

Abstract
Polymers of norbornenyl-modified peptide-based enzyme substrates have been prepared via ring-opening metathesis polymerization (ROMP). Peptides displayed on water-soluble homopolymers retain the ability to be enzymatically processed by a disease-associated enzyme. In contrast, when the peptides are densely arrayed on a nanoparticle derived from a self-assembled amphiphilic block-copolymer, they function poorly as enzymatic substrates.
Functional biohybrid polymers and soft organic nanoparticles (NPs) that display biomolecules on a synthetic skeleton have become increasingly sophisticated in recent years.1-3 In the majority of cases, biomolecules such as peptides are displayed for the purpose of facilitating recognition events, and it is therefore desirable that they be resistant to enzymatic degradation.4 Alternatively, they may be utilized for programming dynamic nanomaterial morphology in response to enzyme-catalyzed reactions.5-12 In this study, we aimed to display peptides on brush polymers and on organic NPs via their direct polymerization in order to qualitatively assess whether such peptides are made resistant to proteolytic degradation, or whether they maintain their activity upon incorporation into polymeric materials.
To study the effect of arraying peptides as brushes conjugated to polymeric backbones or polymeric NPs, we desired a well-controlled method for their incorporation. To this end, we were inspired by the work of others who have utilized living polymerization methods for the “graft-through” construction of peptide-bearing polymers synthesized directly from monomers containing polymerizable peptide units.13-19 Importantly, polymers prepared with a graft-through approach are inherently chemically homogenous since every propagation step of polymerization leads to a displayed peptide. This is in contrast to post-polymerization modification strategies wherein less than quantitative modification of the polymer backbone can be naturally expected.7 For this work we chose to optimize ring-opening metathesis polymerization (ROMP) for the purpose of allowing the preparation of well-defined peptide substrate containing polymers and polymeric NPs. To date, ROMP has been shown to enable polymerization of short oligopeptides to variable degrees, but often with high polydispersity.14,18-20 Moreover, the graft-through incorporation of specific peptide based enzyme substrates into polymeric materials has not yet been examined to our knowledge. We reasoned an initial study of such systems would have implications for future polymeric materials designed to display peptides for reaction with enzymes, or alternatively for resistance to their environment. Therefore, we sought to answer the following key questions regarding polymeric peptide-based synthetic materials synthesized by ROMP: 1) Can graft-through peptide polymers be generated with relatively high degrees of polymerization and simultaneously low polydispersity? 2) Can peptide-brush copolymers be formulated into NPs of low polydispersity? 3) If these synthetic milestones are met, will the dense display of peptides on the resultant polymers and NPs retain or hinder the ability of the peptides to function as substrates for a class of disease-associated human proteolytic enzymes?
To accomplish our goals, we prepared water-soluble homopolymers and amphiphilic block copolymers capable of being formulated into nanoparticles (vide infra), utilizing the three model norbornenyl-peptide monomers shown in Scheme 1. All three contain a polymerizeable norbornenyl group conjugated to the N-terminus of a peptide sequence. Peptides 1 and 2 contain sequences that are known substrates of the disease-associated enzymes matrix metalloproteinases-2 and -9,21 with a water-solubilizing group (Ebes) included in the structures to promote micelle formation. Peptide 3 is a hydrophobic sequence chosen to ensure generality of the approach toward the formation of NPs as will be described (vide infra).
Scheme 1.
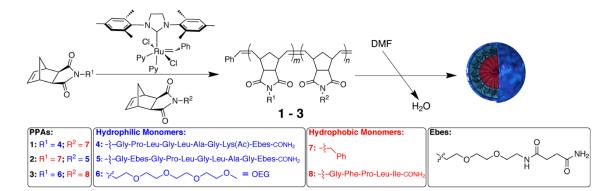
Synthesis of peptide-polymer amphiphiles (PPAs) and formulation into nanoparticles. Hydrophobic components of the PPAs are color-coded in red. Hydrophilic components of the PPAs are color-coded with blue.
We first set out to determine if peptides of this class can be efficienty polymerized via ROMP to prepare homopolymers of high degree of polymerization and with simultaneously low polydispersity. A norbornenyl-peptide monomer (Norbornene-GPLGLAG-Ebes, see Scheme 1 for Ebes structure) was polymerized using a modified 2nd generation Grubbs’ catalyst at room temperature (see ESI).16 NMR spectroscopy demonstrated that the polymerization reaction was complete as evidenced by the conversion of the norbornenyl olefinic protons to polynorbornenyl olefinic protons (see ESI). Size exclusion chromatography coupled to a multiangle laser light scattering detector (SEC-MALS) demonstrated that both high degree of polymerization (DP>100) and favorably low polydispersity (PDI=1.012) were achieved. Importantly, we found that optimized conditions, including the use of an air-free dinitrogen atmosphere, were required for optimal results with either dimethylformamide (DMF) or dichloromethane/methanol mixtures performing as effective solvents.
Next, we set out to determine if graft-through peptide ROMP could be used to prepare peptide containing amphiphilic block copolymers (peptide-polymer amphiphiles, PPAs) of low-polydispersity that could be formulated into NPs. The one-pot synthesis of PPA 1 was accomplished upon the addition of a modified 2nd generation Grubbs’ catalyst (Scheme 1) to a solution of hydrophilic peptidyl monomer 4 followed by the addition of the hydrophobic monomer 7. In addition, PPA 2 was prepared via the reverse order of addition of monomers, that is, polymerization of the hydrophobic monomer 7 followed by the hydrophilic norbornenyl peptide monomer 5, demonstrating generality in the polymerization process with respect to order of addition (Scheme 1). SEC-MALS was utilized to determine the absolute number-average molecular weight (Mn), weight-average molecular weight (Mw), degree of polymerization (DP), and polydispersity index (PDI) of PPAs 1 and 2 prior to generation of NPs (Table 1). Furthermore, we set out to determine if hydrophobic peptide-containing norbornenyl monomer 8, once polymerized, could be utilized to form the hydrophobic block of a well-defined polymer, PPA 3, which is not a specific enzyme substrate, but is structurally related to a previously published system for comparison (Scheme 1).20 Norbornenyl monomer 6, containing a short oligoethyleneglycol (OEG) chain, was used to form the hydrophilic block of PPA 3.22 SEC-MALS analysis of PPA 3, formed from block copolymerization of hydrophilic monomer 6 followed by hydrophobic peptide-containing monomer 8, indicated a low PDI (Table 1). These results indicated that the PPAs were of notably low polydispersity via these optimized polymerization conditions and compare favourably with the aforementioned system,14 similar to PPA 3.
Table 1.
Molecular weights and polydispersities of PPAs and NPs
| PPA | Mn (Da)a | (Mw/Mn)b | DPmc | DPnd | Dhe (nm) | |
|---|---|---|---|---|---|---|
| 1 | 44,630 | 1.20 | 13 | 116 | 28 | 0.062 |
| 2 | 23,570 | 1.06 | 74 | 5 | 124 | 0.020 |
| 3 | 28,130 | 1.05 | 59 | 11 | 282 | 0.053 |
Mn denotes number-average molecular weight.
Mw denotes weight-average molecular weight.
DPm denotes degree of polymerization of the first block.
DPn denotes degree of polymerization of the second block.
Dh denotes hydrodynamic diameter of the nanoparticles from each PPA.
PDI denotes polydispersity of the nanoparticles.
To formulate the PPAs into NPs, each polymer was dissolved separately in DMF followed by the slow addition of water to a final concentration of 50% by volume.23 The incipient NP suspensions were then dialyzed against water to remove DMF. Dynamic light scattering (DLS) in water was employed to determine the hydrodynamic diameter of the NPs derived from PPAs 1 through 3 (Fig. 1A). Statistical analysis of the DLS data demonstrated that the size distribution of the nanoparticles is narrow (Table 1). Transmission electron microscopy (TEM) of the nanoparticles validated the DLS data showing the presence of spherical particles (Fig. 1B and ESI). We note that the resulting micelles are observed to be stable without change in DLS, or TEM for at least 6 months at 4 °C in water.
Fig. 1.
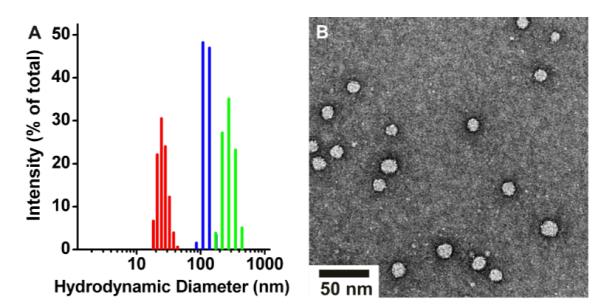
A) DLS size intensity distribution of nanoparticles derived from PPA 1 (red), PPA 2 (blue), and PPA 3 (green). (B) TEM of nanoparticles derived from PPA 1. See supporting information for TEM of other NPs.
Given the high spatial density of peptides displayed on the resultant polymers, we carried out a pilot study to determine if the peptides arrayed covalently on the polymer backbone would retain the ability to function as enzyme substrates. For these studies we chose to compare water soluble homopolymers prepared from monomer 4 to well-defined spherical micellar NPs formed from PPA 1 (Fig. 1B). Following incubation of the water soluble polymer prepared from monomer 4 with MMP-2, RP-HPLC revealed the appearance of the proteolysis product NH2-LAGK(Ac)-Ebes-CONH2 (Fig. 2, iv). Proteolysis did not occur when MMP-2 was heat denatured prior to incubation with the peptide polymer (Fig. 2B, ii) and proteolysis was markedly blunted when carried out in the presence of EDTA (Fig. 2B, iii), which is known to inhibit the enzyme via chelation of the catalytic Zn2+ ion. This demonstrated that the enzyme was indeed responsible for proteolysis of the water soluble peptidehomopolymer prepared by graft-through polymerization of the substrate.
Fig. 2.
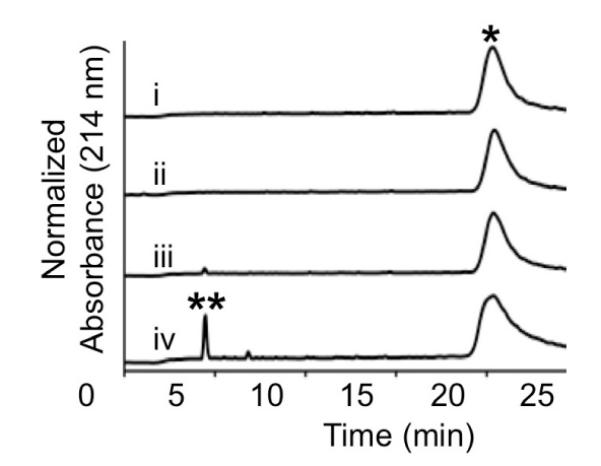
RP-HPLC chromatograms of homopolymer prepared from monomer 4 alone (i) as well as following incubation with heat-treated MMP-2 (ii), EDTA-treated MMP-2 (iii), and active MMP-2 (iv). Peak due to peptide-based brush copolymer (*); peak due to peptide cleavage product NH2-LAGK(Ac)-Ebes-CONH2 (**).
Finally, micellar NPs derived from PPA 1 were incubated with active MMP-2 and no proteolysis product could be identified by RP-HPLC under the same conditions used for processing homopolymers (see ESI). This implies that the peptide substrates arrayed on the particle are protected from protease-mediated cleavage. We hypothesize that this is due to the steric crowding of the displayed peptides on the NP scaffold that forbids the enzyme from accessing the scissile peptide bond within the displayed peptides. This is in contrast to the high activity we have observed for related systems that are responsive to MMP catalyzed cleavage when prepared via post-polymerization modification of polymers with peptides.7 Notably, these related systems were characterized by a lower density of peptides displayed, due to low conjugation efficiency inherent to such reactions. These results hint at the possibility of arranging peptides to capitalize on resistance to proteolytic degradation versus optimizing for activity and responsiveness. An extensive study focusing on the relationships between substrate spatial density, polymer/particle structure/morphology, reaction conditions, sequence identity and enzymatic activity is underway in our laboratories.
In summary, we have demonstrated several important principles crucial to the future use of peptide containing polymers and NPs prepared by graft-through polymerization procedures. Namely, we have demonstrated that homopolymers and block copolymers of high degree of polymerization and simultaneously low polydispersity can be prepared. The resulting NPs formulated via self-assembly of 3 different peptide-containing amphiphilic block co-polymers are also of low polydispersity. Blocks of appropriately chosen peptides can function as either the hydrophilic or hydrophobic block of a block-copolymer PPA. Peptide substrates of disease-associated enzymes (MMPs) displayed on a water-soluble polymer are able to be enzymatically processed. In contrast, when displayed on the NP scaffold, the same peptide is made less susceptible to proteolysis. Ongoing studies aim to address the mechanism of peptide protection when displayed on the NP scaffold and to leverage this potential for stability in the delivery of otherwise proteolytically labile peptides in an in vivo context.
Supplementary Material
Acknowledgments
We acknowledge A. Rush for TEM analysis and S. Nguyen for helpful discussions. MEH acknowledges NIH T32 (5T32EB005970) and the Dept. of Radiology for support. We acknowledge generous support through AFOSR-PECASE (FA9550-11-1-0105), ARO (W911NF-11-1-0264), NIH (NIBIB 1R01EB011633), NIH Transformative Research Award (R01HL117326) and NIH Director’s New Innovator Award (1DP2OD008724). NCG acknowledges the Henry & Camille Dreyfus Foundation for a New Faculty Award and the Alfred P. Sloan Foundation. † Electronic Supplementary Information (ESI) available.
References
- 1.Gauthier MA, Klok HA. Chem Commun. 2008:2591–2611. doi: 10.1039/b719689j. [DOI] [PubMed] [Google Scholar]
- 2.Lutz J-F, Borner HG. Prog. in Pol. Sci. 2008;33:1–39. [Google Scholar]
- 3.Gauthier MA, Gibson MI, Klok HA. Angew. Chem. Int. Ed. 2008;48:48–58. doi: 10.1002/anie.200801951. [DOI] [PubMed] [Google Scholar]
- 4.Ruoslahti E, Bhatia SN, Sailor MJ. J. Cell. Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chien MP, Rush AM, Thompson MP, Gianneschi NC. Angew. Chem. Int. Ed. 2010;49:5076–5080. doi: 10.1002/anie.201000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hahn ME, Gianneschi NC. Chem Commun. 2011;47:11814–11821. doi: 10.1039/c1cc15220c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ku TH, Chien MP, Thompson MP, Sinkovits RS, Olson NH, Baker TS, Gianneschi NC. J. Am. Chem. Soc. 2011;133:8392–8395. doi: 10.1021/ja2004736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Randolph LR, Chien MP, Gianneschi NC. Chem. Sci. 2012;3:2690–2694. doi: 10.1039/C2SC20165H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Börner HG. Macromol. Rapid Commun. 2011;32:115–126. doi: 10.1002/marc.201000646. [DOI] [PubMed] [Google Scholar]
- 10.Hughes M, Birchall LS, Zuberi K, Aitken LA, Debnath S, Javid N, Ulijn RV. Soft Matter. 2012;8:11565–11574. [Google Scholar]
- 11.Amir RJ, Zhong S, Pochan DJ, Hawker CJ. J. Am. Chem. Soc. 2009;131:13949–13951. doi: 10.1021/ja9060917. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Chen Q, Wang Z, Zhang X. Angew. Chem. Int. Ed. 2010;49:8612–8615. doi: 10.1002/anie.201004253. [DOI] [PubMed] [Google Scholar]
- 13.Ayres L, Koch K, Adams HHM, van Hest JCM. Macromolecules. 2005;38:1699–1704. [Google Scholar]
- 14.Biagini SCG, Parry AL. J. Poly. Sci. A. 2007;45:3178–3190. [Google Scholar]
- 15.Breitenkamp RB, Ou Z, Breitenkamp K, Muthukumar M, Emrick T. Macromolecules. 2007;40:7617–7624. [Google Scholar]
- 16.Conrad RM, Grubbs RH. Angew. Chem. Int. Ed. 2009;48:8328–8330. doi: 10.1002/anie.200903888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Trillo F, Dureault A, Bayley JPM, van Hest JCM, Thies JC, Michon T, Weberskirch R, Cameron NR. Macromolecules. 2007;40:6094–6099. [Google Scholar]
- 18.Maynard HD, Okada SY, Grubbs RH. Macromolecules. 2000;33:6239–6248. [Google Scholar]
- 19.Roberts KS, Sampson NS. J, Org. Chem. 2003;68:2020–2023. doi: 10.1021/jo0265737. [DOI] [PubMed] [Google Scholar]
- 20.Parry AL, Bomans PH, Holder SJ, Sommerdijk NA, Biagini SC. Angew Chem Int Ed Engl. 2008;47:8859–8862. doi: 10.1002/anie.200802834. [DOI] [PubMed] [Google Scholar]
- 21.Kessenbrock K, Plaks V, Werb Z. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertin PA, Gibbs JM, Shen CK-F, Thaxton CS, Russin WA, Mirkin CA, Nguyen ST. J. Am. Chem.Soc. 2006;128:4168–4169. doi: 10.1021/ja056378k. [DOI] [PubMed] [Google Scholar]
- 23.Riess G. Prog. in Pol. Sci. 2003;28:1107–1170. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.