

Abstract
Background
The human 9p21.3 chromosome locus has been shown to be an independent risk factor for atherosclerosis in multiple large scale genome-wide association studies, but the underlying mechanism remains unknown. We set out to investigate the potential role of the 9p21.3 locus neighboring genes, including Mtap, the two isoforms of Cdkn2a, p16Ink4a and p19Arf, and Cdkn2b in atherosclerosis using knockout mice models.
Methods and Results
Gene targeted mice for neighboring genes, including Mtap, Cdkn2a, p19Arf, and Cdkn2b, were each bred to mice carrying the human APO*E3 Leiden transgene which sensitizes the mice for atherosclerotic lesions through elevated plasma cholesterol. We found that the mice heterozygous for Mtap developed larger lesion compared to wild-type mice (49623±21650 vs. 18899±9604 μm2/section (Mean±SD); p=0.01), with similar morphology as wild type mice. The Mtap heterozygous mice demonstrated changes in metabolic and methylation profiles and CD4+ cell counts. The Cdkn2a knockout mice had smaller lesions compared to wild-type and heterozygous mice and there were no significant differences in lesion size in p19Arf and Cdkn2b mutants as compared to wild type. We observed extensive, tissue-specific compensatory regulation of the Cdkn2a and Cdkn2b genes among the various knockout mice, making the effects on atherosclerosis difficult to interpret.
Conclusions
Mtap plays a protective role against atherosclerosis, whereas Cdkn2a appears to be modestly proatherogenic. However, no relation was found between the 9p21 genotype and the transcription of 9p21 neighboring genes in primary human aortic vascular cells in vitro. There is extensive compensatory regulation in the highly conserved 9p21 orthologous region in mice.
Keywords: Atherosclerosis, 9p21 risk locus, Gene targeted mice, Methylthioadenosine Phosphorylase
The 9p21.3 region of the genome has been identified as the locus with strongest association to coronary artery disease (CAD) and myocardial infarction (MI) in multiple independent large scale genome-wide association studies (GWAS).1-3 The locus is within a 58kb region that is devoid of protein coding genes, suggestive of a regulatory function (Figure 1). Interestingly, the neighboring genes in the region include well-known tumor suppressor genes, including CDKN2A and CDKN2B.4-6 The CDKN2A locus encodes a cyclin-dependent protein kinase (CDK) inhibitory protein (CKI), p16INK4A, and a p53-regulatory protein, p19ARF. The CDKN2B gene encodes another CKI, p15INK4B. Another gene in the region is methylthioadenosine phosphorylase (MTAP), which encodes a ubiquitously expressed metabolic enzyme S-methyl-5′-thioadenosine phosphorylase7 that processes the polyamine biosynthesis byproduct in the methionine salvage pathway. Loss or inactivation of MTAP has frequently been observed in a number of different human tumors, and it has been shown to have a tumor suppressive role in a mice model.8
Figure 1. The landscape of the 9p21.3 region.

The 400kb region containing the 9p21 CAD risk region (transparent red bar) and the neighboring genes, including MTAP, P16INK4A, P19ARF, CDKN2B, and the non-coding RNA CDKN2BAS are shown along with the orthologous region of the mice genome. The exons are noted as a vertical bar and arrows denote the direction of transcription. Distance in kb (not to scale). The blue arrow marks the third intron of Mtap where the gene trap was introduced with the En2 splice acceptor with beta-geo (see Methods). The red triangle marks where the targeted mutation was introduced and exons were replaced with neo cassette in different mice models used in this study. (2=Cdkn2a KO (Serrano et al.4); 3 = p19ArfKO (Kamijo et al5); 4 = Cdkn2b KO (Latres et al.6))
Multiple studies demonstrated a potential role for cell cycle regulatory mechanisms in atherosclerosis progression. Previously, the master tumor suppressor gene p53 has been implicated in the development of atherosclerosis in apolipoprotein E (ApoE)-null mice9, 10, affecting both cell proliferation and apoptosis within the atheroma. Another tumor suppressor gene, p21Waf1, was also shown to increase the atheroma size in ApoE-null mice11, whereas the tumor suppressor p27Kip1 was shown to protect against atherosclerosis.12
Correlations of the 9p21 locus SNP genotype to differential expression of the neighboring genes have been observed in several studies with inconsistent findings.13-15 A knockout (KO) mouse model involving the entire region orthologous to the 9p21.3 CAD locus showed significant decreases in the expressed levels of Cdkn2a and Cdkn2b, and increased proliferation of primary smooth muscle cells (SMC) and mouse embryonic fibroblasts (MEF), although an effect on atherosclerosis in vivo was not demonstrated.16 Mice deficient in the p19Arf gene were found to have increased atherosclerotic lesions in an ApoE null background with significant attenuation of apoptosis in lesions as well as in cultured primary macrophages and vascular smooth muscle cells.17 However, to date no observation regarding atherosclerotic phenotype has been made involving the other neighboring genes.
We set out to survey the 9p21.3 orthologous region using knockout mice models to systematically address the role of the neighboring protein-coding genes in atherosclerosis. We chose the APOE*3 Leiden sensitizing model because it is dominant, simplifying the construction of the models, and also because it exhibits relatively modest elevations of cholesterol, more realistically modeling the human disease than other widely used models.
Methods
Detailed methods can be found in the supplemental material. Primers used for the genotyping and qPCR assays are listed in supplemental table 1.
Mice
All mouse protocols were approved by the UCLA Animal Review Committee. APOE*3-Leiden transgenic mice were maintained on a C57BL/6 background were obtained from TNO (Leiden, Netherlands)18 and re-derived at the UCLA Division of Laboratory Animal Medicine. The Cdkn2a KO mice4, and the p19Arf KO mice5 were obtained from the National Cancer Institute (NCI) Mice Repository, and re-derived at UCLA. The Cdkn2a KO mice were generated by targeted knock-out of the exons 2 and 3 of the Cdkn2a gene, which are shared by both isoforms of the Cdkn2a gene, p16Ink4a and p19Arf. For the p19Arf KO mice, the alternate reading frame of p19Arf gene was selectively mutated and hence the expression of p16Ink4a isoform was maintained (Figure 1). Both strains were created on a mixed background of 129/Sv and C57BL/6 then backcrossed to the C57BL/6 for more than 5 generations.
The Cdkn2b KO mice were a generous gift from the Licia Selleri's lab at Cornell University. They were originally derived from the Barbacid lab in Spain.6 For the Cdkn2b KO mice, the second exon of Cdkn2b gene was replaced with a Neor cassette using 129/Sv DNA then transfected to CJ7 ES cells (Figure 1). The ES cells were then injected into C57BL/6 blastocysts and subsequently bred to C57BL/6 mice for more than 5 generations.
The KO mice for each strain were initially bred to an APOE*3 Leiden mice to generate F1 mice heterozygous for the mutation. F1 mice were mated with each other where one of the pair carried the Leiden transgene. The F2 generation resulted in homozygous knock-out (KO), wild-type mice (WT), and heterozygous mice (Het). Only female mice carrying the APOE*3 Leiden transgene were selected for the atherosclerosis study.
The Mtap heterozygous mice were derived at UCLA with ES cells (MtapGt(RRK081)Byg) obtained from the Mutant Mouse Regional Resource Center (MMRRC) at UC Davis. Briefly, a gene-trap vector encoding the En2 splice acceptor site fused to β-galactosidase/neo fusion gene (β-geo) was inserted between exon 3 and 4 of the mouse MTAP locus. These mice were maintained on a CBA/Ca background. Mice that are homozygous for the MTAP mutation are embryonic lethal, and hence the heterozygotes were mated with APOE*3 Leiden mice, and resulting wildtype and heterozygote female mice carrying the APOE*3 Leiden transgene were selected for the atherosclerosis study.
Diet
A custom diet consisting of 1% cholesterol and 33kcal% fat from mostly cocoa butter was prepared from Research Diets, Inc (product #D10042101). The mice were put on diet between 6 to 8 weeks of age, then kept on the diet for 16 weeks prior to being sacrificed for specimen collection.
Global metabolic profiling assay
100ug of freshly extracted liver tissue was flash frozen and sent to Metabolon, Inc. (Durham, NC) for extraction and analysis.19 The platform for sample analysis has been described in detail.20
Global methylation pattern analysis
We obtained genomic DNA from liver tissue from Mtap Het and WT male mice of 32-weeks of age and used Reduced Representation Bisulfite Sequencing (RRBS) to examine approximately 1% of the genome, comprised of sequences enriched in CpG.21 To determine sites that were differentially methylated between the two samples, we constructed a confidence interval for the percent methylation level of each site using the binomial distribution (binofit in MATLAB). We called sites as differentially methylated between the two samples if the percent methylation level of each sample was outside of the 95% confidence interval.
Statistical Analysis
Two-group comparisons were made using Welch's modified t-test if met normality by the Kolmogorov-Smirnov test and further evaluated by both bootstrapping and permutations22 at the alpha=0.05 level if the Welch's test was significant (See supplemental methods). All three-group comparisons were performed using the non-parametric Kruskal-Wallis ANOVA test, followed by a pairwise comparison using Dunn's post test if found to have a significant Kruskal-Wallis test.
Results
Baseline characteristics of mice
The baseline characteristics, including age, weight, fasting plasma lipids, fasting glucose, and body weight composition, are shown in Table 1 for all groups from each strain studied. There were no statistically significant differences in weight, lipid profile, and body composition (by nuclear magnetic resonance, NMR) results between the groups within each strain. A significant difference was observed in the fasting glucose levels in the Cdkn2a and p19Arf mutants as compared to their respective wild-type groups (P value 0.001 and 0.007, respectively).
Table 1. Baseline Characteristics of All Strains.
Mean ± SD for each box unless otherwise mentioned; LDL was calculated based as LDL ≈ TC- HDL - TG/5. Welch's modified t-test was used to compare the MTAP group; 1-way ANOVA with Kruskal-Wallis test was used to compare all other groups. Insulin levels were only measured for the MTAP mice. *A significant difference existed between the three groups (p<0.005).
| Mtap | Cdkn2a | p19Arf | Cdkn2b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variables | WT | Het | WT | Het | KO | WT | Het | KO | WT | Het | KO |
| N | 5 | 7 | 11 | 16 | 6 | 8 | 11 | 9 | 9 | 15 | 13 |
| Age, median (wks) | 23.1 | 23.1 | 21.7 | 22.3 | 23 | 23.3 | 22.7 | 24.9 | 22.6 | 22.3 | 22.0 |
| Weight (gm) | 23.7 ±1.5 | 23.2 ±1.7 | 21.7 | 21.9 | 21.8 | 24.6 | 24.8 | 23.8 | 22.4 | 20.6 | 21.1 |
| Lipids (mg/dL) | |||||||||||
| Total cholesterol | 954 ±384 | 1013.3 ±148 | 584 ±350 | 603 ±68 | 385 ±249 | 663 ±326 | 540 ±264 | 718 ±344 | 330 ±139 | 464 ±108 | 445 ±112 |
| LDL | 821 ±360 | 884 ±147 | 334 ±350 | 508 ±68 | 276 ±243 | 525 ±324 | 417 ±296 | 587 ±319 | 220 ±123 | 358 ±115 | 342 ±107 |
| HDL | 72 ±12 | 60 ±5 | 66 ±7 | 61 ±8 | 53 ±12 | 64 ±9 | 59 ±12 | 71 ±20 | 63 ±8 | 82 ±12 | 74 ±16 |
| TG | 303 ±187 | 349 ±142 | 310 ±130 | 167 ±73 | 277 ±35 | 371 ±143 | 319 ±147 | 297 ±251 | 109 ±71 | 119 ±33 | 145 ±32 |
| UC | 343 ±158 | 387 ±64 | 180 ±114 | 209 ±37 | 123 ±88 | 294 ±158 | 215 ±107 | 257 ±152 | 85 ±48 | 130 ±38 | 127 ±40 |
| FFA | 49 ±11 | 72 ±24 | 54 ±9 | 53 ±8 | 49 ±6 | 49 ±11 | 57 ±7 | 44 ±10 | 41 ±16 | 50 ±10 | 56 ±15 |
| Glucose | 194 ±78 | 222 ±45 | 133 ±24 | 182 ±28 | 147* ±28 | 148 ±41 | 197 ±50 | 118* ±37 | 120 ±42 | 127 ±31 | 141 ±39 |
| Insulin | 160 ±52 | 128 ± 51 | - | - | - | - | - | - | - | - | - |
| Body Weight Composition by NMR (gm) | |||||||||||
| Fat | 3.3 ±0.6 | 3.2 ±0.8 | 4.2 ±0.9 | 4.5 ±1.1 | 4.4 ±1.0 | 6.0 ±1.1 | 6.8 ±1.5 | 5.3 ±3.5 | 2.2 ±0.5 | 2.0 ±0.5 | 2.1 ±0.7 |
| Muscle | 21.7 ±1.7 | 21.6 ±1.7 | 19.2 ±0.8 | 19.5 ±1.1 | 19.2 ±1.7 | 20.9 ±1.4 | 20.9 ±1.5 | 19.9 ±3.1 | 15.2 ±1.4 | 14.9 ±1.0 | 14.7 ±0.7 |
| Free water | 0.4 ±0.5 | 0.5 ±0.5 | 0.4 ±0.2 | 0.5 ±0.2 | 0.6 ±0.3 | 0.4 ±0.3 | 0.4 ±0.2 | 0.8 ±0.7 | 0.5 ±0.2 | 0.4 ±0.2 | 0.4 ±0.2 |
| %Fat | 0.13 | 0.13 | 0.18 | 0.18 | 0.18 | 0.22 | 0.24 | 0.19 | 0.21 | 0.21 | 0.23 |
Effect of Mtap gene modulation on atherosclerosis
Mtap WT and Mtap Het mice on an APOE*3 Leiden background were compared for aortic lesion development and other characteristics (Mtap KO are not viable). No significant differences in the baseline weight or lipid levels were observed (Figure 2), and other baseline characteristics including fasting glucose, insulin levels and body fat composition, were also comparable (Table 1). As expected the Mtap mRNA expression level was approximately halved in the heterozygous mice compared to the wild-type mice. A significant increase in the aortic sinus lesion size was observed in the heterozygotes compared to the Mtap WT mice (Figure 2). No significant difference in the expression level of the two Cdkn2a isoforms, p16Ink4a and p19Arf, or Cdkn2b was seen between the two groups (Supplemental Figure 1).
Figure 2. Comparison of wild-type vs. heterozygote for the MTAP mutation.

A. The relative Mtap mRNA expression level by qRT-PCR in liver; and B. Aortic sinus lesion size. Lesion area in WT vs Het was 18899±9604 vs. 49623±21650 μm2/section respectively (Mean±SD, p = 0.01). C and D. Oil-red-O staining, and E and F. CD68 staining specific for macrophages in WT vs Het, respectively. The lipid-laden lesions were mostly macrophage in content.
The sections obtained from the aortic sinus were subjected to immuno-staining with antibodies against CD68. Staining showed the lesions to contain >90% CD68+ cells in both Mtap WT and Mtap Het mice (Figure 2). Minority of cells in the lesions stained for SM-Actin and Mason's trichrome and there was no difference in the staining pattern between the WT and Het mice (data not shown).
MTAP deficiency affects metabolic pathways, global methylation dosage and peripheral T-cell composition
We examined the global metabolomics profiles of liver samples from a group of Mtap Het mice compared to WT controls (n=3 each), using the Metabolon, Inc platform. Supplemental Table 2 lists the metabolites assayed in these samples, and raw expression values in each sample.
Mtap Het mice generally had trends toward lower levels of metabolites involved in the methionine processing pathway including S-adenosylmethionine (SAM), S-adenosylhomocysteine (SAH), 5-methythioadenosine (MTA). Plasmas from Mtap WT and Mtap Het mice were assessed for total plasma homocysteine levels. However, no significant difference was found between the two groups (WT 4.64±0.55μM vs. Het 4.43±0.45μM, Mean±SD). Interestingly, taurine, a cysteine metabolite was significantly decreased (0.6 fold) in the Mtap Het mice compared to WT mice.
Given the role of MTA as a major methyl donor, we compared the global methylation pattern between the WT and Het mice at a single-base methylome level. There was a significant difference in the genome wide % methylation levels -- 5565 sites had significant difference in methylation level on binomial distribution test, with over 2000 sites showing greater than 20% change in methylation levels (Supplemental Figure 2).
We also compared complete blood counts using the HemaTrue Hematology analyzer device (Heska) in Mtap-het compared to controls (n=3 vs. 3). We noted no changes in counts of red blood cells, hemoglobin content, platelets, and white blood cells (WBC), including lymphocytes, monocytes, and granulocytes (Data not shown). Further analysis of the WBC population using FACS analysis of the peripheral blood showed that there was a significant decrease in the level of CD4+ T-cells in the MTAP Het mice compared to the WT mice (11.8±4.0 vs. 32.9±13.2 %CD4+CD3+ leukocytes, p<0.05). There was no significant difference in other population of cells including CD8+ T-cells, NK cells, CD11+ or CD115+ monocytes, or neutrophils (Supplemental Figure 3).
Effect of Cdkn2a deficiency on atherosclerosis
Three groups, including Cdkn2a WT, Het and KO mice all on an APOE*3 Leiden background were compared. There were no statistically significant difference in weight, body fat composition by NMR, and plasma lipid levels (Table 1 and Supplemental Figure 4). There was a significant difference in the plasma glucose level between the three groups, where the Het group had the higher levels than the rest. The expression levels of the 9p21 orthologous genes showed decreased p19Arf and p16Ink4a expression as expected, although in the heterozygotes, p16Ink4a levels were maintained at levels similar to WT mice (Supplemental Figure 5). Unexpectedly, Cdkn2b and Mtap expression levels were inversely correlated to the Cdkn2a deletion status (Figure 3). The KO mice had decreased aortic sinus lesion size compared to WT and heterozygous mice (Figure 3), suggesting a recessive model of inheritance. Immunostaining of the frozen sections with CD68 showed that the lesions were predominantly macrophage in content (Figure 3).
Figure 3. Comparison of Cdkn2a wild-type vs. heterozygote vs. KO.
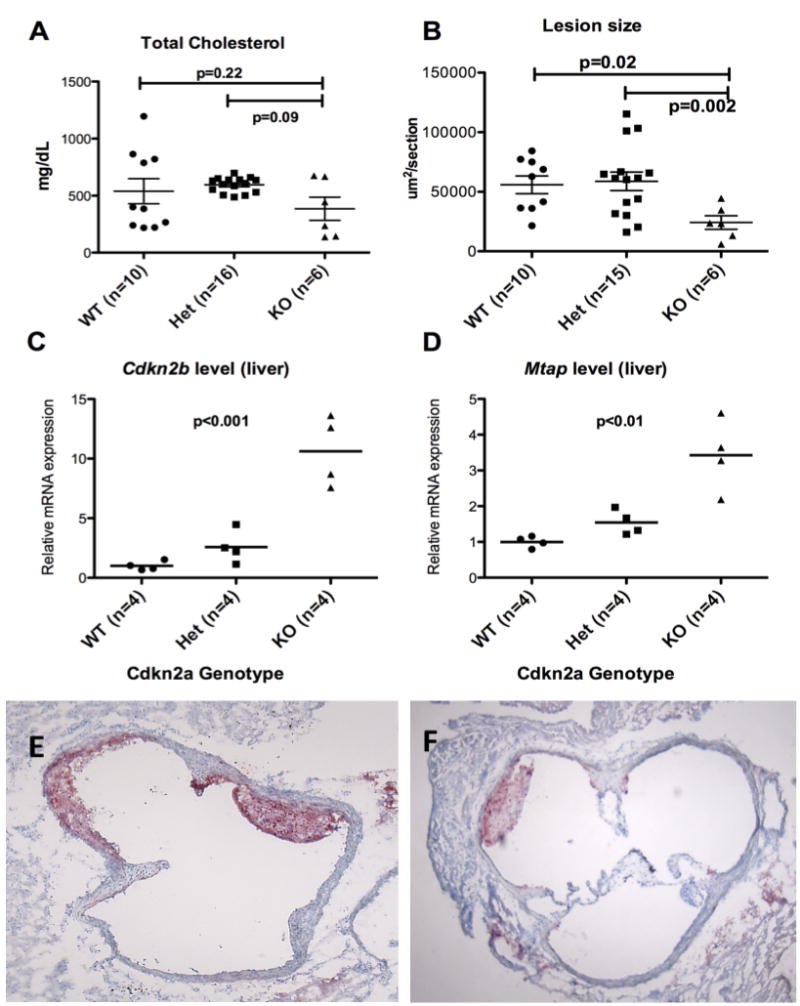
A. Total cholesterol levels; B. Aortic sinus lesion size on Oil red O staining. The KO group had smaller lesion size compared to the WT and Het groups; C. The relative Cdkn2b mRNA expression levels in liver by qPCR (p<0.001); D. The relative Mtap mRNA expression levels in liver by qPCR (p<0.01). The levels of Cdkn2b and Mtap are inversely correlated to the level of p16Ink4a and p19Arf, compensating for the loss of Cdkn2a gene; E and F. Oil red O staining of WT and KO lesions, respectively. Staining of lesions with CD68 showed that the lesions were nearly all macrophages in content (Data not shown).
Effect of p19Arf deficiency on atherosclerosis
Three groups, including p19Arf WT, Het and KO were compared. There were no significant differences in the lesion sizes between all 3 groups on an APOE*3 Leiden background (Figure 4). A similar reciprocal trend in Cdkn2b expression was seen as in the Cdkn2a mutant mice, but there was also significant over-expression of p16Ink4a in the KO mice group in all 3 tissues examined (Figure 4, Supplemental Figure 6). There were no differences between the 3 groups in weight, plasma lipids, fat % by weight, and spleen weight, although the fasting glucose levels were significantly lower in the KO group compared to heterozygotes (p=0.009) (Table 1). Interestingly, the reciprocal activation of the Cdkn2b gene was only seen in the liver and resident peritoneal macrophages and not in the aorta, demonstrating a tissue specific cis-regulatory mechanism within the region. The Mtap level was increased in the heterozygotes compared to both the WT and the KO mice (Figure 4, Supplemental Figure 6). We examined the expression levels for 36B4 in the aorta samples from p19 WT vs. KO. There was no significant change in the average Cp between the two groups, suggesting that the changes noted in gene expression are not the result of a difference in the population of cells within lesions of these aortae samples (Data not shown).
Figure 4. Comparison of p19Arf WT vs. heterozygote vs. KO.

A. Total cholesterol levels. There was no significant difference between the three groups (p=NS).; B. Aortic sinus lesion size. There was no significant difference between the three groups (p=NS); C and D. p16Ink4a (left) and Cdkn2b (right) expression levels in aorta and resident peritoneal macrophages, respectively. There was significant increase in p16Ink4a level with p19Arf KO loss in both aorta (p=0.016) and macrophages (p<0.0001).
Effect of Cdkn2b deficiency on atherosclerosis
The three groups, including Cdkn2b WT, Het and KO were generated from the same littermate and compared. No difference in baseline characteristics were observed including weight at start or end of high cholesterol diet, lipid profile, fasting glucose and fat% by weight (Table 1). There were no differences in the aortic sinus lesion size between the three groups (Figure 5). Again, the expression levels of the neighboring 9p21 orthologous genes were modulated in a compensatory and tissue specific fashion. We observed that the p16Ink4a and p19Arf expression levels were increased in the KO mice in the peritoneal macrophages, but not in the aorta (Figure 5). There was a trend toward a difference in the Mtap expression in liver; however, an opposite relationship was observed in liver and aorta (Figure 5).
Figure 5. Comparison of Cdkn2b WT vs. heterozygote vs. KO.

A. Total fasting cholesterol at time of sacrifice; B. Aortic sinus lesion size; C. MTAP expression in aorta (left) and liver (right); D and E. p16Ink4a (left) and p19Arf (right) expression levels in aorta and peritoneal macrophages, respectively.
Correlation of 9p21 haplotype with 9p21 neighboring gene expression in primary cell culture
Using primary human aortic smooth muscle cells (SMC) from 79 donors, we searched for any correlation between the 9p21 genotype (rs4977574) and the expression levels of the 9p21 neighboring genes (Supplemental Figure 7). No significant correlation was found. Also, we did not find any expression quantitative trait loci (eQTL) that mapped to the 9p21 region using an already published dataset generated using primary human aortic endothelial cells.23
Discussion
We have explored the effects of the multiple protein coding genes near the 9p21 CAD locus on atherosclerosis in a hypercholesterolemic murine model. To our knowledge, this is the first report that systematically describes the effect of these neighboring genes on atherosclerosis including p16Ink4a, p19Arf, Cdkn2b, and Mtap. These included assessment of each of the genes on atherosclerosis and the identification of complex regulatory interactions of the neighboring tumor suppressor genes.
Several novel conclusions emerged. Mtap gene expression clearly conferred a protective phenotype against diet-induced atherosclerosis. Deficiency of the Mtap gene did not affect any of the other 9p21 neighboring genes, unlike other genes in the region that showed significant levels of compensatory modulations, suggesting that the effect on lesion size was solely due to the difference in Mtap expression levels. Little is known about the function of MTAP gene product. One study found Mtap heterozygous mice to have a propensity to develop T-cell lymphoma, acting as a tumor suppressor8. MTAP is the first enzyme in a salvage pathway that allows reutilization of sulfur from MTA (a by-product of the polyamine biosysnthetic pathway) for methionine biosynthesis. The exact mechanism is unclear, but the polyamine metabolism is known to affect cell cycle regulation and apoptosis via the Akt/PKB and Rb pathways, respectively.24 A major pathway of methionine metabolism is the methylation pathway, which involves the conversion of Met to S-adenosylmethionine (SAM), a universal methyl donor. During methylation reactions, SAM is converted to S-adenosylhomocysteine (SAH), which is subsequently hydrolyzed to homocysteine (Hcy).
Total Hcy levels have long been implicated in humans as an independent risk factor for atherosclerosis.25, 26 The mechanism behind this association is unclear, but protein N-homocysteinylation27, changes in redox state, endothelial dysfunction, and smooth muscle cell proliferation have all been implicated.28 To date, there have been no studies that looked at whether the 9p21 risk locus was independent of the homocysteine levels in humans. Although we did not see any difference in the total plasma Hcy levels between the Mtap WT and Mtap Het mice, it is conceivable that there is a dynamic imbalance in the sulfur salvage for methionine biosynthesis that is induced by the modulation in Mtap levels.
Indeed a further examination of the levels of metabolites using a global metabolite panel showed that there were perturbations in the levels of metabolites linked to the methionine and cysteine processing pathways. Overall, Mtap haplo-insufficiency caused a decrease in metabolites upstream of this enzyme within the methionine salvage pathway in liver, plausibly due to feed back inhibition, and likely contributing to reduced levels of taurine (2-aminoethanesulfonic acid). Interestingly, taurine has previously been implicated in atherosclerosis. Taurine concentrations are very high in mammalian tissues29including lymphocytes. There is evidence for reduction in atherosclerosis in animal models following administration of taurine.30-32 Taurine is processed to taurine-chloramine which is formed by reaction between taurine and neutrophil-derived hydroclorous acid, and there is evidence that both substances protect against oxidant stress, regulate inflammatory processes, and inhibit lymphocyte proliferation and cytokine production.33, 34 Other evidence suggests that taurine may have a protective role by increasing T-regulatory cells which are known to protect against atherosclerosis.35 Future studies should include examination of taurine levels in different tissues including plaques, and measuring the cytokine levels involved with T-cell activation and NF-kB pathways.
Interestingly, the choline metabolite trimethylglycine is a source of methyl groups for the synthesis of SAM.36 We have previously shown that serum choline, trimethylamine–N-oxide and trimethylglycine (betaine) levels are positively correlated with atherosclerosis.37 These data are of interest, as we noted changes in metabolites of choline in Mtap het mice, although the levels of choline and betaine were not changed. These data suggest complex interactions between the MTAP pathway of methionine salvage, choline metabolism and atherosclerosis.
A look at the global methylation pattern also showed a significant difference between the Mtap wildtype and heterozygous mice. These changes are consistent with the extensive role of the methionine processing pathway in methyl-donation, although we saw both significant increase or decrease in methylation levels at large number of sites.
We also observed a significant decrease in the level of CD4+ T-lymphocytes in Mtap Het mice compared to the WT mice. The Mtap Het mice develop T-cell lymphomas at a higher incidence with increase in activated CD4+ T-cell expansion beginning around 2 years of age.8 In this study, the mice were young at time of sacrifice, and there was no evidence of general lymphoproliferation. There are numerous studies that report an association of low CD4+ T-cell counts with increased incidence of cardiovascular events, especially in immuno-suppressed or -compromised hosts.38-40 A potential explanation includes the role of T-lymphocytes in inhibiting or modulating the vascular response to injury, protecting against atherogenesis.41
Collectively, these data suggest several plausible mechanisms that could relate MTAP insufficiency with pathogenesis of cardiovascular disease. In mice, homozygous deletion reveals a critical role for Mtap during the developmental process, and suggests an important enzymatic activity. Further studies of these pathways may reveal interesting mechanisms linking MTAP and human CVD phenotype. These data also highlight the value of examinations of heterozygous-deficient mouse models with respect to proteins having critical functions. Further studies to investigate the effect of MTAP on pathways related to cell proliferation in tissues involved in atherosclerosis are warranted.
The involvement of the methionine and cysteine processing pathway also have implications in therapeutics. Deletion of MTAP either in conjunction with CDKN2A has been observed frequently in different types of cancers, and treatment with toxic adenine analogs have been suggested based on in-vitro experiments and mouse models.42 Along the same line, a potential strategy would be to target cells with MTAP deficiency for therapy using toxic adenine analogs and MTA. One report has also demonstrated treatment of mice with an MTAP inhibitor: methylthio-DADMe-Immucillin-A43 and showed increased apoptosis and suppressed tumor growth -- it would be of interest to examine the effects of this compound in a mouse model of atherosclerosis.
The knockout of Cdkn2a also conferred protection against atherosclerosis, in spite of the gene compensation from the other flanking genes, Cdkn2b and Mtap. The mechanism underlying this finding is unclear, but it appears independent of lipids, total body fat, and glucose. In the Cdkn2a KO mice, there was absence of both p16Ink4a and p19Arf expression, hence it is difficult to conclude that the difference in lesion size seen between the WT/Het and KO group were due to p19Arf or p16Ink4a. However, there was sustained expression of p16Ink4a level in the Cdkn2a Het mice compared to the WT mice. A potential explanation includes that the decrease in p19Arf expression increased the expression of p16Ink4a, as suggested by Figure 4C. There was also a compensatory increase in Cdkn2b and MTAP expression levels (Figure 3) further complicating the interpretation of which gene is predominantly affecting the phenotype. The Mtap level was unchanged or decreased in the p19Arf KO mice, suggesting that p16Ink4a has an inhibitory role against Mtap (Supplemental Figure 6). Since the differences in endpoints are seen only in the KO group vs. WT and Het groups, as in the p16Ink4a expression levels, this may suggest that the p16Ink4a expression is the primary driver of atherosclerosis. This hypothesis could potentially be tested using an independent p16Ink4a KO system.
Our findings differ in some respects from the previously published study of Gonzalez-Navarro et al.17 This study demonstrated increased atherosclerosis in p19Arf knockout mice on an ApoE null background, as compared to wild type. The authors noted a decrease in apoptosis in lesions in the plaques of the p19Arf knockout mice. We did not find any difference in p19Arf wild type or knockouts in our study with respect to atherosclerosis, a difference that may be attributable to background (ApoE null vs. APOE*3 Leiden), diet (2.8% vs. 1% cholesterol), or differences in total knockouts examined (n=17 vs. n=9) in the Gonzalez-Navarro study vs. the current study respectively, despite the similar levels of total cholesterol among the mice in both studies. Of note, similar to Gonzalez-Navarro et al., we observed significant gene compensation of other flanking genes in our p19Arf knockout model.17 More recently, the same group published a study examining the role of Cdkn2a over-expression in transgenic ApoE null mice.44 Contrary to expectation, there was no difference in the lesion size between the transgenic and wild type mice. Our results showing no difference in lesion size between the Cdkn2a WT and Het mice are consistent with these findings from the Cdkn2a transgenic study.
Interestingly, recent studies in human and human tissue culture cells have failed to demonstrate a consistent relationship between the levels of the genes flanking 9p21, the non-coding transcript ANRIL, and the 9p21 risk alleles, raising the question that these genes may not be involved at all.13-15, 45 However, as noted above, cell specificity may be important, and studies of cell-specific expression in macrophages, smooth muscle cells, and endothelial cells in atherosclerotic lesions have not been published to date. We have used primary cells extracted from aorta of heart donors, however, did not find correlation of 9p21 SNPs with expression level of neighboring genes in aortic endothelial and smooth muscle cells. It is possible that the differences in expression of these genes with respect to the risk alleles may be subtle, requiring a much higher number of plaques studied to have statistical power to detect this difference. Finally, similar to genes active in the process of development, expression of the genes flanking 9p21 may be more important during earlier time points than those measured in the aforementioned studies. Expression in chronic, late-stage plaques would therefore not reflect the significance of the role they play in the development of atherosclerosis.
Another novel observation in this study was the complexity of gene regulation that exists in this region. The Cdkn2a, p19Arf and Cdkn2b mutations all showed little to no effect on atherosclerosis. Surprisingly, there was a significant compensatory modulation of the expression levels of the 9p21 orthologous genes in all strains except the Mtap mutant mice. This trend was observed in the liver and resident peritoneal macrophages, as well as in the aorta, but tissue specific modulations were also observed in some cases. To our knowledge, this is the first report that describes such an extensive reciprocal regulation within this region. One report suggested a potential inverse relation or lack of relation between the methylation pattern of promoters of p16Ink4a and Cdkn2b in cancers from an observational study.46 p16Ink4a and Cdkn2b are both CKIs that bind to the CDK4 and may explain such redundancy. It is surprising that the functionally distinct protein p19Arf causes not only a significant over-expression of p16Ink4a (>20 fold) in its absence as previously reported17, but also an increase in Cdkn2b (>3 fold). Given the complexity inherent in this locus and modulation of the different neighboring genes, the effect on atherosclerosis of these individual genes is difficult to interpret. By inference, these findings suggest a mechanism to protect the critical function of this region. The high functional redundancy in this locus also points to the likelihood that the 9p21 risk locus affects a cis-regulatory element of this region that is critical for compensatory mechanism activated in response to stress.
The mechanism of this compensatory regulation is unclear, but can potentially involve changes in methylation patterns, histone modification, and common binding motif to a transcription factor. It is interesting that the mutation status of the Cdkn2a, p19Arf, and Cdkn2b genes affected the Mtap gene expression levels as well, whereas the vice versa is not true. The promoter region of Mtap is relatively distant from the promoter regions of Cdkn2a and Cdkn2b, at about 200 kb, and this may suggest a trans- effect such as a transcription factor mediated over-expression as a more likely mechanism of this modulation. There were no statistically significant differences in the Cdkn2a, Cdkn2b genes between Mtap wild type and heterozygotes, which allowed a direct comparison of the effect of Mtap on atherosclerosis.
In summary, we identified the MTAP gene as a novel regulator of atherosclerotic lesion development potentially by modulating the methionine and cysteine processing pathways, the T-lymphocytes, and the global methylation pattern. We also observed significant compensatory regulation of this region, which corroborates previous findings of dense regulatory components inherent to this locus.47 Because of this compensatory regulation, the effects of the other genes in this locus, Cdkn2a, p19Arf, and Cdkn2b, were difficult to assess. However, despite this regulation, an effect of Cdkn2a on atherosclerosis was observed. Further studies are needed to define the regulation of the 9p21 genes, and the mechanisms by which these genes modulate an effect on atherosclerosis.
Supplementary Material
Atherosclerotic coronary artery disease (CAD) is the leading cause of death in the developed world. Epidemiological data show that environmental and genetic factors have roughly equal weight in determining the susceptibility to CAD and the rate of disease progression, although the genes that control this variability are not well characterized. Recently, large genome-wide association (GWA) studies have identified over 30 CAD risk loci. Some of the CAD risk loci work through known CAD risk factors such as lipids, however, this is not true for the majority of loci. This study systematically dissects the highly replicated 9p21.3 locus using different knock-out mice models of the neighboring genes, including CDKN2A, CDKN2B, and MTAP. We describe the complex molecular regulation within the region and show that MTAP (methylthioadenosine phophorylase) affects the progression of atherosclerosis through several potential mechanisms including lymphocyte activation, and changes in metabolic and methylation profiles. This study is one of the first to show a change in atherosclerosis phenotype independent of lipids in a mice model generated as a follow up of new locus identified from GWA studies. This discovery suggests a novel pathway for atherosclerosis and hence opens the door to develop new markers of predicting the risk of atherosclerosis at the genomic, epigenomic, and metabolomic levels, and eventually new strategies for primary and secondary prevention of coronary artery disease.
Acknowledgments
None
Funding Source: The project was funded by the NIH grants HL095154, HL030568 (to A.J.L.) and HL094709-03 (to I.N.M.). Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards CA-16042 and AI-28697, and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, Konig IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson DF, Magnusson KP, Andersen K, Levey AI, Backman VM, Matthiasdottir S, Jonsdottir T, Palsson S, Einarsdottir H, Gunnarsdottir S, Gylfason A, Vaccarino V, Hooper WC, Reilly MP, Granger CB, Austin H, Rader DJ, Shah SH, Quyyumi AA, Gulcher JR, Thorgeirsson G, Thorsteinsdottir U, Kong A, Stefansson K. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 4.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the ink4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 5.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse ink4a locus mediated by the alternative reading frame product p19arf. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 6.Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, Flores JM, Cordon-Cardo C, Barbacid M. Limited overlapping roles of p15(ink4b) and p18(ink4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–3506. doi: 10.1093/emboj/19.13.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shmueli O, Horn-Saban S, Chalifa-Caspi V, Shmoish M, Ophir R, Benjamin-Rodrig H, Safran M, Domany E, Lancet D. Genenote: Whole genome expression profiles in normal human tissues. C R Biol. 2003;326:1067–1072. doi: 10.1016/j.crvi.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Kadariya Y, Yin B, Tang B, Shinton SA, Quinlivan EP, Hua X, Klein-Szanto A, Al-Saleem TI, Bassing CH, Hardy RR, Kruger WD. Mice heterozygous for germ-line mutations in methylthioadenosine phosphorylase (mtap) die prematurely of t-cell lymphoma. Cancer Res. 2009;69:5961–5969. doi: 10.1158/0008-5472.CAN-09-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guevara NV, Kim HS, Antonova EI, Chan L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat Med. 1999;5:335–339. doi: 10.1038/6585. [DOI] [PubMed] [Google Scholar]
- 10.Mercer J, Figg N, Stoneman V, Braganza D, Bennett MR. Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in apoe knockout mice. Circ Res. 2005;96:667–674. doi: 10.1161/01.RES.0000161069.15577.ca. [DOI] [PubMed] [Google Scholar]
- 11.Merched AJ, Chan L. Absence of p21waf1/cip1/sdi1 modulates macrophage differentiation and inflammatory response and protects against atherosclerosis. Circulation. 2004;110:3830–3841. doi: 10.1161/01.CIR.0000148681.01282.89. [DOI] [PubMed] [Google Scholar]
- 12.Diez-Juan A, Andres V. The growth suppressor p27(kip1) protects against diet-induced atherosclerosis. FASEB J. 2001;15:1989–1995. doi: 10.1096/fj.01-0130com. [DOI] [PubMed] [Google Scholar]
- 13.Holdt LM, Beutner F, Scholz M, Gielen S, Gabel G, Bergert H, Schuler G, Thiery J, Teupser D. Anril expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol. 2010;30:620–627. doi: 10.1161/ATVBAHA.109.196832. [DOI] [PubMed] [Google Scholar]
- 14.Jarinova O, Stewart AF, Roberts R, Wells G, Lau P, Naing T, Buerki C, McLean BW, Cook RC, Parker JS, McPherson R. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. 2009;29:1671–1677. doi: 10.1161/ATVBAHA.109.189522. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, Ibrahim JG, Thomas NE, Sharpless NE. Ink4/arf transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One. 2009;4:e5027. doi: 10.1371/journal.pone.0005027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, Blow MJ, Cohen JC, Rubin EM, Pennacchio LA. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–412. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Navarro H, Abu Nabah YN, Vinue A, Andres-Manzano MJ, Collado M, Serrano M, Andres V. P19(arf) deficiency reduces macrophage and vascular smooth muscle cell apoptosis and aggravates atherosclerosis. J Am Coll Cardiol. 2010;55:2258–2268. doi: 10.1016/j.jacc.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 18.van Vlijmen BJ, van den Maagdenberg AM, Gijbels MJ, van der Boom H, HogenEsch H, Frants RR, Hofker MH, Havekes LM. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein e3-leiden transgenic mice. J Clin Invest. 1994;93:1403–1410. doi: 10.1172/JCI117117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fong MY, McDunn J, Kakar SS. Identification of metabolites in the normal ovary and their transformation in primary and metastatic ovarian cancer. PLoS One. 2011;6:e19963. doi: 10.1371/journal.pone.0019963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 21.Gu Y, Shore RE, Arslan AA, Koenig KL, Liu M, Ibrahim S, Lokshin AE, Zeleniuch-Jacquotte A. Circulating cytokines and risk of b-cell non-hodgkin lymphoma: A prospective study. Cancer causes & control: CCC. 2010;21:1323–1333. doi: 10.1007/s10552-010-9560-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox J Bootstrapping regression models. Applied regression analysis and generalized linear models. McMaster University; 2008. [Google Scholar]
- 23.Romanoski CE, Che N, Yin F, Mai N, Pouldar D, Civelek M, Pan C, Lee S, Vakili L, Yang WP, Kayne P, Mungrue IN, Araujo JA, Berliner JA, Lusis AJ. Network for activation of human endothelial cells by oxidized phospholipids: A critical role of heme oxygenase 1. Circ Res. 2011;109:e27–41. doi: 10.1161/CIRCRESAHA.111.241869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koomoa DL, Yco LP, Borsics T, Wallick CJ, Bachmann AS. Ornithine decarboxylase inhibition by alpha-difluoromethylornithine activates opposing signaling pathways via phosphorylation of both akt/protein kinase b and p27kip1 in neuroblastoma. Cancer Res. 2008;68:9825–9831. doi: 10.1158/0008-5472.CAN-08-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med. 1998;338:1042–1050. doi: 10.1056/NEJM199804093381507. [DOI] [PubMed] [Google Scholar]
- 26.Antoniades C, Antonopoulos AS, Tousoulis D, Marinou K, Stefanadis C. Homocysteine and coronary atherosclerosis: From folate fortification to the recent clinical trials. Eur Heart J. 2009;30:6–15. doi: 10.1093/eurheartj/ehn515. [DOI] [PubMed] [Google Scholar]
- 27.Perla-Kajan J, Jakubowski H. Paraoxonase 1 protects against protein n-homocysteinylation in humans. FASEB J. 2010;24:931–936. doi: 10.1096/fj.09-144410. [DOI] [PubMed] [Google Scholar]
- 28.Steed MM, Tyagi SC. Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid Redox Signal. 2011;15:1927–1943. doi: 10.1089/ars.2010.3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wojcik OP, Koenig KL, Zeleniuch-Jacquotte A, Costa M, Chen Y. The potential protective effects of taurine on coronary heart disease. Atherosclerosis. 2010;208:19–25. doi: 10.1016/j.atherosclerosis.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murakami S, Kondo Y, Tomisawa K, Nagate T. Prevention of atherosclerotic lesion development in mice by taurine. Drugs under experimental and clinical research. 1999;25:227–234. [PubMed] [Google Scholar]
- 31.Zulli A, Lau E, Wijaya BP, Jin X, Sutarga K, Schwartz GD, Learmont J, Wookey PJ, Zinellu A, Carru C, Hare DL. High dietary taurine reduces apoptosis and atherosclerosis in the left main coronary artery: Association with reduced ccaat/enhancer binding protein homologous protein and total plasma homocysteine but not lipidemia. Hypertension. 2009;53:1017–1022. doi: 10.1161/HYPERTENSIONAHA.109.129924. [DOI] [PubMed] [Google Scholar]
- 32.Sethupathy S, Elanchezhiyan C, Vasudevan K, Rajagopal G. Antiatherogenic effect of taurine in high fat diet fed rats. Indian journal of experimental biology. 2002;40:1169–1172. [PubMed] [Google Scholar]
- 33.Schuller-Levis GB, Park E. Taurine and its chloramine: Modulators of immunity. Neurochem Res. 2004;29:117–126. doi: 10.1023/b:nere.0000010440.37629.17. [DOI] [PubMed] [Google Scholar]
- 34.Kanayama A, Inoue J, Sugita-Konishi Y, Shimizu M, Miyamoto Y. Oxidation of ikappa balpha at methionine 45 is one cause of taurine chloramine-induced inhibition of nf-kappa b activation. J Biol Chem. 2002;277:24049–24056. doi: 10.1074/jbc.M110832200. [DOI] [PubMed] [Google Scholar]
- 35.Miao J, Zhang J, Zheng L, Yu X, Zhu W, Zou S. Taurine attenuates streptococcus uberis-induced mastitis in rats by increasing t regulatory cells. Amino Acids. 2012;42:2417–2428. doi: 10.1007/s00726-011-1047-3. [DOI] [PubMed] [Google Scholar]
- 36.Schwab U, Torronen A, Toppinen L, Alfthan G, Saarinen M, Aro A, Uusitupa M. Betaine supplementation decreases plasma homocysteine concentrations but does not affect body weight, body composition, or resting energy expenditure in human subjects. The American journal of clinical nutrition. 2002;76:961–967. doi: 10.1093/ajcn/76.5.961. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fyfe AI, Qiao JH, Lusis AJ. Immune-deficient mice develop typical atherosclerotic fatty streaks when fed an atherogenic diet. J Clin Invest. 1994;94:2516–2520. doi: 10.1172/JCI117622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ducloux D, Challier B, Saas P, Tiberghien P, Chalopin JM. Cd4 cell lymphopenia and atherosclerosis in renal transplant recipients. J Am Soc Nephrol. 2003;14:767–772. doi: 10.1097/01.asn.0000048718.43419.44. [DOI] [PubMed] [Google Scholar]
- 40.Kaplan RC, Kingsley LA, Gange SJ, Benning L, Jacobson LP, Lazar J, Anastos K, Tien PC, Sharrett AR, Hodis HN. Low cd4+ t-cell count as a major atherosclerosis risk factor in hiv-infected women and men. AIDS. 2008;22:1615–1624. doi: 10.1097/QAD.0b013e328300581d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansson GK, Holm J, Holm S, Fotev Z, Hedrich HJ, Fingerle J. T lymphocytes inhibit the vascular response to injury. Proc Natl Acad Sci U S A. 1991;88:10530–10534. doi: 10.1073/pnas.88.23.10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lubin M, Lubin A. Selective killing of tumors deficient in methylthioadenosine phosphorylase: A novel strategy. PLoS One. 2009;4:e5735. doi: 10.1371/journal.pone.0005735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basu I, Locker J, Cassera MB, Belbin TJ, Merino EF, Dong X, Hemeon I, Evans GB, Guha C, Schramm VL. Growth and metastases of human lung cancer are inhibited in mouse xenografts by a transition state analogue of 5′- methylthioadenosine phosphorylase. The Journal of biological chemistry. 2011;286:4902–4911. doi: 10.1074/jbc.M110.198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuster JJ, Molina-Sanchez P, Jovani D, Vinue A, Serrano M, Andres V. Increased gene dosage of the ink4/arf locus does not attenuate atherosclerosis development in hypercholesterolaemic mice. Atherosclerosis. 2012;221:98–105. doi: 10.1016/j.atherosclerosis.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 45.Holdt LM, Sass K, Gabel G, Bergert H, Thiery J, Teupser D. Expression of chr9p21 genes cdkn2b (p15(ink4b)), cdkn2a (p16(ink4a), p14(arf)) and mtap in human atherosclerotic plaque. Atherosclerosis. 2011;214:264–270. doi: 10.1016/j.atherosclerosis.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 46.Zhou JX, Niehans GA, Shar A, Rubins JB, Frizelle SP, Kratzke RA. Mechanisms of g1 checkpoint loss in resected early stage non-small cell lung cancer. Lung Cancer. 2001;32:27–38. doi: 10.1016/s0169-5002(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 47.Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG, Frazer KA. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.