

Abstract
With the recent interest of protease-activated receptors (PAR) 1 and PAR4 as possible targets for the treatment of thrombotic disorders, we compared the efficacy of protease-activated receptor (PAR)1 and PAR4 in the generation of procoagulant phenotypes on platelet membranes. PAR4-activating peptide (AP)–stimulated platelets promoted thrombin generation in plasma up to 5 minutes earlier than PAR1-AP–stimulated platelets. PAR4-AP–mediated factor V (FV) association with the platelet surface was 1.6-fold greater than for PAR1-AP. Moreover, PAR4 stimulation resulted in a 3-fold greater release of microparticles, compared with PAR1 stimulation. More robust FV secretion and microparticle generation with PAR4-AP was attributable to stronger and more sustained phosphorylation of myosin light chain at serine 19 and threonine 18. Inhibition of Rho-kinase reduced PAR4-AP–mediated FV secretion and microparticle generation to PAR1-AP–mediated levels. Thrombin generation assays measuring prothrombinase complex activity demonstrated 1.5-fold higher peak thrombin levels on PAR4-AP–stimulated platelets, compared with PAR1-AP–stimulated platelets. Rho-kinase inhibition reduced PAR4-AP–mediated peak thrombin generation by 25% but had no significant effect on PAR1-AP–mediated thrombin generation. In conclusion, stimulation of PAR4 on platelets leads to faster and more robust thrombin generation, compared with PAR1 stimulation. The greater procoagulant potential is related to more efficient FV release from intracellular stores and microparticle production driven by stronger and more sustained myosin light chain phosphorylation. These data have implications about the role of PAR4 during hemostasis and are clinically relevant in light of recent efforts to develop PAR antagonists to treat thrombotic disorders.
Introduction
Thrombin activates platelets through proteolytic cleavage of protease-activated receptors (PARs), resulting in the generation of a tethered ligand. Human platelets express two PARs (PAR1 and PAR4). PAR1 contains a hirudin-like sequence in its exodomain that interacts with thrombin’s anion-binding exosite-1 (Liu et al., 1991; Vu et al., 1991). Because of this high-affinity interaction, PAR1 is engaged at lower concentrations of thrombin than is PAR4, which lacks the hirudin-like domain (Xu et al., 1998; Hammes and Coughlin, 1999; Faruqi et al., 2000). PAR1 and PAR4 differ not only in temporal engagement but also in downstream signaling pathways (Coughlin, 2000; Covic et al., 2000; Ma et al., 2005; Holinstat et al., 2006; Bilodeau and Hamm, 2007; Holinstat et al., 2007; Voss et al., 2007; Holinstat et al., 2009).
Monroe et al. (Monroe et al., 2002) described a model of hemostasis implicating platelets in the amplification/priming and propagation of thrombin generation. Platelet activation results in the expression of a procoagulant surface and assembly of the prothrombinase and intrinsic Xase complexes, leading to cleavage of fibrinogen to fibrin and formation of a hemostatic clot. In addition to the provision of phosphatidyl-serine (PS)–rich membranes for the assembly of coagulation complexes, platelets possess a unique, APC-resistant, preactivated form of factor V (FV) (Alberio et al., 2000; Duckers et al., 2010), which is concentrated in α-granules from plasma sources. Platelet-FV represents 20% of the total amount of FV circulating in whole blood, and FV concentrations in the platelet exceed that of the plasma by 100-fold (Weiss et al., 2001). The importance of platelet-FV has been shown in clinical cases of bleeding associated with defects in FV storage or release (Tracy et al., 1984; Nesheim et al., 1986; Grigg et al., 1989; Weiss and Lages, 1997; Weiss et al., 2001; Diamandis et al., 2008). More recently, patients with severe congenital FV deficiency were shown to be protected against a loss of thrombin generation and severe bleeding by residual FV concentrated in their platelet granules (Duckers et al., 2010).
Activated platelets shed microparticles that mediate a number of processes involved in coagulation, platelet adhesion, angiogenesis, and vascular smooth muscle cell proliferation (Flaumenhaft et al., 2010; Italiano et al., 2010). The concentrations of circulating platelet-derived microparticles correlate with the severity of a number of cardiovascular diseases, including acute coronary syndrome (Mallat et al., 2000), atherosclerosis (Tan and Lip, 2005) hypertension (Preston et al., 2003), peripheral artery disease (Zeiger et al., 2000; Tan and Lip, 2005; van der Zee et al., 2006), aortic valve stenosis (Diehl et al., 2008), metabolic syndrome (Diamant et al., 2002), and type II diabetes (Nomura et al., 1995; Cohen et al., 2002; Tan et al., 2005; Koga et al., 2006). Platelet-derived microparticles possess binding sites for coagulation factors FVa (Alberio et al., 2000), FVIIIa (Gilbert et al., 1991), and FIXa (Hoffman et al., 1992a). Platelet-derived microparticles are generated in response to a number of strong platelet agonists, including thrombin (Flaumenhaft et al., 2010); however, the individual capacity of PAR1 and PAR4 to induce microparticle formation has not been explored.
Platelet degranulation involves activation of phospholipase C and subsequent cleavage of phosphatidyl inositol 4, 5-biphosphate (PIP2) into diacylglycerol and inositol 1, 4, 5-triphosphate. Diacylglycerol directly activates PKC, and inositol 1, 4, 5-triphosphate mobilizes intracellular calcium, which synergistically contributes to platelet secretion (Walker and Watson, 1993). Accordingly, impaired platelet secretion has been observed in Gαq- or phospholipase Cβ-deficient mice and humans (Gabbeta et al., 1997; Offermanns et al., 1997). New data are shedding light on a role for the Gα12/13 pathway and RhoA activation in platelet secretion. In addition to defects in hemostasis and protection against thrombosis, Gα13-deficient mice experience defective agonist-induced secretion (Moers et al., 2003). In mice, a megakaryocyte-specific RhoA deficiency causes impaired platelet granule secretion (Pleines et al., 2012). Kunapuli et al. revealed that supplementation of Gαq signaling with Gα12/13 activation is necessary for dense granule secretion, which can be blocked with a RhoA inhibitor (Jin et al., 2009). Active RhoA, through its effector Rho-kinase, maintains phosphorylation of myosin light chain by inactivating myosin light chain phosphatase. Inhibition of Rho-kinase results in impaired thrombin-mediated secretion (Getz et al., 2010).
The signaling pathways involved in platelet microparticle production have not been clearly defined. Several mechanisms are implicated in microparticle production, including destabilization of membrane-cytoskeletal attachments in a Ca2+/calpain-dependent manner and changes in membrane composition, particularly acute decreases in PIP2 membrane composition (O'Connell et al., 2005; Flaumenhaft et al., 2009; Flaumenhaft et al., 2010). Although the possibility of a Rho/Rho-kinase/myosin signaling axis contributing to microparticle production has been proposed (Flaumenhaft et al., 2010), involvement of this pathway has not been demonstrated.
Activated platelets support thrombin generation and, in turn, are stimulated by thrombin through PARs. Therefore, platelet activation and thrombin generation are interdependent biologic processes. We hypothesize that PAR1 and PAR4 play distinct roles in thrombosis and hemostasis and, therefore, present unique procoagulant phenotypes when stimulated individually. A careful assessment of the efficacy of the PARs to modulate biochemical events involved in thrombin generation has not been conducted. We conducted a series of experiments with PAR-activating peptides (AP) that have been extensively used to describe the signaling of PARs (Faruqi et al., 2000; Holinstat et al., 2006; Holinstat et al., 2007; Voss et al., 2007). We revealed that PAR4 stimulation leads to more FV secretion and microparticle generation than does PAR1 stimulation in human platelets. The stronger procoagulant phenotypes exhibited by PAR4 were attributable to more sustained and robust MLC phosphorylation driven by Rho-kinase activity, a previously undocumented observation. We also reveal a novel role for Rho-kinase in platelet microparticle production.
Materials and Methods
Materials.
The thrombin substrate z-GGR-AMC was purchased from Bachem (Basel, Switzerland). Sheep antihuman FV antibody, FII (prothrombin), FX, FXa, and antithrombin (AT) were from Haematologic Technologies (Essex Junction, VT). Anti-PKC (S)-substrate, phosphomyosin light chain (pMLC) T18 and pMLC S19, and p44/p42 ERK antibodies were from Cell Signaling Technologies Inc. (Danvers, MA). Antirabbit and Antimouse secondary antibodies and ECL reagent were from Perkin Elmer (Waltham, MA). Alexa Fluor 488 donkey antisheep were from Invitrogen (Carlsbad, CA). Activating peptides for PAR1 (PAR1-AP; SFLLRN) and PAR4 (PAR4-AP, AYPGKF) were purchased from GL Biochem (Shanghai, China). Human plasma was purchased from George King Bio-medical Inc. (Overland Park, KS). PAR-1 span IgY and PAR-4 span IgY were kind gifts from Dr. Fred Ofosu.
Blood Collection and Platelet Isolation.
Human platelets were obtained from healthy volunteers.The studies were approved by the Vanderbilt University Internal Review Board. Informed consent was obtained from all individuals before the blood sample obtainment. Blood samples were collected into sodium citrate anticoagulant (final concentration, 0.32%) through a 19-gauge needle. PGE1 (5 μg/ml, final concentration) was added to citrated blood, and gel filtered platelets were prepared as described elsewhere (Hoffman et al., 1992b). Blood samples were overlaid onto Accuprep Lymphocyte separation medium (Accurate Chemical and Scientific Corp., Westbury, NY) and centrifuged for 30 minutes at 500g. The middle band containing platelets and mononuclear cells was isolated and mixed with an equal volume of CGS buffer [13 mM citrate (pH 7.4), 123 mM NaCl, 33 mM dextrose] containing 5 μg/ml PGE1 (final concentration). Nucleated cells were removed by centrifugation at 120g for 10 minutes. The platelet-rich supernatant was isolated and layered onto a Sepharose 4B column (Sigma-Aldrich, St. Louis, MO), equilibrated with Tyrode’s Buffer [15 mM HEPES, 0.33 mM NaH2PO4 (pH 7.4), 138 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 5.5 mM dextrose] with 0.1% BSA. Platelets were collected, counted on a Coulter Counter, and diluted in Tyrode’s with 0.1% BSA to the indicated concentrations.
Immunocytochemistry.
Gel-filtered platelets at a density of 1.5 × 107 cells/ml were incubated with agonist or vehicle control for 15 minutes before fixation with 1% paraformaldehyde. Samples were diluted in phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.46 mM KH2PO4) with 0.1% BSA and added to Laboratory-Tek II chamber slides (NUNC, Rochester, NY) precoated with poly-lysine. Chamber slides were incubated overnight at 4°C to allow platelets to adhere. After seeding, chambers were washed once with an equal volume of PBS. Samples were then blocked for 30 minutes at room temperature with 1% BSA in PBS. After blocking, samples were incubated with antibodies diluted in PBS with 1% BSA for 1 hour, followed by three wash cycles with PBS before incubation with the appropriate fluorescent secondary antibody in PBS with 1% BSA for 30 minutes. Samples were washed three more times before mounting in aqua polymount (Polysciences Inc, Arrington, PA). Images were taken with a 63× /1.40 Plan-APOCHROMAT oil objective on a Zeiss LSM 510 Inverted confocal microscope. Microscopy was performed using the VUMC Cell Imaging Shared Resource.
Flow Cytometry.
For detection of FV, gel-filtered platelets at a density of 1.5 × 107 cells/ml were incubated with vehicle control or agonist for 15 minutes before fixation with 1% paraformaldehyde for 20 minutes. After fixation and washing, samples were incubated with the appropriate concentration of primary antibody for 1 hour at room temperature. After washing with PBS, samples were suspended in 2 μg/ml of labeled secondary antibody and incubated for 30 minutes at room temperature. Samples were washed once more with PBS, then suspended in 500 μl PBS. Samples were analyzed on a FACS Canto II (Becton Dickinson, Franklin Lakes, NJ). For detection of P-selectin, platelets at 1.5 × 107 cells/ml were preincubated with APC-conjugated CD62P before stimulation with the appropriate agonist for 15 minutes. Samples were fixed with 1% paraformaldehyde in PBS for 20 minutes before dilution of the samples with Tyrode’s buffer. Data were analyzed using FACS DiVa acquisition software (Becton Dickinson) and Winlist software (Verity Software House, Topsham, MA) for analysis. Mean fluorescence intensity was determined by collecting the fluorescence recordings in the platelet gate for 100,000 events. The percentage of positive cells was determined by gating to unstimulated platelets incubated with an isotype-matched IgG control for each primary antibody.
Microparticle Detection.
Gel-filtered platelets at a density of 2 × 108 cells/ml were supplemented with 2.5 mM CaCl2 before incubation with the various concentrations of PAR agonists for 30 minutes at 37°C. Microparticle-enriched samples were prepared by centrifugation of platelet preparations for 10 minutes at 700g. The supernatant was collected and incubated for 20 minutes with either FITC-conjugated sheep IgG nonimmune isotype control (Affinity Biologicals, Ancaster, ON, Canada), PE-conjugated CD41 (Becton Dickson), and APC-conjugated CD62P (Beckton Dickson) or FITC-conjugated sheep FV (Affinity Biologicals), PE-conjugated CD41, and APC-conjugated CD62P. Samples were diluted in Tyrode’s buffer and analyzed on a FACS Canto II. Events were collected for a total of 1 minute for each sample. Microparticles included CD41 and CD62P dual-positive particles detected below 1 μm in size, as determined by forward and side scatter analysis of 1-μm beads.
Intracellular Calcium Mobilization.
Washed human platelets are prepared as previously detailed (Holinstat et al., 2009) and diluted in Tyrode’s buffer plus 0.1% BSA. One hour before stimulation, platelets were dye-loaded with 1.25 μg/ml (final concentration) Fluo4-AM in calcium assay buffer (HBSS, 20 mM HEPES, 2.5 mM probenecid). Thirty minutes before stimulation, platelets were mixed with a combination of PAR-1 span IgY and PAR-4 span IgY antibodies or chicken IgY isotype control; 60 μl of dye-loaded platelets were added to each well of a standard-surface BD falcon 384-well plate. The final platelet concentration was 0.8 × 108 cells/ml. Fluorescence measurements are taken on a Functional Drug Screening System (Hamamatsu, Japan) at excitation/emission 480/540 for 240 seconds at 25°C.
Enzyme-Linked Immunoabsorbance Assay.
Gel-filtered platelets at a density of 2.0 × 108 cells/ml were stimulated with the indicated concentration of PAR agonist for 15 minutes. Platelets were then pelleted at 13,000g for 10 minutes at room temperature. Supernatants were collected and stored for analysis at a later date. Immulon 2HB 96-well microtitre enzyme immunoassay plates were coated with a FV capture antibody (FV-EIA-C, Affinity Biologicals) in Carbonate buffer (50 mM Carbonate; pH 9.6) overnight at 4°C. Plates were blocked with 1% BSA before application of samples diluted 1:10 in HEPES-buffered saline with 1% BSA and 0.1% Tween-20. Standards were prepared from purified FV (Haematologic Technologies) ranging from 70 ng/ml to 0.5 ng/ml. After washing with PBS-T, bound FV was detected with an antihuman FV antibody (FV-EIA-D; Affinity Biologicals), followed by another wash and incubation with 416 μg/ml OPD substrate (Sigma-Aldrich). Plates were read at 490 nm in a Spectramax 190-plate reader.
Thrombin Generation.
Gel-filtered platelets at a density of 6.25 × 107 cells/ml were applied to prewarmed (37°C) black-sided 96-well plates (Greiner Bio-one, Monroe, NC). Platelets were stimulated with agonist or vehicle control for 5 minutes at 37°C before addition of an equal volume of citrate-anticoagulated human plasma or a mixture of purified coagulation factors in citrated Tyrode’s buffer, all prewarmed to 37°C. Thrombin generation was initiated immediately with the addition of excess calcium (final concentration, 16.4 mM) and fluorogenic thrombin substrate z-GGR-AMC (440 μM). Thrombin generation was monitored in a FlexStation II 384 at 390/460 nm for 4 hours. After correction for substrate depletion and the inner-filter effect, the first-order derivative was taken from raw fluorescent units and converted into concentrations of thrombin (nM) by comparison with a standard curve generated with fixed concentrations of thrombin (Hemker et al., 2003; De Smedt et al., 2008). Concentrations of thrombin were then plotted against time to establish a period of thrombin production. PAR-1 span IgY and PAR-4 span IgY (Ofosu et al., 2008) were preincubated with platelets for 30 minutes before stimulation with PAR1-AP or PAR4-AP and the initiation of the assay.
Western Blot Analysis.
Gel-filtered platelets at 3.0 × 108 cells/ml in Tyrode’s plus 0.1% BSA were stimulated for indicated periods at 37°C. Reactions were stopped with the addition of 0.6N HClO4- to immediately precipitate all protein. Samples were centrifuged at 13,000g for 10 minutes at 4°C to isolate protein pellets. Pellets were resuspended and centrifuged 2 times with ice cold water. Finally, pellets were resuspended in 1× SDS Lamelli buffer and allowed to dissolve overnight. Samples were analyzed by SDS PAGE, followed by Western blotting. An estimated 25 μg was added to each well. Blots were probed with corresponding primary antibodies and HRP-linked secondary antibodies before exposure with ECL reagent. Primary antibodies were used at 1:1000 dilution, as suggested by Cell Signaling.
Results
Thrombin Generation Supported by Platelet Stimulation with PAR Agonists.
We examined the ability of PAR1- and PAR4-stimulated platelets to induce thrombin generation in plasma. Purified platelets were stimulated with increasing concentrations of PAR1-AP or PAR4-AP and subsequently suspended in plasma. Maximal doses of PAR4-AP induced thrombin generation up to 5 minutes before PAR1-AP (Fig. 1, A and B). These data suggest the induction of a more procoagulant platelet phenotype with PAR4 stimulation, compared with PAR1.
Fig. 1.

Thrombin generation supported by platelet stimulation with PAR agonists. Gel-filtered platelets were stimulated with increasing concentrations of PAR1-AP and PAR4-AP for 5 minutes before the addition of an equal volume of human plasma. Thrombin generation was initiated as described in Materials and Methods. (A) Representative tracings of two independent experiments. (B) Decreases in lag time as a function of PAR1-AP and PAR4-AP concentration. Mean of two separate experiments with S.E.M. A nonpaired t test comparing the lowest values for each agonist demonstrated statistically significant differences. *P = 0.0054.
Expression of FV on Platelets in Response to PAR Activation.
Immunocytochemical analysis of unpermeabilized platelets showed a clear increase in FV expression on the platelet surface in response to PAR stimulation (Fig. 2A). Having confirmed the expression of FV on the platelet surface in response to stimulation, we next quantified the amount of FV on PAR1-AP– and PAR4-AP–stimulated platelets, to determine whether this could account for the more rapid thrombin generation observed with PAR4-AP–stimulated platelets.
Fig. 2.
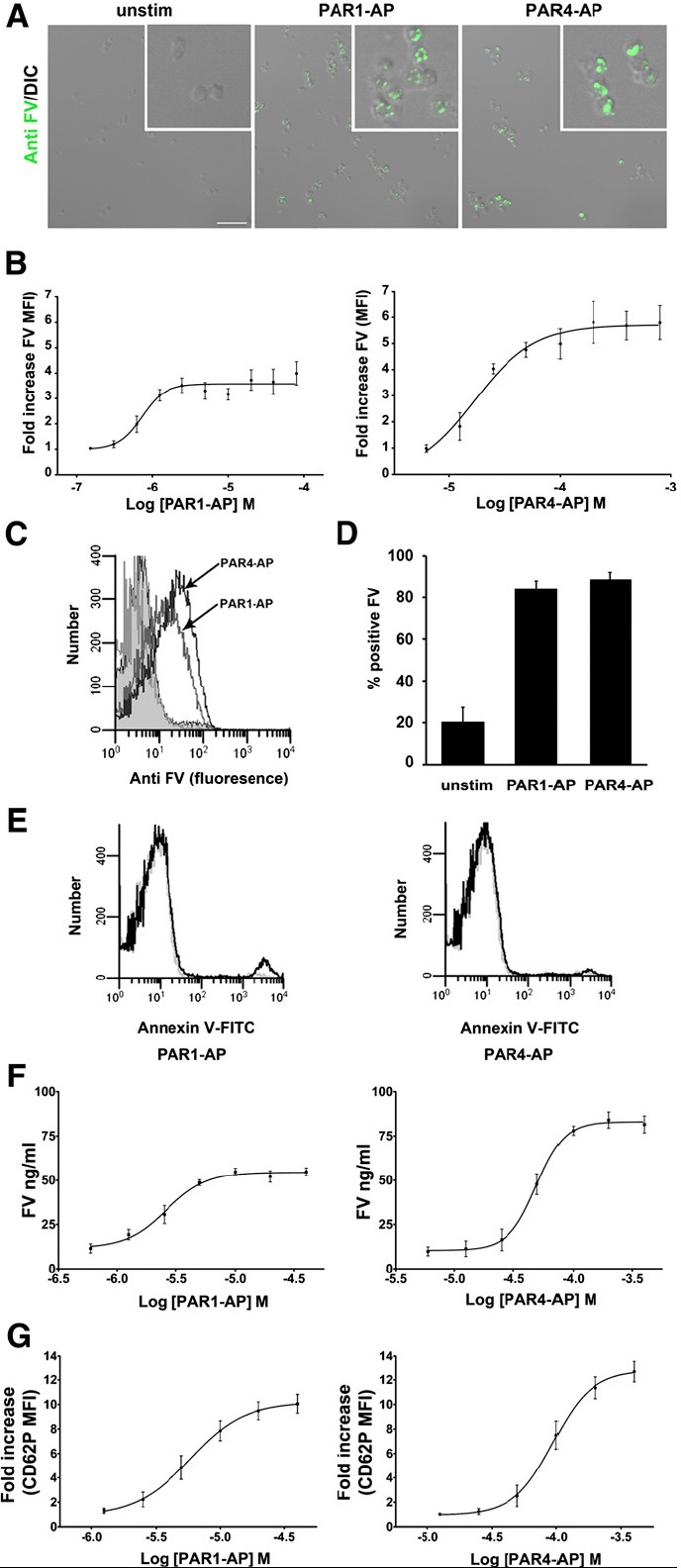
Expression of FV on platelets in response to PAR activation. (A) Immunocytochemical (ICC) analysis of FV surface expression in response to PAR stimulation. Intact, unpermeabilized platelets were stimulated with either 20 μM PAR1-AP or 200 μM PAR4-AP or vehicle control for 15 minutes and stained with 10 μg/ml antifactor V sheep polyclonal antibody and Alexa488 conjugated secondary. Scale bar = 10 μm. Inset = 3× magnification. (B) Flow cytometry analysis of FV surface expression on the platelet surface in response to PAR1-AP and PAR4-AP. Mean with S.E.M. (n = 4). A paired t test comparing maximum values for PAR1-AP and PAR4-AP from each individual nonlinear regression curve (sigmoidal dose response) indicated statistically significant differences (P = 0.021). Raw maximum MFI values ± S.E.M.: unstimulated, 4.6 ± 0.6; PAR1-AP, 18.2 ± 3.9; PAR4-AP, 27.5 ± 5.8. A paired t test comparing PAR1-AP and PAR4-AP MFI values indicated statistical significance (P = 0.018). (C) Histogram representation of FV staining on platelets stimulated with 20 μM PAR1-AP or 200 μM PAR4-AP. Gray unfilled, IgG control (overlaps with unstimulated). Gray filled, unstimulated. Gray unfilled, PAR1-AP and PAR4-AP (as indicated with arrows). (D) Percentage of platelets positive for FV staining. Platelets were stimulated with 20 μM PAR1-AP or 200 μM PAR4-AP. (E) Annexin V binding in response to stimulation with 20 μM PAR1-AP, 200 μM PAR4-AP, or vehicle control. Representative histograms from three repeats. Gray lines, unstimulated platelets. Black lines, stimulated platelets. (F) FV concentrations in the supernatants of platelets stimulated with increasing doses of PAR1-AP and PAR4-AP as determined by sandwich ELISA. Means with S.E.M. (n = 3). A paired t test comparing maximum values for PAR1-AP and PAR4-AP from each individual nonlinear regression curve (sigmoidal dose response) generated with PAR1-AP and PAR4-AP indicated statistically significanct differences (P = 0.0062). (G) P-selectin surface expression on platelets stimulated with increasing doses of PAR1-AP and PAR4-AP, as determined by flow cytometry. Means with S.E.M. (n = 3). A paired t test comparing maximum values for PAR1-AP and PAR4-AP from each individual nonlinear regression curve (sigmoidal dose response) generated with PAR1-AP (maximum, 10.1 ± 0.8 fold) and PAR4-AP (maximum, 12.7 ± 0.8 fold) indicated statistically significant differences (P = 0.0049). Maximal MFI values: unstim, 24.7 ± 4.7; PAR1-AP, 243.4 ± 28; PAR4-AP, 307.2 ± 38.6.
Gel-filtered platelets were treated with increasing concentrations of PAR1-AP and PAR4-AP for 15 minutes before fixation, staining, and analysis by flow cytometry. Dose-response curves were constructed to demonstrate that maximal responses had been reached. All data points comparing PAR1 and PAR4 were collected as matched samples with an unstimulated control from the same patient, allowing for the use of the more powerful paired t test for statistical analysis. Maximal fold-increase in mean fluorescence intensity (MFI) for PAR4-AP–stimulated platelets (5.6 ± 0.6 fold) was significantly higher than that for PAR1-AP–stimulated platelets (3.8 ± 0.3 fold) (P = 0.021, n = 4, paired t test) (Fig. 2B), indicating a 1.6-fold increase in platelet-bound FV on PAR4-stimulated platelets, compared with PAR1-AP–stimulated platelets. Maximal PAR1 stimulation increased the percentage of platelets staining positive for FV from 11.7 to 84.2%, whereas maximal PAR4 stimulation increased the percentage positive to 88.9% (Fig. 2D). Although PAR4 stimulation leads to slightly more positive cells, it does not account for the higher MFI values, indicating that PAR4 stimulation leads to a higher density of FV on the surface of platelets.
To examine whether PS exposure could account for the difference in platelet FV surface expression after PAR1 and PAR4 stimulation, we compared the level of PS exposure by assessing FITC-conjugated Annexin V binding to platelet membranes. Our data show a higher FITC MFI for PAR1-stimulated platelets and generally low PAR1 and PAR4 Annexin V-FITC binding (Fig. 2E). These data indicate that PS exposure does not account for the difference in FV association with the platelet surface.
Release of Platelet-FV from Intracellular Granules in Response to PAR Activation.
We compared the levels of FV in supernatants from PAR1- and PAR4-stimulated platelets as a direct measure of the FV release reaction. Samples were subjected to high-speed centrifugation before assaying to remove membrane debris and any trace of platelets. Dose-response curves constructed from enzyme-linked immunosorbance assays (ELISAs) (Fig. 2F) indicated significantly more FV released from PAR4-stimulated platelets (83 ± 4 ng/ml), compared with PAR1-stimulated platelets (54 ± 3 ng/ml; unstimulated, 10 ± 3 ng/ml; P = 0.0062, n = 3, paired t test). These data indicate that 1.5-fold more FV is released from PAR4-stimulated platelets and suggest that PAR4 is more efficient at liberating FV from intracellular stores.
Platelet-FV is reportedly stored in α-granules (Alberio et al., 2000; Duckers et al., 2010). To determine whether the difference in FV levels on the surface and in the supernatant of PAR1-AP– and PAR4-AP–stimulated platelets was attributable to a difference in each receptor’s ability to induce mobilization and secretion of α-granules, we compared PAR1-AP– and PAR4-AP–induced P-selectin levels on the platelet surface measured by flow cytometry. Dose-response curves constructed with increasing concentrations of PAR1-AP and PAR4-AP indicate a significantly higher maximal response for PAR4-AP (12.7 ± 0.8 fold) than for PAR1-AP–stimulated platelets (10.1 ± 0.8 fold; P =.0049, n = 3, paired t test) (Fig. 2G). These data suggest that the enhanced FV expression in response to PAR4-AP may be attributable to more efficacious mobilization of α-granules by PAR4. The magnitude of the differential between PAR1-AP– and PAR4-AP–mediated responses for FV and P-selectin expression were not identical, suggesting heterogeneity in cargo of the α-granule population.
Platelet Microparticle Production in Response to PAR Activation.
Circulating microparticles represent procoagulant surfaces and have been shown to present binding sites for FV (Sims et al., 1988; Sims et al., 1989; Monkovic and Tracy, 1990; Alberio et al., 2000). We used flow cytometry to quantify the generation of platelet-derived microparticles in response to PAR stimulation. Appropriate gating by size for microparticles was determined using forward and side-scatter analysis with 1 μm beads. Figure 3A (left panel) shows a forward and side-scatter dot plot of 1 μm calibration beads and the size gate based on the forward scatter values used to identify microparticles in subsequent experiments. The right panel shows the dot plot from PAR1-stimulated platelets. Particles above the box are whole platelets, whereas particles in the box were candidates for microparticles. There is an increase in the number of particles under 1 μm in response to both PAR1 and PAR4 stimulation (Fig. 3B). To confirm that these particles were platelet derived, we incubated the microparticle-enriched samples with antibodies against the platelet integrin GPIIb (CD41) and P-selectin (CD62P). Maximal PAR4 stimulation led to a 10.3 ± 0.0–fold increase in GPIIb- and P-selectin–positive microparticles, compared with unstimulated platelets, whereas maximal PAR1 stimulation led to a 4.3 ± 0.3–fold increase (P = 0.0142, n = 2, t test) (Fig. 3C). Consistently, we observed FV expression on the surface of GPIIb- and P-selectin–positive microparticles (Supplemental Fig. 1). The percentage of PAR1-AP– and PAR4-AP–induced particles positive for FV was not significantly different; however, the number of FV-positive microparticles in PAR4-stimulated samples exceeds that in PAR1-stimulated samples by over 2-fold.
Fig. 3.

Platelet microparticle production in response to PAR activation. (A) Representative controls for gating of microparticles against whole platelets. Left panel, forward and side-scatter dot plots of 1-μm beads. Right panel, forward and side-scatter dot plots of platelet sample stimulated with 20 μM PAR1. (B) Forward and side-scatter dot plots of microparticle-enriched samples prepared from unstimulated platelets and platelets stimulated with 20 μM PAR1-AP and 200 μM PAR4-AP showing the depletion of whole platelets from microparticle-enriched samples. (C) Microparticle production in response to increasing concentrations of PAR1-AP and PAR4-AP. Mean fold increase in CD62P (P-selectin) and CD41 (integrin α-IIb) positive particles under 1μm (n = 2). A t test comparing maximal values from each dose-response curve (PAR1-AP, 4.3 ± 0.3 fold; PAR4-AP, 10.3 ± 0.0 fold) indicates statistically significant differences (P = 0.0142).
Regulation of Myosin Light Chain Phosphorylation by PAR-Stimulated Platelets.
Platelet degranulation occurs downstream of Gαq-mediated PKC activation and Ca2+ release and is enhanced by Gα12/13-mediated RhoA activation. The precise mechanism by which microparticle release occurs is largely unknown. It is known that the calcium response downstream of PAR4 is sustained, compared with PAR1. As a possible mechanistic explanation of the difference between PAR1 and PAR4 in the aforementioned procoagulant phenotypes, we examined PAR1- and PAR4-mediated PKC-substrate serine phosphorylation in addition to MLC phosphorylation at both serine 19 (pMLC S19) and threonine 18 (pMLC T18). PAR4-mediated phosphorylation of PKC substrates was more robust and more sustained than was PAR1 (Fig. 4A). Both PAR1 and PAR4 induced phosphorylation of MLC at S19 and T18; however, the PAR4 response was markedly more robust and sustained over the course of 15 minutes (Fig. 4A). Figure 4B shows the quantification of three separate experiments and shows the rapid nature of MLC dephosphorylation downstream of PAR1, in comparison with PAR4.
Fig. 4.

Regulation of myosin light chain phosphorylation by PAR-stimulated platelets. (A) PAR1- and PAR4-mediated PKC (s) substrate phosphorylation and myosin light chain phosphorylation at S19 (pMLC S19) and T18 (pMLC T18). Platelets were stimulated with 20 μM PAR1-AP and 200 μM PAR4-AP for the indicated periods. Reactions were stopped, and samples were prepared for Western blot analysis as indicated in Materials and Methods. Top panel, PKC (s)-substrate; middle panels, pMLC S19 and T18; lower panel, p44/p42 ERK loading controls. (B) Quantification of pMLC S19 and T18. Data were collected from three independent experiments and analyzed using Image J. Data are expressed as fold increase relative to the density of bands in unstimulated lanes. Asterisks indicate statistically significant differences between PAR1-AP– and PAR4-AP–stimulated MLC phosphorylation at the indicated time point with use of a paired t test. P values (S19, 1 minute = 0.0173, 2 minute = 0.008, 5 minute = 0.0133, 10 minute = 0.0222, 15 minute = 0.017, 30 minute = 0.0038; T18, 10 minute, 0.0049, 15 minute = 0.0471, 30 minute = 0.0498)
The Effect of PKC Inhibition and RhoA Pathway inhibition on Platelet Secretion and Microparticle Production.
Because of the sustained nature of the PAR4 PKC activity and MLC phosphorylation, we constructed a time course of FV release from the platelet. Of interest, two distinct phases of secretion were noted: an initial response that occurs within the first 2 minutes and a more sustained response that persists for up to 15 minutes (Fig. 5B). The amount of FV released from PAR1- and PAR4-stimulated platelets at 2 minutes was essentially identical; however, at 10 and 15 minutes, the amount of FV released was markedly different and reflected the differences between PAR1- and PAR4-stimulated platelets previously observed by flow cytometry and ELISA at single times (Fig. 2). To determine whether sustained phosphorylation of MLC downstream of PAR4 could account for the difference in FV secretion between the two agonists, we determined the effect of the Rho kinase inhibitor Y-27632 on MLC phosphorylation and FV secretion. Pretreatment with Y-27632 completely abolished PAR1- or PAR4-mediated MLC phosphorylation at T18 and reduced phosphorylation at S19 (Fig. 5A). Of interest, at 10 and 15 minutes, Y-27632 significantly reduced FV secretion downstream of PAR4 stimulation, close to that observed with PAR1-stimulated platelets, whereas the PAR1 response was not significantly inhibited by Y-27632 at any time (Fig. 5B). The amount of FV released at 1 and 2 minutes by either agonist was not significantly reduced (Fig. 5B). In contrast to Y-27632, the pan PKC inhibitor BIM-1 completely abolished PAR-induced secretion of FV (Fig. 5C). These data indicate biphasic dense granule secretion downstream of PAR stimulation in platelets with an essential role for PKC and an enhancing role for Rho kinase activity and MLC phosphorylation. In addition, these data suggest that PAR4 is capable of inducing more FV secretion from the platelet because of a stronger Rho signaling component initiated by the receptor.
Fig. 5.

The effect of PKC inhibition and Rho-kinase inhibition on platelet secretion and microparticle production. (A) The effect of Y-27632 on S19 and T18 pMLC. Platelets were preincubated with 10 μM Y-27632 or water for 10 minutes before stimulation with 20 μM PAR1-AP or 200 μM PAR4-AP for the indicated periods. (B) The effect of Rho-kinase inhibitor on PAR-mediated FV release. Platelets were preincubated with 10 μM Y-27632 for 10 minutes before stimulation with 20 μM PAR1-AP or 200 μM PAR4-AP for the indicated periods, and supernatants were prepared for sandwich ELISA, as indicated in Materials and Methods. Shown are the means and S.E.M. (n = 3). Paired t test *PAR4-AP versus PAR4-AP + Y-27632, #PAR4-AP versus PAR1-AP, **PAR4-AP versus PAR1-AP + Y-27632. 1 minute *P value = 0.0327; 2 minute *P value = 0.017, **P value = 0.0117; 5 minute *P value = 0.0123, **P value = 0.0463; 10 minute *P value = 0.048, #P value = 0.0145, **P value = 0.0072; 15 minute *P value = 0.0002, #P value = 0.0203, **P value = 0.0031. C. The effect of PKC inhibitor on PAR-mediated FV secretion. Platelets were preincubated with 10 μM BIM-1 or DMSO for 10 minutes before stimulation with 20 μM PAR1-AP or 200 μM PAR4-AP for 15 minutes. Shown are the means and S.E.M. (n = 4). Paired t test, PAR1-AP versus PAR1-AP + BIM-1 *P = 0.0312, PAR4-AP versus PAR4-AP + BIM-1. **P = 0.016. Raw values for FV concentration: unstimulated, 22.7 ± 4.2 ng/ml; unstimulated+BIM-1, 21.4 ± 3.8 ng/ml; PAR1-AP, 68.9 ± 8.1 ng/ml; PAR1-AP+BIM-1, 34.7 ± 12.5 ng/ml; PAR4-AP, 93.3 ± 7.8 ng/ml; PAR4-AP+BIM-1, 30.1 ± 6.3. (D) The effect of Rho-kinase inhibitor on PAR-mediated microparticle production. Platelets were preincubated with 10 μM Y-27632 for 10 minutes before stimulation with 20 μM PAR1-AP or 200 μM PAR4-AP for 30 minutes. Microparticle-enriched supernatants were prepared as indicated in Materials and Methods. Shown are the means with S.E.M. (n = 3). A paired t test comparing PAR4-AP with PAR4-AP+Y-27632 revealed statistically significant differences. *P = 0.0024. A paired t test comparing PAR1-AP with PAR1-AP+Y-27632 did not demonstrate statistically significant differences (P = 0.117). A paired t test comparing PAR1-AP with PAR4-AP showed statistically significant differences (P = 0.002). Raw values for microparticle production (dual positive particles): unstimulated, 79.5 ± 18.4; unstimulated+Y-27632, 77.6 ± 15.6; PAR1-AP, 164.9 ± 31.3; PAR1-AP+Y-27632, 269.4 ± 57.8; PAR4-AP, 769.0 ± 151.2; PAR4-AP+Y27632, 216.8 ± 29.9. (E) The effect of PKC inhibitor on PAR-mediated microparticle production. Platelets were preincubated with 10 μM BIM-1 for 10 minutes before stimulation with 20 μM PAR1-AP or 200 μM PAR4-AP for 30 minutes. Microparticle-enriched platelet supernatants were prepared as indicated in Materials and Methods. Shown are the means with S.E.M. (n = 4). Raw values for microparticle production (dual positive particles): unstimulated, 120.1 ± 15.5; unstimulated+BIM-1, 76.1 ± 8.6; PAR1-AP, 300.6 ± 71.9; PAR1-AP+BIM-1, 5092.2 ± 1662.1; PAR4-AP, 821.5 ± 332.5; PAR4-AP+BIM-1, 139.3 ± 33.6.
We also examined the effects of the PKC inhibitor and the Rho kinase inhibitor on PAR-induced microparticle production. The Rho kinase inhibitor had no significant effect on PAR1-AP–induced microparticle production; however, PAR4-mediated microparticle production was reduced to PAR1-mediated levels in the presence of Y-27632 (Fig. 5D). These data suggest that at least a component of platelet microparticle production is driven by the Rho pathway, in particular Rho-kinase activity, and that the difference in PAR1- and PAR4-mediated microparticle production is attributable to the sustained MLC phosphorylation downstream of PAR4, compared with PAR1. Inhibition of PKC with the pan-PKC inhibitor BIM-1 also had opposing effects on PAR1- and PAR4-mediated microparticle production. Downstream of PAR1, BIM-1 caused a 15-fold enhancement of microparticle production, whereas downstream of PAR4, BIM-1 significantly reduced microparticle production (Fig. 5E).
Prothrombinase Complex Activity on PAR-Stimulated Platelets.
To address the functional relevance of the presentation of the platelet procoagulant phenotype, we evaluated assembly and activity of the prothrombinase complex on platelets with use of a modified thrombin generation assay. FXa and FVa associate on the surface of activated platelets to form the prothrombinase complex. Complex formation greatly enhances the rate of FXa-mediated conversion of FII to thrombin. Purified platelets were reconstituted with the minimal coagulation factors necessary to generate thrombin (FII+FXa+AT), such that FV expression by the platelet was limiting. Plasma was not used in this instance, because the purpose of the experiment was to confirm the function of the excess FV release by PAR4-AP–stimulated platelets as observed in previous experiments.
Platelets were mixed with FII, FXa, and AT and stimulated with either PAR1-AP or PAR4-AP. Unstimulated platelets generated peak thrombin levels of 5.0 ± 1.5 nM, PAR1-AP–stimulated platelets generated 32.0 ± 5.5 nM thrombin, and PAR4-AP–stimulated platelets generated 48.7 ± 7.9 nM thrombin (Fig. 6, A and B). Lag times for PAR4-stimulated platelets were significantly shorter under these conditions (Fig. 6, A and C). These data indicate that PAR4-AP–stimulated platelets support greater prothrombinase complex activity and are capable of generating more thrombin than platelets stimulated with PAR1-AP.
Fig. 6.

Assembly of the prothrombinase complex on PAR-stimulated platelets. (A) Thrombin generation on platelets simulated with 20 μM PAR1-AP or 200 μM PAR4-AP. Stimulated platelets are mixed with coagulation factors: FII (1.4 μM), FXa (100 nM), and AT (100 μg/ml). Representative tracing from three separate experiments. (B) Peak thrombin generation; mean with S.E.M. (n = 3). A paired t test comparing peak thrombin generation in response to PAR1-AP and PAR4-AP indicated statistically significant differences. *P = 0.0129. (C) Lag time, time it takes to reach 10 nM thrombin; mean with S.E.M. (n = 3). A paired t test comparing lag times for PAR1-AP and PAR4-AP stimulated platelets indicated statistically significant differences. *P = 0.0206. (D) Effect of Rho-kinase inhibitor on PAR-induced thrombin generation. Platelets were preincubated with 10 μM Y-27632 or water vehicle control for 10 minutes before stimulation with 20 μM PAR1 or 200 μM PAR4-AP. Thrombin generation was conducted as outlined above and in Materials and Methods. Mean with S.E.M. (n = 3). A paired t test comparing peak thrombin levels for PAR4-AP and PAR4-AP+Y-27632 indicated statistically significant differences. *P = 0.0021.
Of importance, control studies indicate that the thrombin generated in this assay was dependent on platelet activation and expression of the cofactor FV on the platelet surface. No significant amount of thrombin was generated in the absence of FXa (II+AT+activated platelets) or when FII, FXa, and AT were incubated with phospholipid vesicles in place of platelets (Fig. 6A). These control studies indicate that (1) FII conversion to thrombin is minimal on the surface of an activated platelet without FXa and (2) FII is activated poorly by FXa in the absence of platelet stimulation and provision of FV. To confirm that activity observed under these conditions was dependent on FV, platelets were incubated with increasing concentrations of an anti-FV–neutralizing antibody before measuring thrombin generation. Supplemental Fig. 2A shows that 10 μg/ml of the antibody reduced thrombin generation by 25% and increasing concentrations reduced the thrombin generation peak close to unstimulated values.
An alternative explanation for the enhanced thrombin generation observed in PAR4-AP–stimulated platelet samples is the simultaneous engagement of PAR1 by newly generated thrombin and PAR4 by the PAR4-AP. Simultaneous engagement of PAR1 and PAR4 on PAR1-AP–stimulated samples, on the other hand, would not occur until later in the assay, after more thrombin has been generated. To investigate the contribution of PAR activation by newly generated thrombin in the thrombin generation assay, we used PAR-directed thrombin cleavage–blocking antibodies to inhibit any feed-forward PAR activation by thrombin generated in the system. The cleavage-blocking antibodies generated by Ofosu et al. (2008), when used in combination, substantially inhibit peak calcium levels in response to up to 2 nM thrombin and completely inhibit the initial rate of intracellular calcium mobilization in response to 10 nM thrombin (Supplemental Fig. 2B). Conveniently, both PAR1-AP and PAR4-AP are still able to activate platelets in the presence of the cleavage-blocking antibodies (Supplemental Fig. 2B). Platelets were incubated with PAR-1 span IgY and PAR-4 span IgY for 30 minutes before stimulation with PAR1-AP or PAR4-AP. Thrombin generation was conducted under the same conditions as previously indicated. The combination of PAR-1 span IgY and PAR-4 span IgY did not significantly shift the peak or lag time, compared with control conditions (Supplemental Fig. 2C). These data indicate that feed-forward activation of PARs by thrombin generated in the system is not significantly contributing to the level or rate of thrombin generated. Instead thrombin generation is largely reflective of the initial phenotypes established by PAR1-AP and PAR4-AP alone.
Finally, we confirmed the role of Rho kinase in the presentation of procoagulant phenotypes downstream of PAR4 by preincubating platelets with Y-27632 before assessing prothrombinase complex activity with the thrombin generation assay. Incubation of platelets with Y-27632 inhibited PAR4-mediated thrombin generation by 25% but had no significant effect on PAR1-mediated thrombin generation (Fig. 6D). These data confirm the role of sustained MLC phosphorylation in enhancing FV release and provide some insight into the function and implications of this signaling pathway in platelet physiology.
Discussion
We have shown that stimulation of PAR4 on platelets results in the presentation of a procoagulant phenotype exceeding that by stimulation of PAR1 and is characterized by the secretion of more platelet-FV from intracellular stores, the binding of more FV to the platelet surface, and the shedding of dramatically more FV-positive microparticles. The greater procoagulant response results in shorter lag times in the initiation of thrombin generation in full plasma and higher peak thrombin concentrations in a thrombin generation assay measuring activity of the prothrombinase complex. The apparent mechanism through which the PAR4-AP response supersedes the PAR1-AP response is, at least in part, through more sustained phosphorylation of MLC at T18 and S19 downstream of PAR4 activation. The Rho-kinase inhibitor Y-27632 reduced PAR4-AP–mediated FV release, microparticle production, and thrombin generation, but had little effect on PAR1-AP–mediated events. Moreover, we have shown, for the first time to our knowledge, a Rho-kinase component to microparticle production downstream of PAR activation in platelets.
Although PAR1 and PAR4 would not be individually engaged endogenously, it is important to understand the full capacity of each receptor to mediate events involved in hemostasis and thrombosis, to design safe and effective thrombin receptor antagonists. Our data suggest that PAR1- and PAR4-induced procaogulant phenotypes are not redundant and that PAR4 may be playing a unique and yet undescribed role in hemostasis. Currently, there are no data on when PAR4 is engaged during hemostasis. However, because its lower affinity for thrombin, PAR4 should be engaged after PAR1 as the concentration of thrombin at the site of injury increases. This order suggests that the impressive procoagulant response mediated by PAR4 may play a role in supporting continuous thrombin generation during clot formation and propagation. The recently discovered role for RhoA in thrombus stability under flow (Pleines et al., 2012) and the prolonged phosphorylation of MLC downstream of PAR4 are consistent with this hypothesis. Studies are currently being designed to directly address the role of PAR4 in clot formation and stability with use of dynamic rather than static assays that incorporate flow, shear stress, and thrombus formation.
PAR4-AP–stimulated platelets caused the release of more FV and a higher density of FV on the platelet membrane. These data indicate that PAR4 stimulation induces a greater release reaction of FV-containing intracellular granules than does PAR1. We cannot exclude the possibility that FV binding sites on platelets may be differentially regulated by PAR1 and PAR4. However, it seems more likely that the higher density of FV on PAR4-stimulated platelets is the result of higher concentrations of FV released, particularly in light of the lower level of PS exposure observed with PAR4-AP– compared with PAR1-AP–stimulated platelets.
Our group and several others have presented data showing a difference in platelet responses mediated by PAR1 and PAR4 despite being coupled to the same G-proteins, Gαq and Gα12/13. The data presented here in conjunction with work done by Falker et al. (2011) suggest that there is a difference in the regulation of PAR1- and PAR4-initiated signaling pathways instead of an entirely unique signaling pathway initiated by one receptor or the other. We cannot comment on precisely what regulatory element is mediating the enhanced response downstream of PAR4 relative to PAR1; however, we present data that both Gq and G12/13 pathways are stronger and more sustained downstream of PAR4 stimulation. Falker et al. (2011) recently showed that PAR1-mediated Ca2+ mobilization, PKC activity, and granule secretion undergo homologous desensitization, which can be rescued by PAR4 stimulation (Falker et al., 2011). Our data indicate that PKC serine substrate phosphorylation was both stronger and more sustained downstream of PAR4. These data agree with a model of PAR signaling presented by Falker et al. (2011) suggesting that PAR1 signaling desensitizes and PAR4 signaling persists. Falker et al. (2011) also revealed that stimulation of PAR4 could rescue PAR1 signaling desensitization in a Rho-kinase–dependent manner. This suggests that the persistence of the PAR4 signal involves regulation of MLC phosphorylation. RhoA, which activates Rho-kinase, has also been shown to be involved in the full granule release response from platelets. Similar to PKC activation, PAR4-stimulated MLC phosphorylation at both sites (T18 and S19) was more robust and more sustained than was that with PAR1.
As an additional measure of procoagulant potential, we compared PAR1- with PAR4-induced microparticle formation. Recently, Connor et al. showed that Annexin V binding to platelet-derived microparticles relies on the type of buffer and the Ca2+ concentration used to analyze platelet-poor plasma samples (Connor et al., 2010). Because our goal was to compare the efficacy of PAR1 with that of PAR4 in the production of microparticles, we relied on CD41 and CD62P staining to confirm that the microparticles were derived from the platelet membrane. Our study represents the first documentation of microparticle formation in response to selective PAR1 or PAR4 activation and shows that the majority of microparticles generated in response to PAR activation are mediated through PAR4 stimulation, thus revealing an unappreciated role for PAR4 in microparticle generation. Because of the link between circulating microparticles and disease states, these data also have implications for PAR4 as the proper target choice for a thrombin receptor antagonist. The signaling events leading to microparticle formation are not well understood. Elevations in intracellular Ca2+ and activation of the Ca2+-dependent protease calpain have been implicated in microparticle formation, in addition to acute decreases in PIP2 membrane composition, but do not appear to account for all pathways (O'Connell et al., 2005; Flaumenhaft et al., 2009; Flaumenhaft et al., 2010). When we preincubated platelets with Y-27632, PAR4-mediated microparticle production was reduced to PAR1-mediated levels. These data indicate, for the first time, that the Rho pathway and MLC phosphorylation contribute to microparticle production from human platelets. This is plausible because RhoA activity has been implicated in membrane blebbing during apoptosis in other cell lines (Coleman et al., 2001). We also showed that PKC inhibition reduced PAR4-mediated microparticle production but drastically enhanced PAR1-mediated microparticle production. Because of the enhancing effect that BIM-1 has on PAR1-mediated calcium mobilization as documented by Poole et al. (Harper and Poole, 2011) and the established role of intracellular calcium in microparticle production, the differential effect of the PKC inhibitor should be anticipated. These data indicate that, in addition to calcium and acute decreases in PIP2 membrane composition, Rho-kinase is an important signaling component in microparticle production downstream of PAR stimulation. Moreover, because of the role of Myosin IIa in platelet function, the importance of Rho-kinase signaling suggests that platelet contraction may be a precursor to or may enhance microparticle production.
Finally, we showed that PAR4 induced more prothrombinase complex activity than did PAR1-stimulated platelet with use of a thrombin generation assay consisting of purified components. Vretenbrant et al. revealed that PAR4 is involved in the initiation of thrombin generation and the development of clot elasticity with use of thrombin generation assays in plasma and PAR inhibitors (Vretenbrant et al., 2007). Our data showing prolonged signaling and more robust FV secretion point to a role for PAR4 in continuous thrombin generation during clot development, which would be consistent with a role for PAR4 in regulating clot dynamics. We cannot comment on which receptor would be involved in initiating thrombin generation, because we did not use antagonists to explore their relative roles, because of a lack of a potent small molecule antagonist for PAR4. Vretenbrant et al. also revealed a more robust response for PAR4, compared with PAR1, when measuring fibrinogen binding, paralleling our observations of more FV release and more microparticle production.
Our data establish that PAR4 stimulation is more efficacious than is PAR1 stimulation in the induction of procoagulant phenotypes on platelets. Recently, clinical trials of the PAR1 antagonist Vorapaxar were curtailed because of a high number of intracranial hemorrhagic events (Tricoci et al., 2011). Because of the high affinity of PAR1 for thrombin and its engagement by newly generated thrombin early in the process of hemostasis, its inhibition may suppress the platelet’s ability to respond to newly generated thrombin and amplify thrombin generation, a critical process in hemostasis. Therefore, unwanted bleeding adverse effects are an anticipated complication of therapy targeting PAR1. Because thrombin has a lower affinity for PAR4 than for PAR1, PAR4 would be engaged only after higher concentrations of thrombin are reached at a vascular site of injury. Specific inhibition of PAR4 would leave PAR1 signaling intact, allowing platelets to respond to low concentrations of thrombin, perhaps facilitating the initial amplification of thrombin generation and preserving hemostasis.
Supplementary Material
Acknowledgments
The authors thank Dr. Fred Ofosu for the provision of the PAR-AP span and PAR4-AP span IgY thrombin cleavage blocking antibodies; Nancy Colowick for technical assistance; and Catherine Alford for technical advice and assistance.
Abbreviations
- AP
activating peptide
- ELISA
enzyme-linked immunosorbent assay
- F
factor
- FII
prothrombin
- FV
factor V
- MFI
mean fluorescence intensity
- PAR
protease-activated receptor
- PBS
phosphate-buffered saline
- PIP2
phosphatidyl inositol 4,5-biphosphate
- PS
phosphatidyl-serine
Authorship Contributions
Participated in research design: Duvernay, Young, Schoenecker, Hamm.
Conducted experiments: Duvernay, Young.
Performed data analysis: Duvernay, Young.
Wrote or contributed to the writing of the manuscript: Duvernay, Young, Schoenecker, Gailani, Hamm.
Footnotes
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
This work was supported by the National Institutes of Health [Grant R01 HL084388]; the National Institutes of Health National Heart, Lung, and Blood Institute [Grant P50 HL081009]; and the National Institutes of Health [Grants CA68485, DK20593, DK50404, HD15052, DK59637, and EY008126] (for VUMC Cell Imaging Shared Resource).
References
- Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. (2000) Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood 95:1694–1702 [PubMed] [Google Scholar]
- Bilodeau ML, Hamm HE. (2007) Regulation of protease-activated receptor (PAR) 1 and PAR4 signaling in human platelets by compartmentalized cyclic nucleotide actions. J Pharmacol Exp Ther 322:778–788 [DOI] [PubMed] [Google Scholar]
- Cohen Z, Gonzales RF, Davis-Gorman GF, Copeland JG, McDonagh PF. (2002) Thrombin activity and platelet microparticle formation are increased in type 2 diabetic platelets: a potential correlation with caspase activation. Thromb Res 107:217–221 [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. (2001) Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 3:339–345 [DOI] [PubMed] [Google Scholar]
- Connor DE, Exner T, Ma DD, Joseph JE. (2010) The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thromb Haemost 103:1044–1052 [DOI] [PubMed] [Google Scholar]
- Coughlin SR. (2000) Thrombin signalling and protease-activated receptors. Nature 407:258–264 [DOI] [PubMed] [Google Scholar]
- Covic L, Gresser AL, Kuliopulos A. (2000) Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 39:5458–5467 [DOI] [PubMed] [Google Scholar]
- De Smedt E, Al Dieri R, Spronk HM, Hamulyak K, ten Cate H, Hemker HC. (2008) The technique of measuring thrombin generation with fluorogenic substrates: 1. Necessity of adequate calibration. Thromb Haemost 100:343–349 [PubMed] [Google Scholar]
- Diamandis M, Veljkovic DK, Maurer-Spurej E, Rivard GE, Hayward CP. (2008) Quebec platelet disorder: features, pathogenesis and treatment. Blood Coagul Fibrinolysis 19:109–119 [DOI] [PubMed] [Google Scholar]
- Diamant M, Nieuwland R, Pablo RF, Sturk A, Smit JW, Radder JK. (2002) Elevated numbers of tissue-factor exposing microparticles correlate with components of the metabolic syndrome in uncomplicated type 2 diabetes mellitus. Circulation 106:2442–2447 [DOI] [PubMed] [Google Scholar]
- Diehl P, Nagy F, Sossong V, Helbing T, Beyersdorf F, Olschewski M, Bode C, Moser M. (2008) Increased levels of circulating microparticles in patients with severe aortic valve stenosis. Thromb Haemost 99:711–719 [DOI] [PubMed] [Google Scholar]
- Duckers C, Simioni P, Spiezia L, Radu C, Dabrilli P, Gavasso S, Rosing J, Castoldi E. (2010) Residual platelet factor V ensures thrombin generation in patients with severe congenital factor V deficiency and mild bleeding symptoms. Blood 115:879–886 [DOI] [PubMed] [Google Scholar]
- Fälker K, Haglund L, Gunnarsson P, Nylander M, Lindahl TL, Grenegård M. (2011) Protease-activated receptor 1 (PAR1) signalling desensitization is counteracted via PAR4 signalling in human platelets. Biochem J 436:469–480 [DOI] [PubMed] [Google Scholar]
- Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, Coughlin SR. (2000) Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J Biol Chem 275:19728–19734 [DOI] [PubMed] [Google Scholar]
- Flaumenhaft R, Dilks JR, Richardson J, Alden E, Patel-Hett SR, Battinelli E, Klement GL, Sola-Visner M, Italiano JE., Jr (2009) Megakaryocyte-derived microparticles: direct visualization and distinction from platelet-derived microparticles. Blood 113:1112–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaumenhaft R, Mairuhu AT, Italiano JE. (2010) Platelet- and megakaryocyte-derived microparticles. Semin Thromb Hemost 36:881–887 [DOI] [PubMed] [Google Scholar]
- Gabbeta J, Yang X, Kowalska MA, Sun L, Dhanasekaran N, Rao AK. (1997) Platelet signal transduction defect with Galpha subunit dysfunction and diminished Galphaq in a patient with abnormal platelet responses. Proc Natl Acad Sci USA 94:8750–8755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz TM, Dangelmaier CA, Jin J, Daniel JL, Kunapuli SP. (2010) Differential phosphorylation of myosin light chain (Thr)18 and (Ser)19 and functional implications in platelets. J Thromb Haemost 8:2283–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert GE, Sims PJ, Wiedmer T, Furie B, Furie BC, Shattil SJ. (1991) Platelet-derived microparticles express high affinity receptors for factor VIII. J Biol Chem 266:17261–17268 [PubMed] [Google Scholar]
- Grigg AP, Dauer R, Thurlow PJ. (1989) Bleeding due to an acquired inhibitor of platelet associated factor V. Aust N Z J Med 19:310–314 [DOI] [PubMed] [Google Scholar]
- Hammes SR, Coughlin SR. (1999) Protease-activated receptor-1 can mediate responses to SFLLRN in thrombin-desensitized cells: evidence for a novel mechanism for preventing or terminating signaling by PAR1’s tethered ligand. Biochemistry 38:2486–2493 [DOI] [PubMed] [Google Scholar]
- Harper MT, Poole AW. (2011) PKC inhibition markedly enhances Ca2+ signaling and phosphatidylserine exposure downstream of protease-activated receptor-1 but not protease-activated receptor-4 in human platelets. J Thromb Haemost 9:1599–1607 [DOI] [PubMed] [Google Scholar]
- Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, Lecompte T, Béguin S. (2003) Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb 33:4–15 [DOI] [PubMed] [Google Scholar]
- Hoffman M, Monroe DM, Roberts HR. (1992a) Coagulation factor IXa binding to activated platelets and platelet-derived microparticles: a flow cytometric study. Thromb Haemost 68:74–78 [PubMed] [Google Scholar]
- Hoffman M, Monroe DM, Roberts HR. (1992b) A rapid method to isolate platelets from human blood by density gradient centrifugation. Am J Clin Pathol 98:531–533 [DOI] [PubMed] [Google Scholar]
- Holinstat M, Preininger AM, Milne SB, Hudson WJ, Brown HA, Hamm HE. (2009) Irreversible platelet activation requires protease-activated receptor 1-mediated signaling to phosphatidylinositol phosphates. Mol Pharmacol 76:301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holinstat M, Voss B, Bilodeau ML, Hamm HE. (2007) Protease-activated receptors differentially regulate human platelet activation through a phosphatidic acid-dependent pathway. Mol Pharmacol 71:686–694 [DOI] [PubMed] [Google Scholar]
- Holinstat M, Voss B, Bilodeau ML, McLaughlin JN, Cleator J, Hamm HE. (2006) PAR4, but not PAR1, signals human platelet aggregation via Ca2+ mobilization and synergistic P2Y12 receptor activation. J Biol Chem 281:26665–26674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano JE, Jr, Mairuhu AT, Flaumenhaft R. (2010) Clinical relevance of microparticles from platelets and megakaryocytes. Curr Opin Hematol 17:578–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Mao Y, Thomas D, Kim S, Daniel JL, Kunapuli SP. (2009) RhoA downstream of G(q) and G(12/13) pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem Pharmacol 77:835–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H, Sugiyama S, Kugiyama K, Fukushima H, Watanabe K, Sakamoto T, Yoshimura M, Jinnouchi H, Ogawa H. (2006) Elevated levels of remnant lipoproteins are associated with plasma platelet microparticles in patients with type-2 diabetes mellitus without obstructive coronary artery disease. Eur Heart J 27:817–823 [DOI] [PubMed] [Google Scholar]
- Liu LW, Vu TK, Esmon CT, Coughlin SR. (1991) The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J Biol Chem 266:16977–16980 [PubMed] [Google Scholar]
- Ma L, Perini R, McKnight W, Dicay M, Klein A, Hollenberg MD, Wallace JL. (2005) Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc Natl Acad Sci USA 102:216–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat Z, Benamer H, Hugel B, Benessiano J, Steg PG, Freyssinet JM, Tedgui A. (2000) Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 101:841–843 [DOI] [PubMed] [Google Scholar]
- Moers A, Nieswandt B, Massberg S, Wettschureck N, Grüner S, Konrad I, Schulte V, Aktas B, Gratacap MP, Simon MI, et al. (2003) G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med 9:1418–1422 [DOI] [PubMed] [Google Scholar]
- Monković DD, Tracy PB. (1990) Functional characterization of human platelet-released factor V and its activation by factor Xa and thrombin. J Biol Chem 265:17132–17140 [PubMed] [Google Scholar]
- Monroe DM, Hoffman M, Roberts HR. (2002) Platelets and thrombin generation. Arterioscler Thromb Vasc Biol 22:1381–1389 [DOI] [PubMed] [Google Scholar]
- Nesheim ME, Nichols WL, Cole TL, Houston JG, Schenk RB, Mann KG, Bowie EJ. (1986) Isolation and study of an acquired inhibitor of human coagulation factor V. J Clin Invest 77:405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura S, Suzuki M, Katsura K, Xie GL, Miyazaki Y, Miyake T, Kido H, Kagawa H, Fukuhara S. (1995) Platelet-derived microparticles may influence the development of atherosclerosis in diabetes mellitus. Atherosclerosis 116:235–240 [DOI] [PubMed] [Google Scholar]
- O’Connell DJ, Rozenvayn N, Flaumenhaft R. (2005) Phosphatidylinositol 4,5-bisphosphate regulates activation-induced platelet microparticle formation. Biochemistry 44:6361–6370 [DOI] [PubMed] [Google Scholar]
- Offermanns S, Toombs CF, Hu YH, Simon MI. (1997) Defective platelet activation in G alpha(q)-deficient mice. Nature 389:183–186 [DOI] [PubMed] [Google Scholar]
- Ofosu FA, Dewar L, Craven SJ, Song Y, Cedrone A, Freedman J, Fenton JW., 2nd (2008) Coordinate activation of human platelet protease-activated receptor-1 and -4 in response to subnanomolar alpha-thrombin. J Biol Chem 283:26886–26893 [DOI] [PubMed] [Google Scholar]
- Pleines I, Hagedorn I, Gupta S, May F, Chakarova L, van Hengel J, Offermanns S, Krohne G, Kleinschnitz C, Brakebusch C, et al. (2012) Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood 119:1054–1063 [DOI] [PubMed] [Google Scholar]
- Preston RA, Jy W, Jimenez JJ, Mauro LM, Horstman LL, Valle M, Aime G, Ahn YS. (2003) Effects of severe hypertension on endothelial and platelet microparticles. Hypertension 41:211–217 [DOI] [PubMed] [Google Scholar]
- Sims PJ, Faioni EM, Wiedmer T, Shattil SJ. (1988) Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem 263:18205–18212 [PubMed] [Google Scholar]
- Sims PJ, Wiedmer T, Esmon CT, Weiss HJ, Shattil SJ. (1989) Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J Biol Chem 264:17049–17057 [PubMed] [Google Scholar]
- Tan KT, Lip GY. (2005) The potential role of platelet microparticles in atherosclerosis. Thromb Haemost 94:488–492 [DOI] [PubMed] [Google Scholar]
- Tan KT, Tayebjee MH, Lim HS, Lip GY. (2005) Clinically apparent atherosclerotic disease in diabetes is associated with an increase in platelet microparticle levels. Diabet Med 22:1657–1662 [DOI] [PubMed] [Google Scholar]
- Tracy PB, Giles AR, Mann KG, Eide LL, Hoogendoorn H, Rivard GE. (1984) Factor V (Quebec): a bleeding diathesis associated with a qualitative platelet Factor V deficiency. J Clin Invest 74:1221–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van De Werf F, White HD, Aylward PE, Wallentin L, Chen E, et al. (2012) Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med 366:20–33. [DOI] [PubMed] [Google Scholar]
- van der Zee PM, Biró E, Ko Y, de Winter RJ, Hack CE, Sturk A, Nieuwland R. (2006) P-selectin- and CD63-exposing platelet microparticles reflect platelet activation in peripheral arterial disease and myocardial infarction. Clin Chem 52:657–664 [DOI] [PubMed] [Google Scholar]
- Voss B, McLaughlin JN, Holinstat M, Zent R, Hamm HE. (2007) PAR1, but not PAR4, activates human platelets through a Gi/o/phosphoinositide-3 kinase signaling axis. Mol Pharmacol 71:1399–1406 [DOI] [PubMed] [Google Scholar]
- Vretenbrant K, Ramström S, Bjerke M, Lindahl TL. (2007) Platelet activation via PAR4 is involved in the initiation of thrombin generation and in clot elasticity development. Thromb Haemost 97:417–424 [PubMed] [Google Scholar]
- Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. (1991) Domains specifying thrombin-receptor interaction. Nature 353:674–677 [DOI] [PubMed] [Google Scholar]
- Walker TR, Watson SP. (1993) Synergy between Ca2+ and protein kinase C is the major factor in determining the level of secretion from human platelets. Biochem J 289:277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss HJ, Lages B. (1997) Platelet prothrombinase activity and intracellular calcium responses in patients with storage pool deficiency, glycoprotein IIb-IIIa deficiency, or impaired platelet coagulant activity—a comparison with Scott syndrome. Blood 89:1599–1611 [PubMed] [Google Scholar]
- Weiss HJ, Lages B, Zheng S, Hayward CP. (2001) Platelet factor V New York: a defect in factor V distinct from that in factor V Quebec resulting in impaired prothrombinase generation. Am J Hematol 66:130–139 [DOI] [PubMed] [Google Scholar]
- Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. (1998) Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci USA 95:6642–6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiger F, Stephan S, Hoheisel G, Pfeiffer D, Ruehlmann C, Koksch M. (2000) P-Selectin expression, platelet aggregates, and platelet-derived microparticle formation are increased in peripheral arterial disease. Blood Coagul Fibrinolysis 11:723–728 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.