

Abstract
The study objective was to investigate factors that affect the central nervous system (CNS) distribution of elacridar. Elacridar inhibits transport mediated by P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp) and has been used to study the influence of transporters on brain distribution of chemotherapeutics. Adequate distribution of elacridar across the blood-brain barrier (BBB) and into the brain parenchyma is necessary to target tumor cells in the brain that overexpress transporters and reside behind an intact BBB. We examined the role of P-gp and Bcrp on brain penetration of elacridar using Friend leukemia virus strain B wild-type, Mdr1a/b(−/−), Bcrp1(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice. Initially, the mice were administered 2.5 mg/kg of elacridar intravenously, and the plasma and brain concentrations were determined. The brain-to-plasma partition coefficient of elacridar in the wild-type mice was 0.82, as compared with 3.5 in Mdr1a/b(−/−) mice, 6.6 in Bcrp1(−/−) mice, and 15 in Mdr1a/b(−/−)Bcrp1(−/−) mice, indicating that both P-gp and Bcrp limit the brain distribution of elacridar. The four genotypes were then administered increasing doses of elacridar, and the CNS distribution of elacridar was determined. The observed and model predicted maximum brain-to-plasma ratios (Emax) at the highest dose were not significantly different in all genotypes. However, the ED50 was lower for Mdr1a/b(−/−) mice compared with Bcrp1(−/−) mice. These findings correlate with the relative expression of P-gp and Bcrp at the BBB in these mice and demonstrate the quantitative enhancement in elacridar CNS distribution as a function of its dose. Overall, this study provides useful concepts for future applications of elacridar as an adjuvant therapy to improve targeting of chemotherapeutic agents to tumor cells in the brain parenchyma.
Introduction
The blood-brain barrier (BBB) is a complex anatomic and biochemical system of capillary endothelial cells, pericytes, astrocytes, and neurons (also referred to as the neurovascular unit) that together perform a barrier function to protect the central nervous system (CNS) (Hartz and Bauer, 2010). P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp) are two efflux transporters present in the BBB endothelial cells that have been implicated in limiting the brain distribution of several drugs. Consequently, inhibition of these transporters may improve the targeted brain delivery and ensuing efficacy of substrate drugs that are intended for a CNS target.
Many molecularly targeted antitumor agents, including several kinase inhibitors such as imatinib, dasatinib, sorafenib, gefitinib, vemurafenib, sunitinib, cediranib, lapatinib, and dabrafenib, have limited brain distribution due to efflux by P-gp and Bcrp (Dai et al., 2003; Breedveld et al., 2005; Bihorel et al., 2007; Chen et al., 2009; Lagas et al., 2009, 2010; Polli et al., 2009; Agarwal et al., 2010, 2011b; Mittapalli et al., 2012a,b; Tang et al., 2012; Wang et al., 2012). Elacridar is a potent inhibitor of P-gp and Bcrp (Hyafil et al., 1993; Allen et al., 1999), and its coadministration with many of these drugs and other P-gp/Bcrp substrates has been shown to increase their brain distribution by several fold in mice (Bihorel et al., 2007; Chen et al., 2009; Lagas et al., 2009, 2010; Agarwal et al., 2010, 2011b; Tang et al., 2012).
Lack of adequate drug distribution to brain tumors has been thought to hamper effective treatment of lethal malignancies like gliomas (Agarwal et al., 2011a). Effective chemotherapeutic treatment of invasive tumors in the brain requires that the drug traverse an intact BBB and achieve sufficient concentration in the cell of interest in the brain. However, as previously mentioned, several anticancer agents used for treatment of brain tumors fail to effectively enter the brain, often due to P-gp- and Bcrp-mediated active efflux at the BBB.
Elacridar has been used to improve distribution of paclitaxel in brain tumor models (Hubensack et al., 2008). It could potentially be used along with other therapeutic molecules to enhance their CNS distribution and thus improve efficacy, especially in invasive tumor cells that reside behind an intact BBB (Agarwal et al., 2011a). Another important aspect of treatment of brain tumors is the development of resistance to drugs. Expression of P-gp and Bcrp in the tumor cells and tumor-initiating cells forms a second barrier to drug delivery. This causes efflux of drugs from the tumor cells, further lowering their efficacy (Lu and Shervington, 2008). Adequate distribution of elacridar itself into the brain parenchyma would therefore be necessary to inhibit efflux of drugs from such invasive tumor cells and thus overcome this second barrier. This could lead to improved efficacy and possibly reduce recurrence of the tumor. Given that elacridar must effectively inhibit the transporters at both the BBB and invasive tumor cells that reside behind an intact BBB, our primary objective was to examine the dose-dependent factors that influence the distribution of elacridar across the BBB into the brain.
Elacridar efflux by P-gp and Bcrp at the BBB is a saturable process, and therefore its brain distribution would be predicted to be concentration dependent. This was initially suggested by experiments using radiolabeled elacridar and positron emission tomography imaging in mice with genetic deletions of P-gp or Bcrp or both (Dorner et al., 2009; Kawamura et al., 2011a,b). Moreover, elacridar distribution into the brain has been suggested to be influenced by the administered dose (Kawamura et al., 2011b). In a recent study (Sane et al., 2012), we showed that the brain partitioning of elacridar in wild-type mice was a function of its plasma exposure. We therefore anticipated that P-gp and Bcrp at the BBB could be instrumental in affecting the brain distribution of elacridar. In the current study, we examine this phenomenon by using the Friend leukemia virus strain B (FVB) mouse model with genetic deletions of P-gp [Mdr1a/b(−/−)] or Bcrp [Bcrp1(−/−)] or both [Mdr1a/b(−/−)Bcrp1(−/−)].
The brain distribution of elacridar has not been carefully characterized despite its widespread use in preclinical studies examining its effects on transport of substrate drugs across the BBB. We examine in detail the factors, including transporters at the BBB and administered dose, that would work in concert to influence the distribution of elacridar across the BBB into the brain. The results from this study may be useful to formulate an elacridar dosing strategy in preclinical tumor models that could be used to successfully target drug molecules, which are substrates for efflux proteins to invasive tumor cells that reside behind an intact BBB. Experiments such as these would be a proof of concept for employing similar strategies in the clinical arena.
Materials and Methods
Elacridar (GF120918; N-[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]-5-methoxy-9-oxo-10H-acridine-4-carboxamide]) was purchased from Toronto Research Chemicals, Inc. (Toronto, ON, Canada). All other chemicals used were high-performance liquid chromatography (HPLC) or reagent grade and were obtained from Sigma-Aldrich (St. Louis, MO). Radiolabeled [14C]dasatinib was obtained from Bristol-Myers Squibb Co. (Princeton, NJ).
Cell Culture Studies.
Intracellular accumulation of elacridar was studied in epithelial Madin-Darby canine kidney II (MDCKII) cells expressing either human P-gp (MDCKII-MDR1 cell line) or murine Bcrp (MDCKII-Bcrp1 cell line), which were kindly provided by Dr. Piet Borst and Dr. Alfred H. Schinkel (The Netherlands Cancer Institute), respectively. Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin B (250 ng/ml) and were maintained at 37°C with 5% CO2 under humidifying conditions. Confluent monolayers of cells were obtained by seeding at a cell density of 105 cells/well, followed by incubation for 3 days. On the day of the experiment, the cells were washed twice with prewarmed cell assay buffer at 37°C.
Accumulation of Elacridar in MDCKII Cells.
The accumulation of elacridar was examined in MDCKII wild-type, MDR1 transfected, and Bcrp1 transfected cells. The experiment was initiated after a 30-minute preincubation where the cells were equilibrated with 1 ml of cell assay buffer. The cells were then incubated with buffer containing increasing concentrations of elacridar, ranging from 50 nM to 10 μM, for 1 hour in an orbital shaker at 37°C. The experiment was terminated by aspirating the drug solution and washing with ice-cold phosphate-buffered saline. A 1% Triton X-100 (Sigma-Aldrich) solution (0.5 ml) was added to each well to solubilize the cells, and the protein concentration in the solubilized cell fractions was determined by the bicinchoninic acid protein assay (Thermo Fisher Scientific, Waltham, MA). Concentrations of elacridar in 200 µl aliquot of cell lysate were determined by liquid chromatography–tandem mass spectrometry (LC-MS/MS). The amounts of elacridar in the cell lysates were normalized by the protein concentration in the respective wells and expressed as ng/μg of protein. The amount accumulated in the cells was described as a function of the concentration of elacridar in the incubating medium.
Accumulation of [14C]Dasatinib with Increasing Concentrations of Elacridar.
The degree of nonlinearity of elacridar inhibition of the efflux of a prototypical dual substrate, radiolabeled dasatinib, by P-gp and Bcrp was determined by measuring the accumulation of dasatinib in MDCKII-MDR1 cells and MDCKII-Bcrp1 cells, respectively. The experiment was initiated by preincubating the cells for 30 minutes with increasing concentrations of elacridar from 50 nM to 20 µM. The cells were then incubated with trace amounts of [14C]dasatinib in the presence of varying concentrations of elacridar for 1 hour. The experiment was terminated as described in the previous section. The radioactivity (disintegrations per minute) associated with 100 µl of cell lysate was determined by liquid scintillation counting (LS-6500; Beckman Coulter, Fullerton, CA). The observed radioactivity was normalized by the amount of protein in each well, and dasatinib accumulation was expressed as disintegrations per minute/µg of protein. Dasatinib accumulation in absence of elacridar was considered the control, and the accumulation in presence of elacridar was expressed as a percentage of the control.
The nonlinear relationship of dasatinib accumulation in the transfected cells as a function of elacridar concentration was quantitated. Phoenix WinNonlin 6.2 (Pharsight, Mountain View, CA) was used to fit a simple Emax model to the data using eq. 1:
 |
(1) |
where E is the accumulation of dasatinib (ng of dasatinib/µg of protein), Emax (ng/µg) is the maximum accumulation at concentrations of elacridar that saturate the efflux process, EC50 (µM) is the concentration of elacridar at which half-maximal inhibition is seen, and C is the concentration of elacridar in the incubating medium (µM).
In Vivo Studies.
In vivo studies were performed in FVB (wild-type), Mdr1a/b(−/−) (P-gp knockout), Bcrp1(−/−) (Bcrp knockout), and Mdr1a/b(−/−)Bcrp1(−/−) (triple knockout) mice of either sex that were 8 to 10 weeks old (Taconic Laboratories, Hudson, NY). Animals were maintained under a 12-hour light/dark cycle, temperature-controlled environment with free access to food and water at all times. All studies were performed in accordance with guidelines set by Principles of Laboratory Animal Care (National Institutes of Health, Bethesda, MD) and were approved by the Institutional Animal Care and Use Committee of University of Minnesota.
Plasma and Brain Pharmacokinetics of Elacridar in FVB Mice.
An elacridar dosing solution for i.v. administration was prepared in dimethylsulfoxide, propylene glycol, and water (4:4:2 v/v/v) at a concentration of 1.25 mg/ml. FVB mice (P-gp knockout, Bcrp knockout, and triple knockout) received a dose of 2.5 mg/kg elacridar intravenously via tail vein injection. The animals were sacrificed 0.5, 1, 2, 4, and 8 hours after the dose (n = 4 at each time point) by use of a carbon dioxide chamber, and blood and brain were rapidly collected. Blood was collected by cardiac puncture and transferred to heparinized tubes. Brain was removed from the skull, washed with ice-cold saline, and flash-frozen with liquid nitrogen. Plasma was separated by centrifugation at 7500g for 10 minutes at 4°C. Both plasma and brain specimens were stored at −80°C until analysis by LC-MS/MS. Plasma and brain concentration-time profiles from wild-type animals for comparison were obtained from our previously published study (Sane et al., 2012).
The pharmacokinetic parameters for elacridar in plasma and brain for all mouse genotypes were determined by noncompartmental analysis using Phoenix WinNonlin 6.2. The tissue partition coefficient (Kp) for the brain was calculated as the ratio of the area under the concentration-time curve for brain (AUCbrain) to the area under the concentration-time curve for plasma (AUCplasma). Comparisons between the AUCs in plasma and brain were performed using the method described by Nedelman et al. for AUCs measured after destructive sampling (Bailer, 1988; Nedelman et al., 1995).
Dose-Dependent Distribution of Elacridar in FVB Mice.
Dosing solutions for i.v. administration of elacridar were prepared in dimethylsulfoxide, propylene glycol, and water (4:4:2 v/v/v) at elacridar concentrations of 0.25 mg/ml, 0.5 mg/ml, 1.25 mg/ml, 2.5 mg/ml, and 5.0 mg/ml. All four mouse genotypes (wild-type, P-gp knockout, Bcrp knockout, and triple knockout) received i.v. doses of 0.5 mg/kg, 1 mg/kg, 2.5 mg/kg, 5 mg/kg and 10 mg/kg (n = 4 for each dose and for each genotype) by injection into the tail vein. Animals were sacrificed 1 hour after the dose via a carbon dioxide chamber. Blood and brain were collected as described in the previous section.
Calculation of ED50.
The effective dose for half-maximal brain penetration of elacridar was estimated by measuring brain and plasma concentrations of elacridar at various doses and calculating a mean brain-to-plasma ratio at each dose in all mouse genotypes. By use of Phoenix WinNonlin 6.2, a simple Emax model, shown in eq. 2, was fit to the mean brain-to-plasma ratio data as a function of elacridar dose (mg/kg):
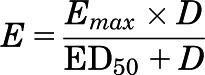 |
(2) |
where E is the brain-to-plasma ratio, Emax is the maximum brain-to-plasma ratio, D is the administered elacridar dose in mg/kg, and ED50 is the dose required to produce a half-maximal brain-to-plasma ratio.
Analysis of Elacridar by Liquid Chromatography–Mass Spectrometry.
The concentration of elacridar in plasma and brain homogenate specimens was determined by HPLC with tandem mass spectrometric detection. Frozen samples stored at −80°C were thawed at room temperature. Brain samples were homogenized with a tissue homogenizer (Thermo Fisher Scientific) along with the addition of three volumes of ice-cold 5% bovine serum albumin solution in distilled water. A 50 µl aliquot of plasma and a 100 µl aliquot of brain homogenate were spiked with 50 ng of the internal standard, tyrphostin (AG1478). The samples were extracted by addition of 100 µl of a pH 11 buffer (0.1 M sodium hydroxide and 0.04 M sodium bicarbonate) and 1 ml of ice-cold ethyl acetate. Samples were centrifuged for 15 minutes at 7500g at 4°C, and 700 µl of the organic supernatant layer was transferred to fresh microcentrifuge tubes and dried under a gentle stream of nitrogen. The dried extracts were reconstituted in mobile phase (20 mM ammonium formate and 1% formic acid: acetonitrile in a ratio of 58:42, v/v) and transferred to glass autosampler vials. We injected 5 µl volumes of reconstituted extract by an autosampler system that was maintained at 10°C into the HPLC system. Analytes were separated by use of an Agilent Technologies Eclipse XBD-C18 column (4.8 × 50 mm, 5-µm particle size; Agilent Technologies, Santa Clara, CA) . The liquid chromatography–mass spectrometry analysis of samples for elacridar concentration was performed as described previously elsewhere (Sane et al., 2012). The free fractions of elacridar in plasma and brain homogenate were not determined due to high nonspecific binding to the dialysis device.
Modeling and Simulation.
A two-compartment pharmacokinetic model with “plasma” (central compartment) and “brain” (peripheral compartment) compartments was created using STELLA (iSEE Systems, Lebanon, NH). Simulations were performed to examine the effect of simultaneous passive diffusion and two active efflux processes from the brain on the brain distribution of a dual substrate such as elacridar (see Fig. 8). Plasma and brain compartments were linked by distributional flows, which represent the rates of drug transfer between compartments, and the effect of active efflux processes from brain into the plasma was examined.
Fig. 8.

Schematic representation of transport processes between plasma (central compartment) and brain (peripheral compartment) for a compound that is a substrate for two different transporters.
The two compartments were connected by a bidirectional passive clearance (CLpass), and two active efflux processes (denoted as A and B) represented clearance due to two efflux transporters such as P-gp and Bcrp. The plasma compartment has an additional clearance due to systemic elimination (CLelim).
The dose was administered as an i.v. bolus into the central compartment at time 0. The rate of elimination from the central compartment is expressed as eq. 3:
| (3) |
The passive diffusion into the brain compartment is described by eq. 4.
| (4) |
The CLpass was assumed to be constant in the models with two active efflux clearances from the brain, and it was consistent with models that described either one active efflux clearance or no active efflux clearance.
The active efflux from the brain compartment was modeled using the Michaelis-Menten equation (eq. 5),
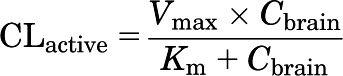 |
(5) |
where CLactive is a nonlinear, saturable efflux clearance, Vmax reflects the transporter capacity or expression of the transporter, and Km is the affinity of the transporter for the compound. The two efflux clearance processes, A and B, were assigned a unique Vmax and Km for each of the two transporters, P-gp and Bcrp, based on their relative expression (Agarwal et al., 2012b) and relative affinity from in vivo results (see Fig. 6).
Fig. 6.

Elacridar brain-to-plasma concentration ratios after a single i.v. dose of 2.5 mg/kg in FVB Mdr1a/b(−/−) mice, Bcrp1(−/−) mice, and Mdr1a/b(−/−)Bcrp1(−/−) mice. The data represent the mean ± S.D. (n = 4). The brain-to-plasma ratio in wild-type, Mdr1a/b(−/−), and Bcrp1(−/−) mice showed an increase followed by a decrease. In Mdr1a/b(−/−)Bcrp1(−/−) mice, the brain-to-plasma ratio showed an increase to a steady state.
Given these assumptions, four models were defined and simulations performed:
Model 1: Passive diffusion and two distinct active efflux processes (A and B) from the brain
Model 2: Passive diffusion and one active efflux process (A)—high expression, low affinity, from the brain
Model 3: Passive diffusion and one active efflux process (B)—low expression, high affinity from the brain
Model 4: Only passive diffusion to and from the brain
These models assume that the concentrations depicted in the plasma (Vp) and brain (Vb) volumes are available for transport. Moreover, they further assume that inhibition or genetic deletion of the transport systems do not influence the systemic elimination (CLelim) of the substrate. Although this was not the case with some early investigations of cytotoxic agents such as doxorubicin and P-gp inhibition (Warren et al., 2000), this is often the case with substrates that are primarily metabolized, such as the tyrosine kinase inhibitors (Mittapalli et al., 2012a; Wang et al., 2012).
For model 1, the rate of change of amount in the plasma can be represented as eq. 6:
 |
(6) |
And the rate of change of amount in the brain can be described as eq. 7:
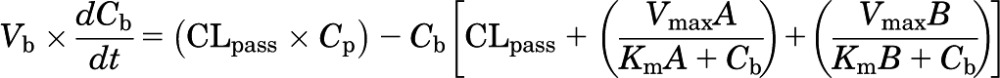 |
(7) |
The brain distribution of a prototypical compound that is a substrate for two transporters (i.e., P-gp and Bcrp) was simulated using this model. The values for Vmax (Vmax A = 150; Vmax B = 50) were chosen to represent differences in the relative magnitude of the capacity of transporters such as P-gp and Bcrp in the FVB mouse (Agarwal et al., 2012b). The Km values for A and B were chosen to reflect possible differences in affinity for the two transporters A and B (Km A = 10; Km B = 0.1) based on results from in vivo studies (Fig. 6). Simulations were run to reflect the changes in the active efflux from the brain with a change in the dose administered (and hence the resultant change in concentration), and how that would result in dose-dependent (saturable) changes in brain distribution. In essence, we examined through simulation the effect of dose escalation and the subsequent increase in plasma concentrations on the brain-to-plasma ratios and the active clearance from the brain in all four models.
Statistical Analysis.
Comparisons between groups were made using SigmaStat, version 3.1 (Systat Software, Inc., San Jose, CA). Statistical differences between two groups were tested by using the two-sample t test, and P < 0.05 was considered statistically significant. Multiple groups were compared by one-way analysis of variance with the Holm-Sidak post hoc test for multiple comparisons at a significance level of P < 0.05. Comparisons between the AUCs in plasma and brain were performed using the method described by Nedelman et al. for AUCs measured after destructive sampling (Bailer, 1988; Nedelman et al., 1995).
Results
Intracellular Accumulation of Elacridar in MDCKII Cells.
Intracellular accumulation of elacridar was examined in MDCKII wild-type cells and polarized epithelial MDCKII cells that overexpressed the transporter proteins, P-gp or Bcrp. In wild-type cells, the accumulation of elacridar increased linearly with the concentration of elacridar in the incubating medium (Fig. 1). In MDCKII MDR1 and Bcrp1 transfected cells, the cellular accumulation showed a nonlinear increase with an increase in concentration in the incubating medium (Fig. 1). The wild-type cells did not reach a maximal cellular accumulation at the concentrations of elacridar used in this experiment. The poor solubility of elacridar limits the use of higher concentrations due to precipitation in the cell assay buffer. The accumulation of elacridar in the transfected cell lines is significantly lower as compared with the wild-type cells, indicating that P-gp and Bcrp limit the cellular delivery of elacridar in an in vitro model system.
Fig. 1.

Accumulation of elacridar in MDCKII wild-type cells and MDR1 transfected and Bcrp1 transfected cell lines with increasing concentrations of elacridar. Accumulation of elacridar is expressed as ng of elacridar/µg of protein in cell lysate (n = 4 wells, mean ± S.D.).
Intracellular Accumulation of Dasatinib in Presence of Elacridar.
Dasatinib is a tyrosine kinase inhibitor that is a substrate for both P-gp and Bcrp (Chen et al., 2009; Lagas et al., 2009). We used radiolabeled dasatinib as a probe substrate to observe the change in accumulation of dasatinib with increasing concentrations of elacridar. The accumulation of [14C]dasatinib increased nonlinearly with increases in the concentration of elacridar (Fig. 2). The maximal dasatinib accumulation when compared with wild-type (Emax) in MDR1 transfected cells was 1323% ± 126% as compared with 1083% ± 154% for Bcrp1 cells. The EC50 for both the cell lines was equivalent, 1.01 ± 0.37 µM for MDR1 transfected cells and 1.01 ± 0.58 μM for Bcrp1 transfected cells. These results indicate the nonlinearity in inhibition of dasatinib efflux by increasing elacridar concentrations, most likely due to saturation of the inhibitory effect on the efflux process.
Fig. 2.

Percentage accumulation of [14C]dasatinib in MDCKII MDR1 overexpressing cells and MDCKII Bcrp1 overexpressing cells with varying concentrations of elacridar. The accumulation of radiolabeled dasatinib increased nonlinearly with an increase in concentration of elacridar in the incubating media, described by a simple Emax model (n = 4 wells, mean ± S.D.).
Plasma and Brain Disposition of Elacridar in FVB Mice.
The plasma disposition and the brain distribution of elacridar were studied in FVB mice to understand the role of P-gp and Bcrp on its distribution to the brain. The mouse models used were FVB Mdr1a/b (−/−), Bcrp1 (−/−), and Mdr1a/b(−/−)Bcrp1(−/−); the results from our previously published study using wild-type mice (Sane et al., 2012) were used for comparison.
Elacridar plasma concentrations, measured after a 2.5 mg/kg i.v. dose, varied between the four genotypes (Fig. 3). This difference in plasma concentrations is reflected in the area under the plasma concentration-time curves (AUCplasma). The AUCplasma in triple knockout mice was statistically significantly (P < 0.001) lower than for the other mouse genotypes (Table 1). In wild-type mice, the AUCplasma was statistically significantly higher than in both the single knockout mice [Mdr1a/b (−/−) and Bcrp1 (−/−)]. The absence of P-gp and Bcrp may allow elacridar to reach sites of metabolism in tissues that are otherwise limited by the presence of efflux proteins. This could be one of the possible reasons why the knockout mice have lower plasma exposure than wild-type mice.
Fig. 3.

Elacridar concentrations in plasma after a single i.v. dose of 2.5 mg/kg in FVB Wild-type, Mdr1a/b(−/−) mice, Bcrp1(−/−) mice, and Mdr1a/b(−/−)Bcrp1(−/−) mice. The data represent the mean ± S.D. (n = 4). The plasma concentrations in the Mdr1a/b(−/−)Bcrp1(−/−) mice were lower than in the wild-type mice.
TABLE 1.
Plasma pharmacokinetics determined by noncompartmental analysis after 2.5 mg/kg i.v. bolus dose of elacridar in wild-type, Mdr1a/b(−/−), Bcrp1(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice
AUCplasma in wild-type mice is significantly higher than that calculated in the knockout mice.
| Elimination Rate Constant |
Half-Life |
Volume of Distribution |
Clearance |
AUCtlast |
AUCinf
|
|
|---|---|---|---|---|---|---|
| h−1 | h | ml | ml/min | μg*min/ml | μg*min/ml | |
| Wild typea | 0.155 | 4.4 | 170 | 0.46 | 138 ± 6.5 | 161.0 |
| Bcrp1(−/−) | 0.139 | 4.9 | 248 | 0.57 | 87.4 ± 6.0b | 130.4 |
| Mdr1a/b(−/−) | 0.17 | 4.1 | 306 | 0.86 | 64.3 ± 7.8b | 86.7 |
| Mdr1a/b(−/−)Bcrp1(−/−) | 0.173 | 3.9 | 374 | 1.08 | 51 ± 1.3b | 69.4 |
AUCinf, area under the concentration-time curve to infinity; AUCtlast, area under the concentration-time curve from zero to last measured time point.
Data for wild-type mice from previously published study.
P < 0.001.
As expected for a P-gp and Bcrp substrate, the brain concentration-time profiles varied markedly between the mouse genotypes (Fig. 4) and did not follow the rank order among genotypes of the plasma concentration time profiles. The maximum concentration observed (Cmax brain) was different for various genotypes despite administration of equivalent doses. The Cmax for Mdr1a/b(−/−) mice (1.6 ± 0.2 µg/g) was higher than that in wild-type mice (1 ± 0.4 µg/g) but lower than in Bcrp1(−/−) mice (2.9 ± 0.2 µg/g) and Mdr1a/b(−/−)Bcrp1(−/−) mice (3.2 ± 0.2 µg/g) (Table 2). This difference in peak brain concentrations was also reflected in the area under the brain concentration-time profiles (AUCbrain). The absence of transporters at the BBB influenced the AUC; where the AUCbrain in Mdr1a/b(−/−)Bcrp1(−/−) mice was nearly 10-fold higher than in wild-type mice, the AUCbrain in Bcrp1(−/−) mice was approximately 8-fold higher than in the wild-type mice, and the AUCbrain in Mdr1a/b(−/−) mice was about 3-fold higher than in the wild-type mice. The tissue-to-plasma partition coefficient for brain (Kp) reflects this as well: the values were 0.82 for wild-type mice, 3.5 for Mdr1a/b (−/−) mice, 6.6 for Bcrp1 (−/−) mice, and 15 for Mdr1a/b(−/−)Bcrp1(−/−) mice.
Fig. 4.

Elacridar concentrations in the brain after a single i.v. dose of 2.5 mg/kg in FVB wild-type, Mdr1a/b(−/−) mice, Bcrp1(−/−) mice, and Mdr1a/b(−/−)Bcrp1(−/−) mice. The data represent the mean ± S.D. (n = 4). The brain concentrations in the wild-type mice were much lower than in the Mdr1a/b(−/−)Bcrp1(−/−) mice.
TABLE 2.
Brain pharmacokinetics of elacridar as determined by noncompartmental analysis after 2.5 mg/kg i.v. bolus dose of elacridar in wild-type, Mdr1a/b(−/−), Bcrp1(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice
AUCbrain in the knockout animals is significantly higher than in the wild-type mice.
| Elimination Rate Constant |
Half-Life |
Cmax |
AUCtlast |
AUCinf
|
Kp | |
|---|---|---|---|---|---|---|
| h−1 | h | μg/g | μg*min/ml | μg*min/ml | ||
| Wild typea | 0.474 | 1.5 | 1 ± 0.4 | 128 ± 19 | 131 | 0.82 |
| Bcrp1(−/−) | 0.237 | 2.9 | 2.9 ± 0.2 | 727 ± 24b | 863 | 6.6 |
| Mdr1a/b(−/−) | 0.213 | 3.25 | 1.6 ± 0.2 | 253 ± 12b | 303 | 3.5 |
| Mdr1a/b(−/−)Bcrp1(−/−) | 0.126 | 5.4 | 3.2 ± 0.2 | 735 ± 27b | 1042 | 15.03 |
AUCinf, area under the concentration-time curve to infinity; AUCtlast, area under the concentration-time curve from zero to last measured time point; Kp, tissue partition coefficient.
Data for wild-type mice from previously published study.
P < 0.001.
These results are similar to the “greater than additive” efflux effect seen for several drugs that are substrates for both P-gp and Bcrp (Polli et al., 2009; Kodaira et al., 2010; Agarwal and Elmquist, 2012), which confirms that the two transporters, P-gp and Bcrp, work together to keep elacridar out of the brain. It was initially hypothesized that these disproportionate increases might be because of compensatory upregulation of Pgp or Bcrp at BBB of Bcrp knockout and P-gp knockout mice, respectively. However, recent quantitative proteomic analysis data have shown that there are no compensatory changes in the BBB expression of relevant transporters in the single and combined knockout mice (Agarwal et al., 2012b). Further, the pharmacokinetic model of Kodaira et al. (2010), which discussed relative affinities and capacities of the two efflux clearance processes, provides a simple theoretical explanation for the observed disproportionate effects in combined knockout mice.
These plasma and brain concentration time profiles and the resultant AUC ratios (Kp values) were not described as the ratio of free concentrations in the plasma and brain because nonspecific binding issues prevented us from accurately and precisely determining the respective free fractions of elacridar. Nevertheless, it is clear that elacridar is highly bound to plasma proteins, as shown by Ward and Azzarano (2004) in species other than the mouse, and it is also anticipated that, given its physicochemical characteristics, it will be highly bound to brain parenchymal binding sites. Therefore, the actual free concentrations achieved in the brain will be quite low compared with the total concentrations. It is not likely that the binding patterns either in plasma or brain will be influenced by transporter genotype. In this regard, we showed in a previous study examining dabrafenib brain distribution (Mittapalli et al., 2012a) that the plasma and brain homogenate-free fractions in wild-type and Mdr1a/b(−/−)Bcrp1(−/−) mice were not significantly different between the genotypes. Therefore, our conclusions based on total levels are valid regarding transport saturation, even while keeping in mind that the actual free concentration in the brain will be much lower than the total.
The half-life of elimination from the brain was found to be longer in Mdr1a/b(−/−)Bcrp1(−/−) mice (5.4 hours) compared with the Mdr1a/b(−/−) mice (3.2 hours), Bcrp1(−/−) mice (2.9 hours), and wild-type mice (1.5 hours). The half-life in the brain corresponds to the brain Cmax in the different genotypes. A longer half-life generally corresponds to a higher Cmax in the brain (see Table 2). The presence of P-gp and Bcrp at the BBB decreases the apparent half-life of elacridar in the brain. Figure 5 shows the brain and plasma concentration time profiles in the four different genotypes, plotted on different graphs with the same scale for ease of visual comparison. The brain concentrations in the Mdr1a/b(−/−)Bcrp1(−/−) mice were statistically significantly greater throughout the time course than those in the wild-type or either Mdr1a/b(−/−) or Bcrp1(−/−) mice.
Fig. 5.

Elacridar concentrations in the plasma and brain after a single i.v. dose of 2.5 mg/kg in FVB wild-type (A), Bcrp1(−/−) mice (B), Mdr1a/b(−/−) mice (C), and Mdr1a/b(−/−)Bcrp1(−/−) mice (D). The data represent the mean ± S.D. (n = 4).
The brain-to-plasma concentration ratios with respect to time for the four genotypes are shown in Fig. 6. These brain-to-plasma concentration ratios, when plotted with respect to time, show an increase and then plateau to a distributional steady state in Mdr1a/b(−/−)Bcrp1(−/−) mice. However, in wild-type, Mdr1a/b(−/−), and Bcrp1(−/−) animals, the brain-to-plasma ratios increased to a maximum value, then decreased with time. Although this may be counterintuitive for a peripheral (brain) compartment if it were a completely linear system, it can be explained by the involvement of nonlinear active processes in the distribution of elacridar to the mouse brain. P-gp and Bcrp at the mouse BBB limit the distribution of elacridar into the mouse brain. The peculiar brain-to-plasma ratio versus time profiles can be explained by the active efflux of elacridar out of the brain. The concentrations in the brain decrease more rapidly compared with the plasma concentrations due to the active efflux of elacridar at the BBB, thus resulting in decreasing brain-to-plasma concentration ratios with time. This decrease in brain concentration is also enhanced at later times when the efflux clearance is not saturated by high concentrations, effectively changing the distribution ratio (as outlined in the following section).
Dose-Dependent Brain Distribution of Elacridar in FVB Mice.
These experiments were performed to determine whether the efflux of elacridar from the BBB leads to nonlinear distribution with increasing elacridar concentrations. The brain penetration of elacridar in various mouse genotypes was studied 1 hour after the i.v. injection. Mice were administered doses ranging from 0.5 mg/kg to 10 mg/kg, brain and plasma concentrations were measured, and brain-to-plasma concentration ratios were determined (Table 3). Elacridar brain-to-plasma concentration ratios increased nonlinearly with dose in all four mouse genotypes (Fig. 7). In the wild-type mice, the mean brain-to-plasma concentration ratio was 0.4 ± 0.1 at the lowest dose of 0.5 mg/kg and 12.7 ± 2.8 at the highest dose of 10 mg/kg, an approximately 30-fold increase due to saturation of efflux (Fig. 7A). This shows that at lower doses of elacridar the brain penetration in wild-type mice is restricted, but with increasing doses concentrations of elacridar that are sufficient to saturate the efflux transporters at the BBB are achieved, thus enhancing its own brain distribution. Profiles observed in the Bcrp1(−/−) mice (Fig. 7B) and Mdr1a/b(−/−) mice (Fig. 7C) were similar, demonstrating a nonlinear increase in brain-to-plasma ratio with increasing dose. However, in Mdr1a/b(−/−)Bcrp1(−/−) mice, the brain-to-plasma concentration ratios were much higher than those observed in the other genotypes at the lower doses (Fig. 7D), as would be expected for the situation where there is no dose-dependent change in efflux clearance at the BBB. The brain-to-plasma concentration ratios in the Mdr1a/b(−/−) and Bcrp1(−/−) mice showed an increasing trend toward a maximum value at higher doses, except for wild-type mice. In the wild-type mice, higher doses that could have achieved brain-to-plasma concentration ratios closer to the maximum value could not be tested due to the solubility limitations of elacridar which prevented higher dosing than 10 mg/kg using the same vehicle. At the lowest dose of 0.5 mg/kg, the brain-to-plasma ratio in Mdr1a/b(−/−)Bcrp1(−/−) mice was statistically significantly higher than the other genotypes (P < 0.05). However, at 10 mg/kg this difference was abolished, and the brain-to-plasma ratios at this highest dose were not significantly different from each other in all four genotypes (P > 0.05), suggesting that at high plasma concentrations of elacridar the effect of P-gp and Bcrp on brain distribution is overcome (due to transporter saturation).
TABLE 3.
Brain-to-plasma concentration ratios for wild-type, Mdr1(−/−), Bcrp1(−/−), and Mdr1(−/−)Bcrp1(−/−) mice at different doses
| Dose |
Wild-Type |
Bcrp1(−/−) |
Mdr1a/b(−/−) |
Mdr1a/b(−/−)Bcrp1(−/−) |
|---|---|---|---|---|
| mg/kg | ||||
| 0.5 | 0.4 ± 0.1 | 2.5 ± 1.8 | 5.4 ± 0.5 | 6.8 ± 3 |
| 1.0 | 2.2 ± 0.6 | 4.9 ± 1.5 | 4.6 ± 0.8 | 8.2 ± 0.5 |
| 2.5 | 5.2 ± 0.7 | 8.9 ± 2.9 | 7.3 ± 1.0 | 14.9 ± 1.6 |
| 5.0 | 6.0 ± 2.2 | 11.6 ± 3 | 8.8 ± 1 | 10.7 ± 1.3 |
| 10.0 | 12.7 ± 2.8 | 10.3 ± 2.7 | 10.1 ± 2.3 | 10 ± 1.6 |
Fig. 7.

Brain-to-plasma concentration ratios at 1 hour after the dose in wild-type mice (A), Bcrp1(−/−) mice (B), Mdr1a/b(−/−) mice (C), and Mdr1a/b(−/−)Bcrp1(−/−) mice (D) after i.v. dosing of increasing doses. The brain-to-plasma concentration ratios were lower at low doses, and they increased with the increase in dose in a nonlinear fashion, as described by a simple Emax model. The data represent the mean ± S.D.
These results confirm our hypothesis that brain distribution of elacridar is influenced by saturable active efflux mediated by P-gp and Bcrp at the mouse BBB. The brain-to-plasma concentration ratios versus dose relationship in all genotypes was described by a simple Emax model (Fig. 7, A–D; Table 4). The model predicted maximum brain-to-plasma ratio (Emax) was 21.6 ± 7.0, 14.2 ± 2.2, 10.4 ± 0.8, 11.9 ± 1.1 in the wild-type, Bcrp1(−/−), Mdr1a/b (−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice, respectively. We compared the 95% confidence intervals (CI) for Emax in all four genotypes. In wild-type mice, the Emax and EC50 showed a high coefficient of variation, and a large 95% CI (12.46, 29.11), that overlaps with the 95% CI for Emax for Bcrp1(−/−) (9.6, 18.78) and Mdr1a/b(−/−)Bcrp1(−/−) (9.21, 14.18) mice. The Emax predicted for the wild-type mice is higher as compared with the other genotypes, but it did not reach significance.
TABLE 4.
Parameters for dose-dependent brain penetration of elacridar in wild type, Mdr1a/b(−/−), Bcrp(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice after i.v. doses ranging from 0.5 mg/kg to 10 mg/kg
| Mouse |
Emax |
ED50 |
|---|---|---|
| mg/kg | ||
| Wild-type | 21.6 ± 7.0 | 9.8 ± 5.1 |
| Bcrp1(−/−) | 14.2 ± 2.2 | 1.7 ± 0.7 |
| Mdr1a/b(−/−) | 10.4 ± 0.8 | 0.83 ± 0.24 |
| Mdr1a/b(−/−)Bcrp1(−/−) | 11.9 ± 1.1 | 0.3 ± 0.18 |
Emax, maximum brain-to-plasma ratio.
Simulation of Brain Distribution and Impact of Saturable Active Efflux Processes.
Simulations were performed to examine the factors that affect the brain distribution of a compound that is a substrate for two separate active efflux processes (Fig. 8). Model 1 (equivalent to wild-type mice) had two distinct active efflux processes in addition to a passive process between the central (e.g., plasma) and the peripheral (e.g., brain) compartment. Models 2 and 3 [equivalent to Bcrp1(−/−) and Mdr1a/b(−/−) mice, respectively] had one active efflux process and passive diffusion that linked the brain and plasma compartments. Model 4 [equivalent to Mdr1a/b(−/−)Bcrp1(−/−) mice] had only passive diffusion between the plasma and the brain compartment. The effect of increasing dose on the active clearance was examined in model 1 (Fig. 9). The active clearance from the brain is dependent on the concentration of the compound in the brain, when the clearance is a saturable process of the Michaelis-Menten type (see eq. 5). The increase in dose, and the consequent increase in the brain concentration, saturates the efflux from the brain compartment, leading to a lower CLactive from the brain, as illustrated in Fig. 9.
Fig. 9.

Change in active clearance (CLactive) with respect to time, and with an increase in dose for Model 1. The different symbols indicate different doses as per the legend. The total clearance from the brain due to active transport is a function of the dose administered. At higher doses, the transporters are saturated, and the CLactive from the brain is inhibited. At lower doses, with an increase in time, the brain concentrations decrease, and the CLactive increases.
The brain-to-plasma ratios with respect to time were also simulated for the same dose across the four models (Fig. 10). The brain-to-plasma concentration ratios in model 4 showed an increase to a distributional pseudo-equilibrium. This is consistent with observed data for the Mdr1a/b(−/−)Bcrp1(−/−) mice and is expected for distribution into a peripheral compartment with only passive diffusion. The fact that this ratio is greater than unity, in spite of equivalent clearances into and out of the brain compartment, is related to the inherent two-compartment nature of the model, where the brain-to-plasma concentration ratio will be greater in the postdistributional phase than at steady state. The brain-to-plasma concentration ratios in Models 1–3, when plotted against time, show an increase to a maximum value, followed by a decrease to an eventual distributional pseudo-equilibrium. This simulation shows good concordance to the profiles obtained experimentally for wild-type, Mdr1a/b(−/−), and Bcrp1(−/−) mice (Fig. 6). The distinctive pattern of the observed brain-to-plasma ratios in these mice is therefore a function of both capacity and affinity of the active efflux out of the brain and the subsequent dose that may either partially or fully saturate that efflux clearance.
Fig. 10.

Simulated brain-to-plasma ratio with respect to time for a compound in Models 1–4 that is a substrate for two transporters at a dose of 1000 units. The brain-to-plasma ratio shows an increase followed by a decrease in Models 1–3 where there is one or more active efflux(es) from the brain compartment. In Model 4, the transport is achieved only by passive diffusion, and the brain-to-plasma ratio shows an increase to a maximum plateau.
The brain-to-plasma concentration ratios at peak brain concentrations for Models 1–4, with an increasing dose, were modeled as a function of time (Fig. 11, A and B). The brain-to-plasma concentration ratios for all models (1–4) could be described by a simple Emax model. These simulated ratios correspond well to the data obtained experimentally for wild-type, Mdr1a/b(−/−), Bcrp1(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice. Figure 11A shows the predicted dose-dependent brain-to-plasma concentration ratios using the parameter estimates obtained directly from the model fits of the data in each genotype (see Fig. 7; Table 4). Figure 11B depicts the same dose dependency of the brain distribution using the simulated parameter values from the two transporter model (model 1, equivalent to wild-type), the high capacity-low affinity model [Model 2, equivalent Bcrp1(−/−)], the low capacity-high affinity transporter model [Model 3, equivalent to Mdr1a/b(−/−)], and the model where both transport system have been deleted [model 4, equivalent to Mdr1a/b(−/−)Bcrp1(−/−)]. One can see that there is concordance between Fig. 11, A and B, in that there is an asymptotic rise to a maximum brain-to-plasma ratio, and the relative affinities of each transport system yield the half-maximal value (ED50). This simple nonlinear distribution model is therefore successful in qualitatively describing the distribution of a compound with multiple active effluxes and passive diffusion between the plasma and the brain compartments, such as elacridar.
Fig. 11.

(A) The predicted dose-response relationship that is based on parameters (Emax and ED50) estimated from the data depicted in Fig. 7, A–D. (B) Simulated brain-to-plasma ratio with an increase in unit dose at 1 hour after the dose for a compound that is a substrate for two transporters, in Models 1–4. The brain-to-plasma ratios show a gradual increase to a maximum value for Models 1–3, and can be described by a simple Emax model.
Discussion
The objective of this study was to characterize the influence of P-gp and Bcrp at the mouse BBB on the nonlinear CNS distribution of elacridar. This compound has been widely used as a P-gp and Bcrp inhibitor, both in vitro and in vivo. However, the brain distribution of elacridar has not been fully characterized. Earlier studies using radiolabeled elacridar and positron emission tomography qualitatively examined the CNS distribution of elacridar-associated radioactivity in wild-type, P-gp knockout, Bcrp knockout, and triple knockout mice (Dorner et al., 2009; Kawamura et al., 2011a,b). The results from those studies indicated that P-gp and Bcrp could play a role in limiting the brain distribution of elacridar. Our previous study (Sane et al., 2012) was the first to quantitatively examine the brain distribution of elacridar in wild-type mice using a specific stability-indicating assay (LC-MS/MS). The current study examined various factors that could influence elacridar brain distribution, including P-gp and Bcrp expression at the BBB, relative affinities of the transporters for the substrate, and the dose of elacridar, all of which could influence the nonlinear brain distribution of elacridar through influencing the saturability of the efflux process. Results from this study further enable us to understand the mechanisms that influence the brain distribution of this important transport inhibitor.
Elacridar has been used to improve brain distribution of paclitaxel in mouse brain tumor models (Hubensack et al., 2008). Coadministration of elacridar with chemotherapeutic drugs such as topotecan and paclitaxel has been found to improve the brain distribution of these drugs (Kemper et al., 2003; de Vries et al., 2007). As such, elacridar could be used as an adjuvant in glioma chemotherapy and could be particularly useful in targeting invasive tumor cells that reside behind an intact BBB (Lu and Shervington, 2008; Agarwal et al., 2011a). Some tumor cells also express P-gp and Bcrp in the cell plasma membrane, which contributes to the development of resistance to chemotherapy by active efflux of drugs from the tumor cells (Lu and Shervington, 2008). If a tumor expresses P-gp or Bcrp, elacridar must inhibit the transporters both at the BBB and at the tumor cells themselves in the brain parenchyma to effectively improve drug delivery to brain tumors. To do this, the distribution of elacridar itself into the brain parenchyma is an important consideration, and factors influencing this should be incorporated in designing treatment strategies to improve the targeted delivery of drugs such as the tyrosine kinase inhibitors.
In vitro studies conducted in MDCKII cells have confirmed that elacridar is a substrate for P-gp and Bcrp. The active efflux of elacridar due to P-gp and Bcrp is saturable, and an increase in elacridar concentration causes a decrease in the efflux clearance (see Fig. 1), leading to an increase in cellular accumulation. The transfected cells were found to reach a distributional equilibrium at lower concentrations than the wild-type cells. This is because the efflux clearance from the transfected cells is greater than that in wild-type cells. The wild-type cells should eventually show a maximal cellular accumulation at a high concentration of elacridar in the incubating medium. However, the poor solubility of elacridar in the incubating medium limits the in vitro use of concentrations higher than 10 μM. The inhibitory effect of elacridar is also nonlinearly dependent on its concentration. A maximal possible inhibition of the transporters can be achieved only with high concentrations of elacridar (see Fig. 2). Dasatinib was chosen as a probe substrate because it is a dual substrate for P-gp and Bcrp (Chen et al., 2009; Lagas et al., 2009) and is also being considered as a candidate tyrosine kinase inhibitor for the treatment of glioblastoma (Agarwal et al., 2012a; Reardon et al., 2012). Therefore, to effectively enhance the brain distribution of a substrate drug using elacridar, it is essential to achieve a high concentration of elacridar, one that is sufficient to saturate the transporters at the BBB, thereby improving the partitioning of elacridar into the brain parenchyma.
Elacridar pharmacokinetic studies in mice indicate a lower plasma exposure (AUCplasma) in the transporter-deficient mice compared with wild-type mice. Plasma concentrations of elacridar after an i.v. dose of 2.5 mg/kg in the wild-type mice were significantly higher than the concentrations found in other mouse genotypes (see Fig. 3). Although this result regarding effects on systemic clearance was unexpected, it is possible that the plasma concentrations are lower in the knockout models because genetic deletion of the transporters allows elacridar to reach sites of metabolism in tissues that are otherwise restricted. The specific mechanisms behind this finding are beyond the scope of our current study but may be of interest for future work.
Elacridar brain concentrations in the knockout mice were significantly higher than in the wild-type mice. The half-lives of elacridar in the brains of wild-type, P-gp knockout, and Bcrp knockout mice were shorter than the half-life in plasma (Tables 1 and 2). This can be attributed to the active efflux of elacridar from the brain into the plasma. In the absence of either, or both, of these transporters, the half-life in the brain was prolonged, as seen in the triple knockout mice. The ratio of AUCbrain to AUCplasma (Kp, or tissue partition coefficient) in the wild-type mice was lower than that in the knockout mice. The Kp for P-gp knockout mice was 3.5 as compared with 6.6 for Bcrp knockout mice and 15 for triple knockout mice. This greater than additive effect for brain distribution in mice with genetic deletion or pharmacologic inhibition of both transporters has been observed before with several dual substrates of P-gp and Bcrp such as sorafenib, gefitinib, laptinib, and dasatinib (Chen et al., 2009; Lagas et al., 2009, 2010; Polli et al., 2009; Agarwal et al., 2010, 2011b) Taken together, these results confirm that P-gp and Bcrp work in concert at the mouse BBB to restrict the brain distribution of elacridar.
An important consideration here is the tissue binding of elacridar. Previously published studies indicate that elacridar is nearly 98% bound in plasma (Ward and Azzarano, 2004) in several species other than the mouse. Nonspecific binding of the drug to the dialysis apparatus prevented us from quantifying the free fraction of elacridar in the brain. However, based on prior knowledge of its plasma-binding properties and its physicochemical characteristics, we can assume that elacridar is highly bound in the brain. This is further exemplified by the high brain-to-plasma ratios (i.e., greater than unity).
In our previous study, we observed that the brain-to-plasma concentration ratio in wild-type mice reached a maximum and then decreased with time after different routes of administration (Sane et al., 2012). We hypothesized that the decrease in the brain-to-plasma concentration ratios with time occurred because the brain concentrations of elacridar decrease faster than the corresponding plasma concentrations due to a concentration-dependent (and hence time-dependent after a single i.v. dose) change in the active efflux clearance of elacridar from the brain mediated by P-gp and Bcrp at the BBB. To test this hypothesis, we examined the brain-to-plasma concentration ratios in the various mouse genotypes. The brain-to-plasma ratios in wild-type, P-gp knockout, and Bcrp knockout mice showed an increase to a maximum value and then decreased with time (Fig. 6). However, in the triple knockout mice, the brain-to-plasma ratio increased to a maximum and reached a plateau at the distributional steady-state value. This confirms our hypothesis that the decrease in brain-to-plasma ratios is a function of the active efflux clearance processes mediated by P-gp and Bcrp, and that the distribution of elacridar into the brain could be a nonlinear process, saturable by changes in concentration.
Saturable efflux of elacridar from brain was then evaluated by studying the dose-dependent brain distribution of elacridar after i.v. administration in the four mouse genotypes. The observed brain-to-plasma concentration ratio was modeled as a response to the elacridar dose (Fig. 7). The brain-to-plasma ratios of elacridar were a function of dose, and were found to increase with an increase in dose. A study performed in wild-type mice and P-gp knockout mice with lapatinib, a dual substrate for P-gp and Bcrp, examined the brain partitioning of lapatinib at two different doses (Polli et al., 2008). The brain-to-plasma ratio of lapatinib was found to increase with increase in dose, which supports the hypothesis that, for a substrate of P-gp and Bcrp, the active efflux out of the brain can be overcome with a plasma concentration that can saturate the transporters.
An interesting result from the experiment was that the maximum brain distributional responses (Emax) seen in all mouse genotypes were not statistically different (Tables 3 and 4). This indicates that at higher plasma concentrations there is a saturation of the relevant efflux transporter(s), leading to an increased distribution of elacridar into the brain—that is, the maximum brain distribution when efflux clearances are at their minimum and where brain distribution is determined by passive diffusion, which is theoretically equivalent among the genotypes. We previously showed that the integrity of the BBB tight junctions in these mice is not compromised by the genetic deletion of the transporters. This is true regarding functional assays and the expression of important junctional proteins such as claudin 5 (Agarwal et al., 2010, 2012b).
The ED50 for elacridar distribution into the brain of the different mouse genotypes varied significantly. It was approximately 0.3 mg/kg for triple knockout mice, indicating that a very low dose is required to achieve the half-maximal effect (i.e., brain distribution coefficient of elacridar 1 hour after the dose). This is expected because there will be no saturation of the efflux clearance because both transporters have been deleted. For P-gp knockout mice, the ED50 was predicted to be 0.83 ± 0.24 mg/kg, while for Bcrp knockout mice it was greater (1.7 ± 0.72 mg/kg) (Table 4). This small difference in the ED50 values might be attributable to the relative expression of P-gp and Bcrp at the mouse BBB. In mouse brain microvessel endothelial cells isolated from FVBn mice, the expression of P-gp is greater (∼5-fold) relative to Bcrp, as determined by quantitative proteomics (Agarwal et al., 2012b). As a result, a lower dose is required to achieve half-maximal effect when only Bcrp is present at the BBB (P-gp knockout mice) whereas a larger dose is required to overcome the P-gp efflux in the Bcrp knockout mice. In wild-type mice, the ED50 is the greatest (9.8 ± 5.1 mg/kg) among all mouse genotypes. In the wild-type mice, both P-gp and Bcrp work in tandem to restrict elacridar distribution to the brain, and a greater dose will be required to achieve half-maximal saturation.
A compartmental model was created in STELLA to examine the dose-dependent and saturable behavior of the transporters at the BBB. This simple nonlinear model can be instructive because the low solubility of elacridar limits the number of doses and the amount of elacridar that can be administered to mice; therefore, a simulation will be helpful in exploring distribution behavior over a wide range of doses and concentrations. The brain distribution of a compound that is a dual substrate for two transporters, such as elacridar, was modeled as a function of dose and time. Under these conditions and assumptions, the model simulations show that the active efflux from the brain is a function of the dose administered and that at higher doses the active efflux can be saturated (resulting in behavior depicted in Fig. 9). The brain-to-plasma concentration ratio versus dose profiles in the four genotypes that were determined experimentally showed a distinctive pattern for each genotype (Fig. 6). We expected that these patterns would be a result of active transport of elacridar from the brain. If one compares Fig. 6 (experimentally derived brain-to-plasma ratios for the four genotypes) with Fig. 10 (model predicted brain-to-plasma ratios), it can be readily seen that the simple dual-transporter model recapitulates the changes in brain distribution of elacridar as the dose increases. From the experimental data, the brain-to-plasma coefficient ratio at maximum brain concentration with increasing dose was described by the Emax equation in the various mouse genotypes (Fig. 7). As such, these simulations also correspond to the observed data (upon comparing Figs. 7 and 11), indicating that the equilibrium distribution coefficient (Kp) of a substrate, while limited by the presence of the efflux transporters, can be increased by increasing the dose. Our model was useful in understanding the various transport processes and the role played by the dose and the resulting concentration in influencing brain distribution.
In summary, this study shows that brain distribution of elacridar is influenced by P-gp and Bcrp mediated active efflux at the BBB. We show that there is a greater than additive increase in elacridar transport across the BBB when these two transporters are absent. Moreover, we show that efflux of elacridar out of the brain is a saturable process such that maximal brain penetration and targeting are seen at higher doses—that is, the doses that yield concentrations high enough to saturate P-gp and Bcrp at the BBB. Thus, the results from this study delineate the various distributional clearance mechanisms that regulate the brain distribution of elacridar. The dose of elacridar and the expression of the efflux pumps at the BBB are two important factors that influence the rate and extent of distribution of elacridar into the brain. The results from this study contribute to our understanding of how factors influencing the distribution of elacridar through the BBB are affected by efflux transport processes. This information will be useful in the formulation of elacridar dosing strategies that may be used to enhance the distribution and targeting of molecularly targeted agents, agents that are substrates for both P-gp and Bcrp, to the sites of action inside the invasive tumor cells that reside behind an intact, functional BBB.
Abbreviations
- AG1478
tyrphostin
- AUC
area under the concentration-time curve
- BBB
blood-brain barrier
- Bcrp
breast cancer resistance protein
- CI
confidence interval
- CNS
central nervous system
- Emax
maximum brain-to-plasma ratio
- FVB
Friend leukemia virus strain B
- HPLC
high-performance liquid chromatography
- Kp
tissue partition coefficient
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MDCKII-Bcrp1
Madin-Darby canine kidney II expressing murine Bcrp
- MDCKII-MDR1
Madin-Darby canine kidney II expressing human P-gp
- P-gp
P-glycoprotein
Authorship Contributions
Participated in research design: Sane, Agarwal, Mittapalli, Elmquist.
Conducted experiments: Sane, Agarwal.
Performed data analysis: Sane, Mittapalli, Elmquist.
Wrote or contributed to the writing of the manuscript: Sane, Agarwal, Mittapalli, Elmquist.
Footnotes
This work was supported by National Institutes of Health National Cancer Institute [Grant CA138437] (to W.F.E.); and a Faculty Development grant at the University of Minnesota (to W.F.E.); a Doctoral Dissertation Fellowship from the University of Minnesota (to S.A.); and Ronald J. Sawchuk Fellowship and Rowell Fellowship (to R.S.).
References
- Agarwal S, Elmquist WF. (2012) Insight into the cooperation of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) at the blood-brain barrier: a case study examining sorafenib efflux clearance. Mol Pharm 9:678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Mittapalli RK, Zellmer DM, Gallardo JL, Donelson R, Seiler C, Decker SA, Santacruz KS, Pokorny JL, Sarkaria JN, et al. (2012a) Active efflux of Dasatinib from the brain limits efficacy against murine glioblastoma: broad implications for the clinical use of molecularly targeted agents. Mol Cancer Ther 11:2183–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. (2010) Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther 334:147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. (2011a) Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med 13:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Ohlfest JR, Elmquist WF. (2011b) The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther 336:223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Uchida Y, Mittapalli RK, Sane R, Terasaki T, Elmquist WF. (2012b) Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug Metab Dispos 40:1164–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JD, Brinkhuis RF, Wijnholds J, Schinkel AH. (1999) The mouse Bcrp1/Mxr/Abcp gene: amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res 59:4237–4241 [PubMed] [Google Scholar]
- Bailer AJ. (1988) Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm 16:303–309 [DOI] [PubMed] [Google Scholar]
- Bihorel S, Camenisch G, Lemaire M, Scherrmann JM. (2007) Influence of breast cancer resistance protein (Abcg2) and p-glycoprotein (Abcb1a) on the transport of imatinib mesylate (Gleevec) across the mouse blood-brain barrier. J Neurochem 102:1749–1757 [DOI] [PubMed] [Google Scholar]
- Breedveld P, Pluim D, Cipriani G, Wielinga P, van Tellingen O, Schinkel AH, Schellens JH. (2005) The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res 65:2577–2582 [DOI] [PubMed] [Google Scholar]
- Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. (2009) P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther 330:956–963 [DOI] [PubMed] [Google Scholar]
- Dai H, Marbach P, Lemaire M, Hayes M, Elmquist WF. (2003) Distribution of STI-571 to the brain is limited by P-glycoprotein-mediated efflux. J Pharmacol Exp Ther 304:1085–1092 [DOI] [PubMed] [Google Scholar]
- de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. (2007) P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res 13:6440–6449 [DOI] [PubMed] [Google Scholar]
- Dörner B, Kuntner C, Bankstahl JP, Bankstahl M, Stanek J, Wanek T, Stundner G, Mairinger S, Löscher W, Müller M, et al. (2009) Synthesis and small-animal positron emission tomography evaluation of [11C]-elacridar as a radiotracer to assess the distribution of P-glycoprotein at the blood-brain barrier. J Med Chem 52:6073–6082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AM, Bauer B. (2010) Regulation of ABC transporters at the blood-brain barrier: new targets for CNS therapy. Mol Interv 10:293–304 [DOI] [PubMed] [Google Scholar]
- Hubensack M, Müller C, Höcherl P, Fellner S, Spruss T, Bernhardt G, Buschauer A. (2008) Effect of the ABCB1 modulators elacridar and tariquidar on the distribution of paclitaxel in nude mice. J Cancer Res Clin Oncol 134:597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. (1993) In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res 53:4595–4602 [PubMed] [Google Scholar]
- Kawamura K, Yamasaki T, Konno F, Yui J, Hatori A, Yanamoto K, Wakizaka H, Ogawa M, Yoshida Y, Nengaki N, et al. (2011a) Synthesis and in vivo evaluation of 18F-fluoroethyl GF120918 and XR9576 as positron emission tomography probes for assessing the function of drug efflux transporters. Bioorg Med Chem 19:861–870 [DOI] [PubMed] [Google Scholar]
- Kawamura K, Yamasaki T, Konno F, Yui J, Hatori A, Yanamoto K, Wakizaka H, Takei M, Kimura Y, Fukumura T, et al. (2011b) Evaluation of limiting brain penetration related to P-glycoprotein and breast cancer resistance protein using [11C]GF120918 by PET in mice. Mol Imaging Biol 13:152–160 [DOI] [PubMed] [Google Scholar]
- Kemper EM, van Zandbergen AE, Cleypool C, Mos HA, Boogerd W, Beijnen JH, van Tellingen O. (2003) Increased penetration of paclitaxel into the brain by inhibition of P-glycoprotein. Clin Cancer Res 9:2849–2855 [PubMed] [Google Scholar]
- Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. (2010) Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther 333:788–796 [DOI] [PubMed] [Google Scholar]
- Lagas JS, van Waterschoot RA, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. (2010) Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther 9:319–326 [DOI] [PubMed] [Google Scholar]
- Lagas JS, van Waterschoot RA, van Tilburg VA, Hillebrand MJ, Lankheet N, Rosing H, Beijnen JH, Schinkel AH. (2009) Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res 15:2344–2351 [DOI] [PubMed] [Google Scholar]
- Lu C, Shervington A. (2008) Chemoresistance in gliomas. Mol Cell Biochem 312:71–80 [DOI] [PubMed] [Google Scholar]
- Mittapalli RK, Vaidhyanathan S, Dudek AZ, Elmquist WF. (2012a) Mechanisms limiting distribution of the BRAFV600E inhibitor dabrafenib to the brain: implications for the treatment of melanoma brain metastases. J Pharmacol Exp Ther DOI: 10.1124/jpet.112.201475 [published ahead of print]. [DOI] [PMC free article] [PubMed]
- Mittapalli RK, Vaidhyanathan S, Sane R, Elmquist WF. (2012b) Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: vemurafenib (PLX4032). J Pharmacol Exp Ther 342:33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelman JR, Gibiansky E, Lau DT. (1995) Applying Bailer’s method for AUC confidence intervals to sparse sampling. Pharm Res 12:124–128 [DOI] [PubMed] [Google Scholar]
- Polli JW, Humphreys JE, Harmon KA, Castellino S, O’Mara MJ, Olson KL, John-Williams LS, Koch KM, Serabjit-Singh CJ. (2008) The role of efflux and uptake transporters in [N-3-chloro-4-[(3-fluorobenzyl)oxy]phenyl-6-[5-([2-(methylsulfonyl)ethyl]aminomethyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos 36:695–701 [DOI] [PubMed] [Google Scholar]
- Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE. (2009) An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-3-chloro-4-[(3-fluorobenzyl)oxy]phenyl-6-[5-([2-(methylsulfonyl)ethyl]aminomethyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos 37:439–442 [DOI] [PubMed] [Google Scholar]
- Reardon DA, Vredenburgh JJ, Desjardins A, Peters KB, Sathornsumetee S, Threatt S, Sampson JH, Herndon JE, 2nd, Coan A, McSherry F, et al. (2012) Phase 1 trial of dasatinib plus erlotinib in adults with recurrent malignant glioma. J Neurooncol 108:499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane R, Agarwal S, Elmquist WF. (2012) Brain distribution and bioavailability of elacridar after different routes of administration in the mouse. Drug Metab Dispos 40:1612–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, Lagas JS, Lankheet NA, Poller B, Hillebrand MJ, Rosing H, Beijnen JH, Schinkel AH. (2012) Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer 130:223–233 [DOI] [PubMed] [Google Scholar]
- Wang T, Agarwal S, Elmquist WF. (2012) Brain distribution of cediranib is limited by active efflux at the blood-brain barrier. J Pharmacol Exp Ther 341:386–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward KW, Azzarano LM. (2004) Preclinical pharmacokinetic properties of the P-glycoprotein inhibitor GF120918A (HCl salt of GF120918, 9,10-dihydro-5-methoxy-9-oxo-N-[4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]phenyl]-4-acridine-carboxamide) in the mouse, rat, dog, and monkey. J Pharmacol Exp Ther 310:703–709 [DOI] [PubMed] [Google Scholar]
- Warren KE, Patel MC, McCully CM, Montuenga LM, Balis FM. (2000) Effect of P-glycoprotein modulation with cyclosporin A on cerebrospinal fluid penetration of doxorubicin in non-human primates. Cancer Chemother Pharmacol 45:207–212 [DOI] [PubMed] [Google Scholar]