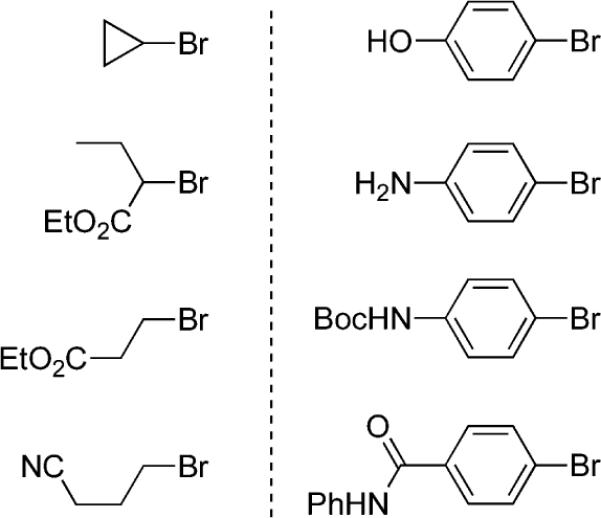
Substrates That Give Low Yields
CONCLUSIONS
In summary, the direct cross-coupling between alkyl and aryl bromides has been achieved in water at room temperature. Required for success in these reactions was a switch in surfactant from PTS to Brij 30, notwithstanding a seemingly modest change in going from alkyl iodides to bromides, an observation that further attests to our limited understanding of the vagaries of micellar catalysis in synthesis. Nonetheless, unprecedented zinc insertion has been found to take place and was used, to great advantage, under remarkably mild room temperature conditions in the presence of surfactant Brij 30. The results obtained from this nanotechnology raise many questions not only with regard to the possibilities of performing reactive metal chemistry in water as the gross reaction medium but also, in a broader sense, just what the new “rules” are for doing transition-metal-catalyzed couplings under the influence of the hydrophobic effect.
EXPERIMENTAL SECTION
General Considerations
Reactions were performed in a 10 mL round-bottom flask under an argon atmosphere containing a Teflon-coated stir bar and septum. All commercially available reagents were used without further purification. Water was degassed with argon. Brij 30 (CAS No. 9002-92-0) is available from Acros (Catalog No. 21672). Zinc powder 99.9% (−325 mesh) and zinc dust 97.5% (−325 mesh) were purchased from Strem Chemicals (Catalog Nos. 93-3060 and 93-3056) and stored in a glovebox. PdCl2(Amphos)2 (CAS No. 887919-35-9) was obtained from Johnson Matthey (Pd-132, Catalog No. C4138). Column chromatography was preformed using Silicycle Silia-P 60 Å flash silica gel. GC analyses were recorded on a Hewlett-Packard HP 6890 chromatograph equipped with a capillary column HP-1 (). 1H and 13C NMR spectra were measured on a Varian Unity Inova-500 (500 and 125 MHz, respectively) spectrometer at ambient temperature. Proton NMR data are given as follows: chemical shift in ppm referenced from residual solvent peak (CDCl3, 7.26 ppm), multiplicity (s = singlet; d = doublet; t = triplet; q = quartet; quintuplet = qt; sext = sextuplet; spt = septuplet; m = multiplet), coupling constant (Hz), and integration. 13C chemical shifts are given in ppm from the solvent resonance employed as the internal standard (CDCl3, 77.00 ppm). Mass spectral data were acquired on either a VF Autospec or an analytical VG-70-250 HF instrument.
Solution of Brij 30
Deionized water (250 mL) was degassed by bubbling argon through it for 4 h. Brij 30 (10 g) was added into an empty 250 mL round-bottom flask, and then degassed water (240 mL) was added to the flask. The solution was stirred overnight under an argon atmosphere.
General Procedure for Cross-Coupling Reactions between Alkyl and Aryl Bromides
In a 10 mL round-bottom flask under argon containing zinc (390 mg, 6 mmol) and PdCl2(Amphos)2 (7 mg, 0.01 mmol) was added a solution of 4 wt % Brij 30 (5 mL). N,N,N′,N′-Tetramethylethylenediamine (TMEDA, 116–696 mg, 1–6 mmol) was added at room temperature followed by the addition of the alkyl halide (3–5 mmol) and the aryl bromide (2 mmol). The flask was stirred vigorously at room temperature for 48 h unless indicated otherwise. The reaction mixture was then filtered through a plug of silica (10 g), completely removing excess zinc, all solids, and the surfactant, which remains at the top of the absorbent. The column was washed with diethyl ether (70 mL) into a 100 mL flask containing 2 g of silica, and the solvents were removed under vacuum. The resulting dry, crude silica was introduced on top of a silica gel chromatographic column to purify the product.
Ethyl 4-Decylbenzoate (1)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 696 mg, 6 mmol), 1-bromodecane (660 mg, 3 mmol), and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (324 mg) was obtained in 56% yield in 72 h. By adding NaCl (224 mg, 4 mmol) to the reaction mixture in water, the product (394 mg) was obtained in 68% yield after 72 h. 1H NMR (400 MHz): δ 7.95 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 4.35 (q, J = 7.2 Hz, 2H), 2.64 (t, J = 7.3 Hz, 2H), 1.62 (m, 3H), 1.39 (t, J = 7.2 Hz, 3H), 1.25 (m, 14H), 0.89 (t, J = 7.2 Hz, 2H). 13C NMR (100 MHz): δ 166.9, 148.6, 129.7, 128.5, 128.1, 60.9, 36.2, 32.1, 31.8, 29.8, 29.7, 29.6, 29.5, 29.4, 22.9, 14.5, 14.3. HRMS (C19H30O2): m/z calcd 290.2246, found 290.2252.
Ethyl 4-Cyclohexylbenzoate (2)
From zinc powder (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 116 mg, 1 mmol), bromocyclohexane (818 mg, 5 mmol), and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (400 mg) was obtained in 86% yield. When NaCl (224 mg, 4 mmol) was added to the reaction mixture in water, the product (417 mg) was obtained in 90% yield. The corresponding spectroscopic data matched those reported in the literature for ethyl 4-cyclohexylbenzoate.21 HRMS (C15H20O2): m/z calcd 232.1463, found 232.1468.
Ethyl 4-[4-(Ethoxycarbonyl)phenyl]butanoate (3; CAS No. 38632-65-4)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 232 mg, 2 mmol), ethyl 4-bromobutyrate (780 mg, 4 mmol), and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (380 mg) was obtained in 72% yield. When NaCl (224 mg, 4 mmol) was added to the reaction mixture in water, the product (443 mg) was obtained in 84% yield. HRMS (C15H20O4): m/z calcd 264.1362, found 263.1355. The corresponding spectroscopic data matched those reported in the literature for ethyl 4-[4-(ethoxycarbonyl)phenyl]butanoate.22
Ethyl 4-[4-(Ethoxycarbonyl)phenyl]hexanoate (4)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 232 mg, 2 mmol), ethyl 6-bromohexanoate (669 mg, 3 mmol), and ethyl 2-bromobenzoate (458 mg, 2 mmol), the product (379 mg) was obtained in 65% yield. 1H NMR (400 MHz): δ 7.95 (d, J = 8.2 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 4.35 (q, J = 7.2 Hz, 2H), 4.11 (q, J = 7.2 Hz, 2H), 2.66 (t, J = 7.3 Hz, 2H), 2.27 (t, J = 7.3 Hz, 2H), 1.66 (m, 4H), 1.37 (m, 5H), 1.24 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz): δ 173.8, 166.8, 148.1, 128.8, 128.5, 128.9, 60.9, 60.4, 35.9, 34.4, 30.9, 28.8, 24.9, 14.5, 14.4. HRMS (C17H24O4): m/z calcd 292.1675, found 292.1670.
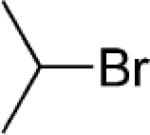
Ethyl 4-Isopropylbenzoate (5; CAS No. 19024-50-1)
From zinc powder (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 116 mg, 1 mmol), 2-bromopropane (615 mg, 5 mmol) and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (265 mg) was obtained in 69% yield as an inseparable mixture from ethyl 4-bromobenzoate. The corresponding spectroscopic data matched those reported in the literature for ethyl 4-isopropylbenzoate.9 HRMS (C12H16O2): m/z calcd 192.1150, found 192.1152.
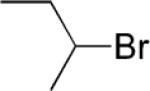
Ethyl 4-sec-Butylbenzoate (6; CAS No. 860695-72-3)
From zinc powder (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 116 mg, 1 mmol), 2-bromobutane (548 mg, 4 mmol) and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (321 mg) was obtained in 78% yield as an inseparable mixture from ethyl 4-bromobenzoate. HRMS (C13H18O2): m/z calcd 206.1307, found 206.1308.
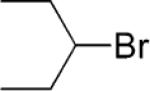
Ethyl 4-(Pent-3-yl)benzoate (7)
From zinc powder (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 116 mg, 1 mmol), 3-bromopentane (596 mg, 4 mmol), and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (378 mg) was obtained in 86% yield as an inseparable mixture from ethyl 4-bromobenzoate. HRMS (C14H20O2): m/z calcd 220.1463, found 220.1460.
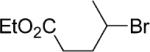
Ethyl 4-[4-(Ethoxycarbonyl)phenyl]pentanoate (8)
From zinc powder (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 116 mg, 1 mmol), ethyl 4-bromovalerate (836 mg, 4 mmol), and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (455 mg) was obtained in 82% yield. 1H NMR (400 MHz): δ 7.95 (d, J = 8.2 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 4.35 (q, J = 7.2 Hz, 2H), 4.07 (q, J = 7.2 Hz, 2H), 2.78 (sxt, J = 7.3 Hz, 1H), 2.14 (m, 2H), 1.90 (m, 2H), 1.38 (t, J = 7.3 Hz, 3H), 1.27 (d, J = 7.2 Hz, 3H), 1.18 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz): δ 173.3, 166.4, 151.6, 129.7, 128.5, 126.9, 60.7, 60.2, 39.3, 32.8, 32.3, 21.8, 14.3, 14.1. HRMS (C16H22O4): m/z calcd 278.1518, found 278.1515.
Ethyl 4-[4-(Ethoxycarbonyl)phenyl]pentanoate (9)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 232 mg, 2 mmol), ethyl 5-bromovalerate (627 mg, 3 mmol), and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (372 mg) was obtained in 67% yield. 1H NMR (400 MHz): δ 7.95 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 4.35 (q, J = 7.2 Hz, 2H), 4.11 (q, J = 7.2 Hz, 2H), 2.66 (t, J = 7.3 Hz, 2H), 2.30 (t, J = 7.3 Hz, 2H), 1.62 (m, 4H), 1.37 (t, J = 7.2 Hz, 3H), 1.22 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz): δ 173.7, 166.8, 147.7, 129.8, 128.5, 128.3, 60.9, 60.4, 35.8, 34.3, 30.7, 24.7, 14.5, 14.4. HRMS (C16H22O4): m/z calcd 278.1518, found 278.1520.
5-[4-(Ethoxycarbonyl)phenyl]chloropentane (10)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 232 mg, 2 mmol), 1-bromo-5-chloropentane (742 mg, 4 mmol), and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (396 mg) was obtained in 78% yield. 1H NMR (400 MHz): δ 7.96 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 4.38 (q, J = 7.2 Hz, 2H), 3.52 (t, J = 7.2 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 1.81 (qt, J = 7.2 Hz, 2H), 1.64 (qt, J = 7.2 Hz, 2H), 1.52 (qt, J = 7.2 Hz, 2H), 1.37 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz): δ 166.9, 148.6, 129.7, 128.5, 128.1, 60.9, 45.2, 36.0, 32.6, 31.1, 28.6, 14.3. HRMS (C14H19ClO2): m/z calcd 254.1074, found 254.1079.
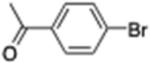
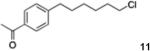
6-(4′-Acetophenone)chlorohexane (11)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 232 mg, 2 mmol), 1-bromo-6-chlorohexane (798 mg, 4 mmol), and 4′-bromoacetophenone (398 mg, 2 mmol), the product (380 mg) was obtained in 80% yield. 1H NMR (400 MHz): δ 7.83 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 3.52 (t, J = 7.2 Hz, 2H), 2.68 (t, J = 7.2 Hz, 2H), 2.57 (s, 3H), 1.75 (qt, J = 7.2 Hz, 2H), 1.64 (qt, J = 7.2 Hz, 2H), 1.47 (qt, J = 7.2 Hz, 2H), 1.35 (qt, J = 7.2 Hz, 2H). 13C NMR (100 MHz): δ 198.0, 148.6, 135.2, 128.6, 128.7, 45.2, 36.0, 32.6, 31.1, 28.6, 26.8, 26.7. HRMS (C14H19ClO): m/z calcd 238.1124, found 238.1120.
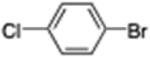
Ethyl 4-(4-Chlorophenyl)butanoate (12; CAS 3435-98-1)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 232 mg, 2 mmol), ethyl 4-bromobutyrate (780 mg, 4 mmol), and 4-bromochlorobenzene (398 mg, 2 mmol), the product (339 mg) was obtained in 75% yield. The corresponding spectroscopic data matched those reported in the literature for ethyl 4-(4-chlorophenyl)-butanoate.12a HRMS (C12H15ClO2): m/z calcd 226.0761, found 226.0767.
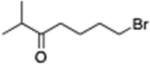
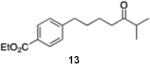
7-[4-(Ethoxycarbonyl)phenyl]2-methylheptan-3-one (13)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 232 mg, 2 mmol), 7-bromo-2-methylheptan-3-one (824 mg, 4 mmol), and ethyl 4-bromobenzoate (458 mg, 2 mmol), the product (463 mg) was obtained in 84% yield. 1H NMR (400 MHz): δ 7.94 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 4.35 (q, J = 7.2 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 2.56 (spt, J = 7.2 Hz, 1H), 2.45 (t, J = 7.2 Hz, 2H), 1.60 (m, 4H), 1.38 (t, J = 7.2 Hz, 3H), 1.08 (d, J = 7.2 Hz, 6H). 13C NMR (100 MHz): δ 216.2, 166.8, 147.9, 129.8, 128.5, 60.9, 41.0, 40.2, 36.0, 30.9, 23.5, 18.4, 14.5. HRMS (C17H24O3): m/z calcd 276.1725, found 276.1722.
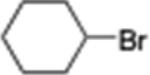
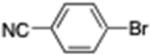
4-Cyclohexylbenzonitrile (14; CAS 27634-88-4)
From zinc powder (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 116 mg, 1 mmol), bromocyclohexane (818 mg, 5 mmol), and 4-bromobenzonitrile (364 mg, 2 mmol), the product (255 mg) was obtained in 69% yield. 1H NMR (400 MHz): δ 7.57 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 2.55 (m, 1H), 1.84 (m, 4H), 1.38 (m, 4H), 1.26 (m, 2H). 13C NMR (100 MHz): δ 153.7, 132.4, 127.8, 119.4, 109.7, 44.9, 34.2, 26.8, 26.1. HRMS (C13H15N): m/z calcd 185.1204, found 185.1201. The corresponding spectroscopic data matched those reported in the literature for 4-cyclohexylbenzonitrile.23
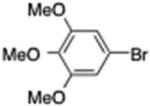
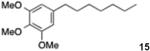
5-Heptyl-1,2,3-trimethoxybenzene (15)
From zinc dust (390 mg, 6 mmol), N,N,N′,N′-tetramethylethylenediamine (TMEDA, 448 mg, 3 mmol), 1-bromoheptane (716 mg, 4 mmol), and 5-bromo-1,2,3-trimethoxybenzene (494 mg, 2 mmol), the product (372 mg) was obtained in 70% yield. 1H NMR (400 MHz): δ 6.54 (s, 2H), 3.83 (s, 9H), 2.40 (t, J = 7.2 Hz, 2H), 1.65–1.55 (m, 2H), 1.48–1.28 (m, 8H), 0.90 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz): δ 153.2, 137.7, 105.8, 60.8, 56.1, 36.0, 31.8, 31.2, 29.3, 29.2, 22.7, 14.3. HRMS (C16H26O3): m/z calcd 266.1881, found 266.1881.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the NIH for support (GM 86485) and Johnson Matthey for generously supplying several palladium catalysts, including PdCl2(Amphos)2 (Pd-132, Catalog No. C4138). Dr. S. Ghorai is warmly acknowledged for providing DLS data.
Biography

Bruce Lipshutz was born on December 10, 1951, in the Washington Heights section of Manhattan. He attended the Bronx High School of Science (1969) and then went upstate as an undergraduate at Harpur College within the State University of New York at Binghamton. His instructor for sophomore organic chemistry was Howard Alper, with whom he eventually did senior honors research, defending his thesis in 1973. Ph.D. studies were subsequently carried out at Yale with Harry Wasserman (1977), prior to a two-year stint as an American Cancer Society Postdoctoral Fellow at Harvard in the labs of E. J. Corey. At age 27, he began his professorial career at UC Santa Barbara in 1979 and was promoted to Associate Professor in 1984 and Professor in 1987. During these early years he received several awards for research, including those in 1984 from the Alfred P. Sloan Foundation, and the Camille & Henry Dreyfus Foundation (Teacher-Scholar Award). In 1997, he received an American Chemical Society Cope Scholar Award. He has also been the recipient of the two top teaching awards at UCSB: the Harold J. Plous Award in 1984 given each year to a single Assistant Professor, and a Distinguished Faculty Teaching Award in 2002. Most of the research done in the Lipshutz group has been in the areas of synthetic methods development and natural products partial or total synthesis. However, over the past five years, much of the group's effort has turned to green chemistry, focused in particular on “designer” surfactants that enable reactions to be run in water at room temperature under micellar catalysis conditions. These reactions include transition-metal-catalyzed cross-couplings, as well as processes based on organocatalysis, where the goal is to get organic solvents out of organic reactions. The promise offered by this technology, that industrial labs will consider switching from massive usage of organic solvents as reaction media to water as the gross medium, has very recently been acknowledged in the form of a 2011 Presidential Green Chemistry Challenge Award, given by the U.S. Environmental Protection Agency.
Footnotes
Supporting Information Figures giving product spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).de Meijere A, Diederich F. Metal Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]
- (2).(a) Netherton MR, Fu GC. Adv. Synth. Catal. 2004;346:1525. [Google Scholar]; (b) Frisch A, Beller M. Angew. Chem., Int. Ed. 2005;44:674. doi: 10.1002/anie.200461432. [DOI] [PubMed] [Google Scholar]; (c) Rudolph A, Lautens M. Angew. Chem., Int. Ed. 2009;2656;48 doi: 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]; (d) Nakamura M, Ito. S. In: Modern Arylation Methods. Ackermann L, editor. Wiley-VCH; Weinheim, Germany: 2009. p. 155. [Google Scholar]
- (3).(a) Zhou J, Fu GC. J. Am. Chem. Soc. 2004;126:1340. doi: 10.1021/ja039889k. [DOI] [PubMed] [Google Scholar]; (b) Fischer C, Fu GC. J. Am. Chem. Soc. 2005;127:4594. doi: 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]; (c) Arp FO, Fu GC. J. Am. Chem. Soc. 2005;127:10482. doi: 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]; (d) Smith SW, Fu GC. J. Am. Chem. Soc. 2008;130:12645. doi: 10.1021/ja805165y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nakamura M, Ito S, Matsuo K, Nakamura E. Synlett. 2005:1794. [Google Scholar]
- (4).(a) Cahiez G, Avedissian H. Synthesis. 1998:1199. [Google Scholar]; (b) Fürstner A, Leitner A, Méndez M, Krause H. J. Am. Chem. Soc. 2002;124:13856. doi: 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]; (c) Fürstner A, Leitner A. Angew. Chem., Int. Ed. 2002;41:609. [Google Scholar]; (d) Martin R, Fürstner A. Angew. Chem., Int. Ed. 2004;43:3955. doi: 10.1002/anie.200460504. [DOI] [PubMed] [Google Scholar]; (e) Nakamura M, Matsuo K, Ito S, Nakamura E. J. Am. Chem. Soc. 2004;126:3686. doi: 10.1021/ja049744t. [DOI] [PubMed] [Google Scholar]; (f) Nagano T, Hayashi T. Org. Lett. 2004;6:1297. doi: 10.1021/ol049779y. [DOI] [PubMed] [Google Scholar]; (g) Bedford RB, Bruce DW, Frost R;M, Hird M. Chem. Commun. 2005:4161. doi: 10.1039/b507133j. [DOI] [PubMed] [Google Scholar]; (h) Bedford RB, Betham M, Bruce DW, Danopoulos AA, Frost R, Hird M. J. Org. Chem. 2006;71:1104. doi: 10.1021/jo052250+. [DOI] [PubMed] [Google Scholar]; (i) Cahiez G, Habiak V, Duplais C, Moyeux A. Angew. Chem., Int. Ed. 2007;46:4364. doi: 10.1002/anie.200700742. [DOI] [PubMed] [Google Scholar]; (j) Cahiez G, Duplais C, Moyeux A. Org. Lett. 2007;9:3253. doi: 10.1021/ol7016092. [DOI] [PubMed] [Google Scholar]; (k) Sherry BD, Fürstner A. Acc. Chem. Res. 2008;41:1500. doi: 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]; (l) Vechorkin O, Hu X. J. Am. Chem. Soc. 2009;131:9756. doi: 10.1021/ja9027378. [DOI] [PubMed] [Google Scholar]; (m) Lou S, Fu GC. J. Am. Chem. Soc. 2010;132:1264. doi: 10.1021/ja909689t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Ishiyama T, Abe S, Miyaura N, Suzuki A. Chem. Lett. 1992:691. [Google Scholar]; (b) Zhou J, Fu GC. J. Am. Chem. Soc. 2004;126:1340. doi: 10.1021/ja039889k. [DOI] [PubMed] [Google Scholar]; (c) González-Bobes F, Fu GC. J. Am. Chem. Soc. 2006;128:5360. doi: 10.1021/ja0613761. [DOI] [PubMed] [Google Scholar]; (d) Duncton MAJ, Estiarte MA, Tan D, Kaub C, O'Mahony DJR, Johnson RJ, Cox M, Edwards WT, Wan M, Kincaid J, Kelly MG. Org. Lett. 2008;10:3259. doi: 10.1021/ol8011327. [DOI] [PubMed] [Google Scholar]; (e) Saito B, Fu GC. J. Am. Chem. Soc. 2007;129:9602. doi: 10.1021/ja074008l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Saito B, Fu GC. J. Am. Chem. Soc. 2008;130:6694. doi: 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]; (g) Lundin PM, Fu GC. J. Am. Chem. Soc. 2010;132:11027. doi: 10.1021/ja105148g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Powell DA, Maki T, Fu GC. J. Am. Chem. Soc. 2005;127:510. doi: 10.1021/ja0436300. [DOI] [PubMed] [Google Scholar]
- (7).(a) Powell DA, Fu GC. J. Am. Chem. Soc. 2004;126:7788. doi: 10.1021/ja047433c. [DOI] [PubMed] [Google Scholar]; (b) Strotman NA, Sommer S, Fu GC. Angew. Chem., Int. Ed. 2007;46:3556. doi: 10.1002/anie.200700440. [DOI] [PubMed] [Google Scholar]; (c) Dai X, Strotman NA, Fu GC. J. Am. Chem. Soc. 2008;130:3302. doi: 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]
- (8).Everson DA, Shrestha R, Weix DJ. J. Am. Chem. Soc. 2010;132:920. doi: 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]
- (9).Cahiez G, Foulgoc L, Moyeux A. Angew. Chem., Int. Ed. 2009;48:2969. doi: 10.1002/anie.200900175. [DOI] [PubMed] [Google Scholar]
- (10).(a) Chen X, Li J-J, Hao X-S, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:78. doi: 10.1021/ja0570943. [DOI] [PubMed] [Google Scholar]; (b) Deng G, Zhao L, Li C-J. Angew. Chem., Int. Ed. 2008;47:6278. doi: 10.1002/anie.200801544. [DOI] [PubMed] [Google Scholar]; (c) Ackermann L, Novak P, Vincente R, Hofman N. Angew. Chem., Int. Ed. 2009;48:6045. doi: 10.1002/anie.200902458. [DOI] [PubMed] [Google Scholar]
- (11).Czaplik WM, Mayer M, von Wangelin AJ. Angew. Chem., Int. Ed. 2009;48:607. doi: 10.1002/anie.200804434. [DOI] [PubMed] [Google Scholar]
- (12).(a) Krasovskiy A, Duplais C, Lipshutz BH. J. Am. Chem. Soc. 2009;131:15592. doi: 10.1021/ja906803t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krasovskiy A, Duplais C, Lipshutz BH. Org. Lett. 2010;12:4742. doi: 10.1021/ol101885t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Duplais C, Krasovskiy A, Wattenberg A, Lipshutz BH. Chem. Commun. 2010:562. doi: 10.1039/b922280d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krasovskaya V, Krasovskiy A, Bhattacharjya A, Lipshutz BH. Chem. Commun. 2011:5717. doi: 10.1039/c1cc11087j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Krasovskaya V, Krasovskiy A, Lipshutz BH. Chem. Asian J. 2011;6:1974. doi: 10.1002/asia.201100153. [DOI] [PubMed] [Google Scholar]
- (14).Krasovskiy A, Malakhov V, Gavryushin A, Knochel P. Angew. Chem., Int. Ed. 2006;45:6040. doi: 10.1002/anie.200601450. [DOI] [PubMed] [Google Scholar]
- (15).Lipshutz BH, Ghorai S, Leong WY, Taft BR, Krogstad DV. J. Org. Chem. 2011;76:5061. doi: 10.1021/jo200746y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ, Reider PJ. Org. Lett. 2006;8:1787. doi: 10.1021/ol060268g. [DOI] [PubMed] [Google Scholar]; (b) Guram AS, Wang X, Bunel EE, Faul MM, Larsen RD, Martinelli MJ. J. Org. Chem. 2007;72:5104. doi: 10.1021/jo070341w. [DOI] [PubMed] [Google Scholar]
- (17).Lipshutz BH, Ghorai S. Aldrichim. Acta. 2008;41:59. [Google Scholar]
- (18).(a) Tamao K, Kiso Y, Sumitani K, Kumada M. J. Am. Chem. Soc. 1972;94:9268. [Google Scholar]; (b) Hayashi T, Konishi M, Kobori Y, Kumada M, Higuchi T, Hirotsu K. J. Am. Chem. Soc. 1984;106:158. [Google Scholar]
- (19).Dreher SD, Dormer P, Sandrock D, Molander GA. J. Am. Chem. Soc. 2008;130:9257. doi: 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Han C, Buchwald SL. J. Am. Chem. Soc. 2009;131:7532. doi: 10.1021/ja902046m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Luo X, Zhang H, Duan H, Liu Q, Zhu L, Zhang T, Lei A. Org. Lett. 2007;9:4571. doi: 10.1021/ol701995t. [DOI] [PubMed] [Google Scholar]
- (22).Matsuoka T, Negi T, Otsubo T, Sakata Y, Misumi S. Bull. Chem. Soc. Jpn. 1972;45:1825. [Google Scholar]
- (23).Itou T, Yoshimi Y, Morita T, Hatanaka M, Tokunaga Y. Tetrahedron. 2009;65:263. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.