

Abstract
A physiologically based pharmacokinetic (PBPK) modeling approach was used to assess the prediction accuracy of propofol hepatic and extrahepatic metabolic clearance and to address previously reported underprediction of in vivo clearance based on static in vitro–in vivo extrapolation methods. The predictive capacity of propofol intrinsic clearance data (CLint) obtained in human hepatocytes and liver and kidney microsomes was assessed using the PBPK model developed in MATLAB software. Microsomal data obtained by both substrate depletion and metabolite formation methods and in the presence of 2% bovine serum albumin were considered in the analysis. Incorporation of hepatic and renal in vitro metabolic clearance in the PBPK model resulted in underprediction of propofol clearance regardless of the source of in vitro data; the predicted value did not exceed 35% of the observed clearance. Subsequently, propofol clinical data from three dose levels in intact patients and anhepatic subjects were used for the optimization of hepatic and renal CLint in a simultaneous fitting routine. Optimization process highlighted that renal glucuronidation clearance was underpredicted to a greater extent than liver clearance, requiring empirical scaling factors of 17 and 9, respectively. The use of optimized clearance parameters predicted hepatic and renal extraction ratios within 20% of the observed values, reported in an additional independent clinical study. This study highlights the complexity involved in assessing the contribution of extrahepatic clearance mechanisms and illustrates the application of PBPK modeling, in conjunction with clinical data, to assess prediction of clearance from in vitro data for each tissue individually.
Introduction
Propofol is a probe substrate for UGT1A9 and is also cleared by cytochrome P450 (P450) enzymes, primarily via CYP2B6 and to a minor extent by CYP2C9, CYP1A2, and CYP3A4 (Guitton et al., 1998; Court et al., 2001; Oda et al., 2001; Court, 2005). Propofol has not been reported to be a substrate for transporters and undergoes minimal renal excretion (Vree et al., 1987; Simons et al., 1988; Veroli et al., 1992), and therefore represents a good candidate for exploring the prediction of clearance due to metabolism alone. However, pronounced underprediction of in vivo clearance has been observed for propofol using static in vitro–in vivo extrapolation (IVIVE) techniques (Kilford et al., 2009; Gill et al., 2012). Use of inappropriate in vitro systems, exclusion of extrahepatic metabolism, inadequacy of scaling factors, and/or models applied to the in vitro data may all contribute to this underprediction trend. Both in vivo and in vitro data indicate that the kidneys play an important role in the glucuronidation of certain drugs, including morphine and propofol (Mazoit et al., 1990; Pichette and du Souich, 1996; Soars et al., 2002; Takizawa et al., 2005a; Gill et al., 2012). Similarly, there is extensive evidence that both P450 and conjugation metabolism in the small intestine represent important contributors to drug clearance (Galetin et al., 2008; Cubitt et al., 2009, 2011; Gertz et al., 2010). We previously showed that the inclusion of renal metabolic clearance data in IVIVE improved prediction of glucuronidation clearance; however, underprediction was still apparent for certain drugs, including propofol (Gill et al., 2012).
Recently, there has been an increased use of dynamic modeling techniques such as physiologically based pharmacokinetic (PBPK) models to predict drug exposure and clearance (Rowland et al., 2011; Huang and Rowland, 2012). The application of PBPK models by the pharmaceutical industry and regulatory bodies together with some of the limitations of this approach have been highlighted recently (Poulin et al., 2011; Zhao et al., 2011; Huang and Rowland, 2012: Jones et al., 2012). A variety of physiologic and drug-specific parameters, including in vitro and in vivo clearance and tissue binding data, can be incorporated to optimize these models and improve prediction of in vivo pharmacokinetics (Nestorov, 2007; Huang and Rowland, 2012). The relative accuracy of predictions of hepatic versus extrahepatic clearance from in vitro data has not previously been assessed as the availability of in vivo data to allow such analysis is very limited. However, the use of a dynamic approach such as PBPK modeling in conjunction with suitable in vivo data would be expected to improve the understanding of the predictive capacity of in vitro clearance data for different tissues in isolation.
Previous PBPK modeling for propofol has not incorporated in vitro hepatic clearance data and has ignored the contribution of renal metabolic clearance (Levitt and Schnider, 2005; Upton and Ludbrook, 2005). In vivo propofol concentration-time data for subjects during liver transplantation have been reported in the literature (Veroli et al., 1992); under these conditions, the clearance observed is solely mediated by extrahepatic metabolism. Such in vivo data used in conjunction with mechanistic PBPK models allow independent assessment of the predictive capacity of in vitro clearance data for hepatic and renal metabolism.
The aim of this study was to apply a PBPK model to assess the prediction of propofol systemic clearance using in vitro metabolism data from the kidney and liver. Propofol blood concentration-time profiles and systemic clearance after intravenous dosing were predicted using intrinsic clearance (CLint) data obtained in different in vitro systems (microsomes and hepatocytes); the predictive ability of hepatic in vitro CLint data from different sources was assessed. The rich in vivo data available for propofol were used to optimize the PBPK model and to bridge the gap in the IVIVE of propofol clearance. In vivo data from anhepatic patients allowed the analysis of the prediction success of renal glucuronidation clearance in isolation from the liver; in conjunction with data from intact patients, these data were used to refine the prediction of renal and hepatic clearance.
Materials and Methods
Chemicals.
Propofol, alamethicin (from Trichoderma viride), UDP-glucuronic acid, EDTA, bovine serum albumin (BSA), saccharic acid lactone, MgCl2, NADP+, isocitric acid, and isocitric acid dehydrogenase were purchased from Sigma-Aldrich (Gillingham, UK). Propofol glucuronide was purchased from Toronto Research Chemicals (North York, ON, Canada). All other reagents were of the highest grade available.
Source of the Microsomes.
BD UltraPool human liver microsomes (HLMs) were purchased from BD Gentest (Woburn, MA). HLMs were pooled from 150 Caucasian donors, 50% of whom were female, with a mean age of 53 years (range, 18–79 years). Pooled human kidney and intestinal microsomes (HKMs and HIMs, respectively) were purchased from XenoTech (Tebu-Bio Ltd., Peterborough, UK). HKMs were pooled from eight donors, 88% of whom were Caucasian and 50% were female, and a mean age of 61 years (range, 48–69 years). HIMs were pooled from 13 donors, 92% of whom were Caucasian and 46% were female, with a mean age of 40 years (range, 18–55 years). Microsomes were stored at −80°C. Activity for pooled HLMs was reported as 0.054 nmol/min per milligram protein for CYP2B6 and 2.00 nmol/min per milligram protein for UGT1A9, using specific probe substrates. For HKM and HIM pools, glucuronidation capacity was characterized for 4-methylumbelliferone (a substrate of multiple uridine diphosphate glucuronosyltransferases [UGTs]), with reported activity of 125 and 7.22 nmol/min per milligram protein, respectively.
Experimental Conditions for Microsomal P450 Depletion Assays.
Incubations were performed in duplicate in Eppendorf vials (Eppendorf, Hamburg, Germany) using a substrate depletion approach. A final substrate concentration of 3 µM was used, which is >3-fold below the reported Km (Michaelis-Menten constant) value in HLMs (Guitton et al., 1998; Court et al., 2001; Al-Jahdari et al., 2006), ensuring suitable conditions to determine the intrinsic clearance of propofol. Propofol was preincubated at a microsomal protein concentration of 0.5 mg/ml in 0.1 M phosphate buffer, pH 7.4, for 5 minutes at 37°C shaken at 900g using an Eppendorf thermomixer (Kilford et al., 2009). The reaction was initiated by addition of the NADPH regenerating system, containing 1 mM NADP+, 7.5 mM isocitric acid, 1 U/ml isocitric acid dehydrogenase, and 10 mM MgCl2 in a final incubation volume of 1 ml (Kilford et al., 2009). Concentration of the organic solvent used (methanol) was 0.5% v/v of the incubation media. The total length of the incubation was 45 minutes. To terminate the reaction, 100-µl samples of the incubation were removed at each time point and added to an equal volume of ice-cold methanol containing 1 µM tolbutamide as the internal standard. Samples were kept at −20°C for at least 1 hour and then centrifuged (MSE Mistral 3000i centrifuge; MSE, London, UK) at 4°C and 2500g for 30 minutes. An aliquot of the supernatant (30 µl) was analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS) for propofol concentration. Experiments were repeated on three separate occasions. No P450 clearance was observed in HKMs and HIMs in the absence of BSA.
Experiments performed in the presence of BSA used the same methodology, with the exception of the addition of 2% BSA to the incubation. Samples of 100 µl of the incubation were removed at each time point and added to double volume of ice-cold acetonitrile containing 1 µM tolbutamide to terminate the reaction. Samples were refrigerated for at least 10 minutes and then centrifuged (Eppendorf Mini Spin) at 13,400g and room temperature for 5 minutes prior to analysis by LC-MS/MS for propofol concentration. Similarly to data in the absence of BSA, no P450 clearance was observed in HKMs and HIMs in the presence of BSA. Nonenzymatic depletion of propofol was monitored and clearance estimates were corrected for the observed nonenzymatic loss.
Experimental Conditions for Propofol Glucuronide Formation in Microsomes.
Incubations were performed in duplicate using Eppendorf vials. Initial experiments showed propofol glucuronide formation was linear up to 10 min at protein concentrations ≤0.5 mg/ml in all three tissues, both in the presence and absence of 2% BSA. The time period and protein concentration for each tissue were selected to ensure that metabolite formation was within the linear range. Substrate concentrations ranging from 0.5 to 400 µM were used to assess the kinetic parameters Km and Vmax (maximum rate of reaction), with the exception of HIMs in which a concentration range of 2.5 to 600 µM was employed. Microsomal protein concentrations for experiments performed in the absence of BSA were 0.2, 0.1, and 0.4 mg/ml for HLMs, HKMs, and HIMs, respectively. Microsomes were treated with alamethicin (50 µg/mg protein) for 15 minutes on ice as reported previously (Fisher et al., 2000; Cubitt et al., 2009; Kilford et al., 2009). Activated microsomes were preincubated with propofol and 0.1 M phosphate buffer, pH 7.1, containing 3.45 mM MgCl2, 1.15 mM EDTA, and 115 µM saccharic acid lactone, for 5 minutes at 37°C shaken at 900g (Fisher et al., 2001; Cubitt et al., 2009; Kilford et al., 2009). The reaction was initiated by the addition of UDP-glucuronic acid (5 mM in incubation), to give a final incubation volume of 0.1 ml. Organic solvent (methanol) made up 1% v/v of the incubation media. Control incubations were performed with no cofactor present to account for any potential cofactor independent formation of the metabolite over the incubation time. In the absence of BSA, the reaction was terminated after 10 minutes by addition of an equal volume ice-cold methanol containing 1 µM of the internal standard tolbutamide. Samples with starting propofol concentrations in excess of 10 µM were diluted in ice-cold blank 1:1 matrix:methanol to give a final propofol concentration of ≤10 µM. Samples were kept at −20°C for at least 1 hour and then centrifuged (MSE Mistral 3000i centrifuge) at 4°C and 2500g for 30 minutes. An aliquot of the supernatant (20 µl) was analyzed by LC-MS/MS for propofol glucuronide concentration.
Incubation conditions for experiments including BSA were comparable with those without BSA, with the exception of the addition of 2% BSA. Optimal conditions of 0.1 mg/ml microsomal protein for HLMs and HKMs or 0.2 mg/ml for HIMs were employed for the kinetic assessment in the presence of BSA. The reaction was terminated after 10 minutes by addition of double volume ice-cold acetonitrile containing 1 µM of the internal standard tolbutamide. Samples with starting propofol concentrations in excess of 10 µM were diluted in ice-cold blank 1:2 matrix:acetonitrile to give a final propofol concentration of ≤10 µM. Samples including BSA were refrigerated for at least 10 minutes and then centrifuged (Eppendorf Mini Spin) at 13,400g and room temperature for 5 minutes. An aliquot of the supernatant (20 µl) was analyzed by LC-MS/MS for propofol glucuronide concentration. Experiments were repeated on three separate occasions.
Propofol intrinsic clearance due to glucuronidation (CLint,UGT) data in HLMs, HKMs, and HIMs obtained by the depletion method and corresponding incubation details were reported previously (Gill et al., 2012). The batch of HLMs and HKMs used were the same as those used herein for determination of intrinsic clearance due to P450s (CLint,P450) and metabolite formation CLint,UGT. A different pool of HIMs was used due to the limited supply of the batch used previously; the donor demographics and activity characterization were comparable between the two HIM pools.
In addition to microsomal data, use of in-house unbound CLint data obtained in hepatocytes at a single propofol concentration of 5 µM was also investigated. CLint data were generated by the substrate depletion approach using pooled cryopreserved human hepatocytes from 10 donors purchased from In Vitro Technologies (Baltimore, MD). Experiments were performed in the absence and presence of nonspecific P450 inhibitor 1-aminobenzotriazole (ABT) at 2.5 mM to determine total CLint (combined CLint,P450 and CLint,UGT) and CLint,UGT, respectively.
LC-MS/MS.
Propofol and propofol glucuronide were analyzed on a Waters 2790 (Waters, Milford, MA) or an Agilent 1100 high-performance liquid chromatography system (Agilent Technologies, Santa Clara, CA), respectively, with a Micromass Quattro Ultima (Waters) triple quadruple mass spectrometer in negative mode. Source temperature was 125°C, desolvation temperature was 350°C, and the desolvation gas flow rate was 600 l/h. Cone gas flow rate was 150 l/h for propofol and 50 l/h for propofol glucuronide. The capillary voltage was 3.25 kV. Propofol and propofol glucuronide were analyzed using single ion recording due to their limited fragmentation. The transitions for propofol, propofol glucuronide, and tolbutamide were 176.90, 353.55, and 269.00 m/z, respectively. Analytes were separated using a Luna C18 (3 μm, 50 × 4.6 mm) column (Phenomenex, Macclesfield, UK). Four mobile phases were used, with varying gradients for each compound: 1) 90% water, 10% methanol, and 0.05% formic acid; 2) 10% water, 90% methanol, and 0.05% formic acid; 3) 90% water, 10% methanol, and 1 mM ammonium acetate; and 4) 10% water, 90% methanol, and 1 mM ammonium acetate. The flow rate was 1 ml/min, splitting to 0.25 ml/min prior to entry into the mass spectrometer. Cone voltage was set to 60 V for propofol and 75 V for tolbutamide, and the corresponding retention times were 3.40 and 2.80 minutes. When analyzing for the metabolite, the cone voltage was set to 30 and 45 V for propofol glucuronide and tolbutamide, with retention times of 3.40 and 3.20 minutes, respectively. The lower limit of quantification for propofol and its glucuronide metabolite were 0.039 and 0.020 µM.
Data Analysis.
The mean propofol concentrations of the duplicate samples from the P450 depletion assays at each time point were analyzed using GraFit 5 (Erithacus Software, Horley, UK) to determine the elimination rate constant (k) by fitting a single exponential equation to the data. This rate constant was used to calculate the CLint,P450 (Eq. 1). The nonenzymatic loss was taken into account when analyzing the experimental data. The CLint,P450 values were corrected for nonspecific binding (CLint,P450/fu,inc), using the experimentally determined fraction unbound in the microsomal incubation (fu,inc) in the presence and absence of 2% BSA reported previously (Gill et al., 2012), to generate the unbound intrinsic clearance (µl/min per milligram protein). The unbound CLint,P450 values have been reported.
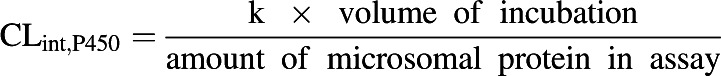 |
(1) |
The mean propofol glucuronide formation rates of the duplicate samples across the employed propofol concentration range were analyzed using nonlinear regression in GraFit 5 software (Erithacus Software). Km and Vmax were determined by fitting the Michaelis-Menten equation to the data. Binding of propofol to microsomal protein and albumin has been shown to be independent of substrate concentration over the range of propofol concentrations used herein (Rowland et al., 2008, 2009). The Km values were corrected for nonspecific binding in the presence and absence of 2% BSA, using data reported previously (Gill et al., 2012), to give unbound Km. The ratio of Vmax and unbound Km values were used to calculate the CLint,UGT. Unbound CLint,UGT and Km values have been reported.
In vitro CLint data obtained using HLMs in the presence of either P450 or UGT cofactors were used to determine the fraction of metabolism due to glucuronidation (fm,UGT) (Eq. 2) (Kilford et al., 2009). Similarly, CLint data derived in hepatocytes in the presence or absence of ABT were used to determine the fm,UGT.
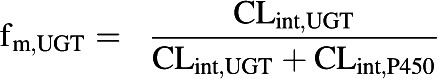 |
(2) |
In vitro microsomal CLint data from the various assays were scaled with the microsomal protein yields: 40, 12.8, and 20.6 mg protein per gram tissue were used for hepatic, renal, and intestinal data, respectively (Al-Jahdari et al., 2006; Barter et al., 2007; Cubitt et al., 2009). Hepatocyte unbound CLint data were scaled with a hepatocellularity value of 120 × 106 cells/g tissue (Brown et al., 2007). The scaled CLint data were compared between the in vitro systems.
Development of the Propofol PBPK Model.
A previously reported in-house whole-body PBPK model (Gertz et al., 2011) was adapted to predict propofol concentration-time profiles and pharmacokinetics. The resultant model contained 14 organ compartments connected by arterial and venous blood supplies (Fig. 1). An additional compartment representing the rest of the body was included, which accounted for <5% of the total body weight and <8% the total blood flow. Propofol plasma binding, blood to plasma partition coefficient, and renal excretion of unchanged drug were collated from the literature, as reported previously (Gill et al., 2012). Tissue to plasma concentration ratios (Kp) were predicted using the Rodgers and Rowland (2006) method, and are detailed in Fig. 1 and Supplemental Table 1. Tissue blood flow and volume were collated from the literature (ICRP, 2002) (Fig. 1; Supplemental Table 1).
Fig. 1.

The structure of the whole-body PBPK model used for prediction of propofol blood concentration-time profiles and clearance. Blood flow and tissue mass are presented for each compartment as a percentage of the whole-body flow/mass for an average man. Remaining body mass and blood flow were accounted for in the “rest of body” compartment. Kp values for each compartment were predicted using the Rodgers and Rowland method and normalized for observed Vss (Vss,obs) by a uniform scalar, scaling factor for volume = Vss,obs – blood volume / Vss,pred – blood volume. Kp for the “rest of body” compartment was assumed to be the same as muscle.
All tissues were assumed to be well stirred compartments where unbound tissue concentration is at equilibrium with the unbound concentration in the emerging blood (Pang and Rowland, 1977). Eq. 3 was used for noneliminating organs (Nestorov, 2003).
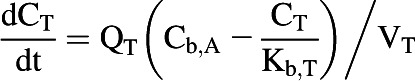 |
(3) |
where CT, QT, Cb,A, Kb,T, and VT represent unbound tissue concentration, tissue blood flow, unbound concentration in arterial blood, tissue to blood concentration ratio, and tissue volume.
The liver, kidney, and enterocytes were considered to potentially contribute to systemic drug clearance. The liver and kidney were separated into cellular tissue and blood compartments to allow assessment of the extraction ratio across each tissue. Eq. 4 represents the tissue compartment and Eq. 5 represents the blood compartment for the kidney and the liver.
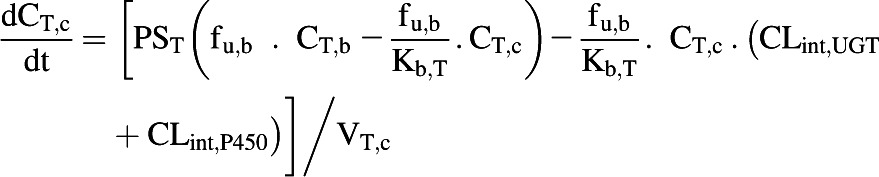 |
(4) |
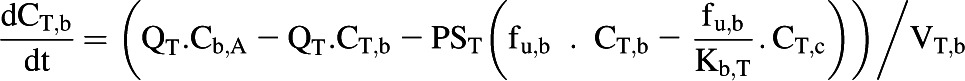 |
(5) |
where CT,c, PST, CT,b, fu,b, VT,c, and VT,b represent tissue cell concentration, permeability surface area (set to >10,000 × tissue blood flow to ensure perfusion limited kinetics), concentration in blood residing in the tissue, blood binding, volume of tissue cells, and volume of blood residing in tissue. Microsomal CLint,UGT and CLint,P450 were scaled with the microsomal recovery and mass of the relevant tissue, as detailed above. Hepatocyte CLint (combined CLint,UGT and CLint,P450) data were scaled with the hepatocellularity value and the mass of the liver. Kidney and liver blood volumes were set to 100 ml (ICRP, 2002). The portal vein concentration represents the differential of emergent blood concentrations from the large and small intestine, enterocytes, stomach, spleen, and pancreas.
The extraction ratio for the enterocytes was not calculated and therefore this tissue was defined as a single compartment (Eq. 6).
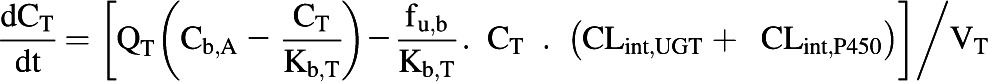 |
(6) |
The blood flow to the small intestine represents approximately 10% of the cardiac output (ICRP, 2002) and the enterocytic blood flow represents approximately 50% of the small intestinal blood flow (Gertz et al., 2011). The rate equations were solved in MATLAB software (version 7.12.0; MathWorks, Natick, MA) using the ODE15s solver. The dose recovery over time was assessed using mass balance equations.
Assumptions Applied to the Propofol PBPK Model.
The following assumptions were made: 1) there is no active uptake or efflux of propofol in any tissue and therefore tissue distribution/elimination is perfusion rate limited; this assumption seems justifiable because, together with the high lipophilicity of propofol (Reiner et al., 2009), there is a lack of in vitro or in vivo data suggesting that propofol is a substrate of transporters; 2) propofol does not affect the cardiac output (Grounds et al., 1985; Price et al., 1992); and 3) the volume of distribution at steady state (Vss) for the anhepatic patients was not expected to be markedly different from that in healthy subjects. This is supported by a number of studies where changes in Vss for patients with varying grades of liver cirrhosis were not apparent in comparison with patients with healthy livers (mean values ranging from 202 to 637 l in subjects with mild liver cirrhosis) (Servin et al., 1988a, b, 1990). Similarly, no changes in propofol Vss during chronic hepatic cirrhosis were stated in the AstraZeneca propofol product monograph (http://www.astrazeneca.ca/documents/ProductPortfolio/DIPRIVAN_PM_en.pdf). In addition, no change in blood binding of propofol was observed in patients with mild liver cirrhosis (Servin et al., 1990).
Validation of Propofol PBPK Model.
The PBPK model was validated using data from two clinical studies, covering a propofol dose range of 2–18 mg/kg (Gepts et al., 1987; Doenicke et al., 1997). The study details and subject demographics for these studies are presented in Table 1. The cardiac output was corrected for the mean age of each dose group (Brown et al., 1997). One of the studies (Gepts et al., 1987) included elderly subjects but no alterations were made to the tissue volumes for this population. Plasma binding was assumed to be equivalent in young and elderly subjects for which evidence could be found in the literature (Kirkpatrick et al., 1988). Minimal age and sex effects have been observed for in vitro microsomal UGT activity for a range of probe substrates including propofol (Court, 2010); therefore, no alterations were made to microsomal protein yields for the different populations.
TABLE 1.
Study details for clinical data used during validation and optimization of a whole-body PBPK model for prediction of propofol in vivo clearance and blood concentration-time profiles from in vitro CLint data
Data are presented as mean (± S.D.) unless otherwise specified.
| Study | Status of Patients | Dose Type | Dose Level | Subjects | Body Weight | Age | Male |
|---|---|---|---|---|---|---|---|
| mg/kg | n | kg | yr | % | |||
| Doenicke et al. (1997) | Intact | Intravenous bolus | 2 | 12 | ND | 24–42a | 100 |
| 2 | 12 | ND | 24–42a | 100 | |||
| Gepts et al. (1987) | Intact | Intravenous infusion over 2 h | 6 | 6 | 75.0 (7.2) | 60.3 (4.9) | 100 |
| 12 | 6 | 65.5 (7.9) | 50.6 (14.7) | 67 | |||
| 18 | 6 | 61.6 (8.8) | 39.8 (10.0) | 50 | |||
| Veroli et al. (1992) | Anhepatic | Intravenous bolus | 0.5 | 10 | 60.0 (7.0) | 38.0 (ND) | ND |
ND, not detailed; PBPK, physiologically based pharmacokinetic.
Mean age not detailed; age range of subjects presented instead
Vss for propofol is highly variable in the literature, with mean values reported in 14 studies collated from the literature (including 194 subjects) ranging from 121 to 722 l; no trends were observed regarding sex, age, dose level, length of infusion, or disease status (details provided in Supplemental Fig. 1). The Vss data considered for this analysis were taken from studies in which the propofol blood concentrations were available over at least 8 hours, which is considered adequate to describe its pharmacokinetics. Where necessary, the predicted Kp values were optimized using a universal scalar for all tissues to ensure that the reported Vss was recovered correctly for each dose group used in the model validation and for the prediction of propofol blood concentration-time profiles.
Contradicting reports exist in the literature over the potential propofol metabolism and/or sequestration in the lung (Dawidowicz et al., 2000; He et al., 2000; Hiraoka et al., 2005; Takizawa et al., 2005a). Reduction in propofol concentration across the lung in one study was proportional to the formation of the quinol metabolite and assessment of the blood concentration data suggested a lung extraction ratio of 0.40 (Dawidowicz et al., 2000). Assumption of that extent of lung extraction would predict clearance in excess of the observed values for the in vivo studies used in the current analysis. In contrast, the majority of other available data proposed that the reduction in propofol concentration across the lung was not due to metabolism but binding, with subsequent slow release from the lung (He et al., 2000; Hiraoka et al., 2005; Levitt and Schnider, 2005; Takizawa et al., 2005a; Upton and Ludbrook, 2005). Previous studies showed that sequestration of propofol was not apparent after intravenous infusion dosing, particularly for patients aged >35 years (He et al., 2000; Levitt and Schnider, 2005). The majority of the in vivo data used in the current analysis were reported in subjects of similar or greater age after an infusion dose, which may explain the lack of evidence for sequestration in the lung. With the lack of suitable data to define association and dissociation rate constants for potential propofol binding in the lung, a truly physiologic model to account for this process could not have been included within the current PBPK model and therefore no adjustment for lung sequestration was employed. Accumulation within lysosomes, one potential route of sequestration within tissues such as lungs, is not anticipated for propofol due to its physicochemical properties. The potential for propofol sequestration in other tissues or via other mechanisms could not be investigated, although this cannot be ruled out.
In Vitro Clearance Data Used as Inputs for the PBPK Model to Predict in Vivo Clearance of Propofol.
In vitro CLint,P450 and CLint,UGT data for HLMs, HKMs, and HIMs were used within the PBPK model to assess the success of in vivo clearance predictions. The prediction success of systemic clearance was investigated using CLint,UGT data from substrate depletion assays, obtained previously from Gill et al. (2012), and metabolite formation assays (current study). CLint,P450 data were obtained using the substrate depletion approach (detailed herein). Clearance prediction success for in-house data determined via the substrate depletion method in hepatocytes (combined CLint,P450 and CLint,UGT) in conjunction with CLint,UGT data from HKMs and HIMs from either the substrate depletion or metabolite formation methods was also investigated. Details of the in vitro data used for prediction of in vivo clearance are given in Table 2.
TABLE 2.
Kinetic parameters from different in vitro assays used to characterize propofol CLint,P450 and CLint,UGT in the presence and absence of 2% BSA and used in a whole-body PBPK model to predict propofol in vivo clearance and blood concentration-time profiles
Data are the mean (± S.D.) of n = 3 experiments, each performed in duplicate, with the exception of CLint,UGT in HIM substrate depletion assays where n = 1.
| Assay Type | Parameter | Without BSA |
With 2% BSA |
|||||
|---|---|---|---|---|---|---|---|---|
| HLMs | HKMs | HIMs | Hepatocytes | HLMs | HKMs | HIMs | ||
| Metabolite formation | Vmax (pmol/min per milligram protein) | 1460 (244) | 5220 (726) | 1280 (147) | NT | 1390 (101) | 4310 (1406) | 1470 (457) |
| Km (µM) | 107 (39.9) | 91.0 (12.8) | 458 (65.4) | NT | 5.22 (0.360) | 3.45 (0.931) | 133 (55.6) | |
| CLint,UGT (ml/min per gram tissue)a | 0.594 (0.215) | 0.735 (0.0247) | 0.0581 (0.00652) | NT | 10.6 (0.936) | 17.0 (7.62) | 0.237 (0.0551) | |
| Substrate depletion | CLint,UGT (ml/min per gram tissue)a,b | 2.71 (0.18) | 1.17 (0.41) | 1.29 (—) | 7.63 (1.68)c | 8.05 (0.46) | 13.1 (1.66) | 2.13 (—) |
| CLint,P450 (ml/min per gram tissue)a | 9.12 (2.03) | ND | ND | 10.2d | 18.1 (0.455) | ND | ND | |
| CLint,P450 & UGT (ml/min per gram tissue)a | NT | NT | NT | 17.9 (3.36)c | NT | NT | NT | |
BSA, bovine serum albumin; HIM, human intestinal microsomes; HKM, human kidney microsomes; HLM, human liver microsomes; (—), not applicable; ND, no depletion observed; NT, not tested; PBPK, physiologically based pharmacokinetic.
In vitro CLint data scaled with microsomal recovery factors (40, 12.8, and 20.6 mg protein/g tissue for HLMs, HKMs, and HIMs, respectively) or hepatocellularity (120 × 106 cells/g tissue) to give scaled CLint/g tissue.
Data presented previously in Gill et al. (2012).
In-house data, where CLint,P450 and CLint,UGT were determined in hepatocytes in the absence of ABT and CLint,UGT was determined in hepatocytes in the presence of ABT.
Hepatocyte CLint in absence of ABT – hepatocyte CLint in presence of ABT.
In addition, the in vitro data were used in the PBPK model to predict hepatic and renal extraction ratios (EH and ER, respectively) at steady state. The predicted extraction ratios were compared with values reported in a clinical study (Hiraoka et al., 2005), which was independent to the clinical studies used for model validation. The reported EH (0.93) and ER (0.69) were obtained from blood concentration measurements in the renal vein, hepatic vein, and radial artery at steady state. The extraction ratios for each tissue (ET) were calculated by Eq. 7.
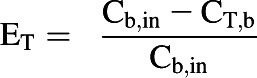 |
(7) |
where Cb,in represents the concentration in blood entering the tissue.
Optimization of Predictions of Renal and Hepatic Clearance from in Vitro Data.
Initial analysis indicated underprediction of hepatic and renal CLint, regardless of the in vitro system used. Therefore, optimization was performed to bridge the gap in IVIVE and determine the empirical scaling factors required for the in vitro data to accurately recover in vivo clearance, by fitting the PBPK model to the in vivo propofol blood concentration-time profiles. Data from three dose levels (6–18 mg/kg) in intact patients (Gepts et al., 1987) and from anhepatic patients (Veroli et al., 1992) (who received a 0.5 mg/kg dose) were fitted simultaneously in MatLab using a nonlinear least-squares regression analysis (lsqnonlin function). For the anhepatic patients, the PBPK model was adapted removing the liver compartment to reflect the in vivo situation. Data from the anhepatic patients were used to delineate the contribution of the renal CLint,UGT and to refine the PBPK model with respect to renal metabolism, whereas data from intact patients provided input for optimization of the hepatic CLint and Kp. Propofol concentration-time profiles in the anhepatic patients were only available for 1 hour; therefore, Vss for these patients could not be determined from the profile. The data from intact patients were used to inform the optimization of the Vss for both intact and anhepatic patients in the simultaneous fitting routine. Optimization of hepatic and renal CLint in the PBPK model allowed the assessment of the degree of underprediction of in vitro CLint for each tissue individually.
The in vitro CLint data for the intestinal microsomes were low in comparison with that in the kidney and the liver. Considering their lower tissue mass and blood flow, the enterocytes were not expected to contribute extensively to the metabolism of propofol. There is also a lack of suitable in vivo data for assessment of the predictive capacity of the intestinal in vitro CLint,UGT data. For these reasons, the optimization was performed for the kidney and liver metabolism only.
Results
Propofol P450 Depletion Assays in Human Hepatic, Renal, and Intestinal Microsomes.
No P450-mediated clearance was observed in renal or intestinal microsomes; however, in the liver, CLint,P450 in the presence of BSA was 2.2-fold higher than CLint,UGT obtained via substrate depletion (Table 2). BSA increased propofol CLint,P450 by 2-fold in HLMs (Table 2), whereas a 3-fold change in CLint,UGT in the presence of BSA was reported previously (Gill et al., 2012). Depletion profiles in HLMs over time are shown in the Supplemental Fig. 2.
Propofol Glucuronide Formation Assays in Human Hepatic, Renal, and Intestinal Microsomes.
Propofol Km and Vmax were determined in hepatic, renal, and intestinal microsomes both in the presence and absence of BSA; corresponding kinetic profiles are shown in Fig. 2. Use of a protein concentration of 0.1 mg/ml for HLMs and HKMs in combination with 2% BSA had no impact on propofol Vmax. However, use of lower microsomal protein concentrations (0.05 and 0.075 mg/ml) in the presence of BSA reduced Vmax by up to 70% (Supplemental Fig. 3, A and B). The extent of the decrease in Vmax observed at the lowest protein concentration was more pronounced in the kidney than the liver (3-fold versus 2-fold). Use of a 1% BSA concentration produced comparable results to data obtained in the presence of 2% BSA (Supplemental Fig. 3, C and D).
Fig. 2.
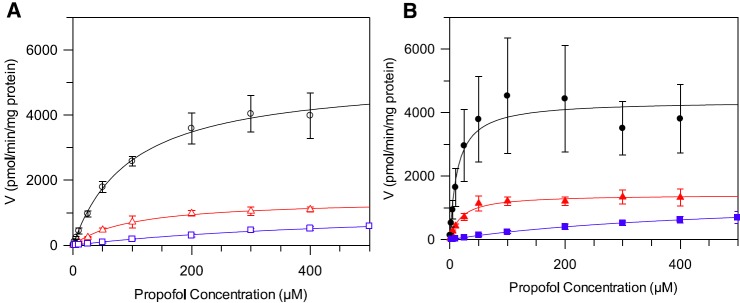
Formation rate plots for propofol glucuronide as a function of substrate concentration in alamethicin activated human hepatic, renal, and intestinal microsomes in the presence and absence of 2% BSA. Data represent the mean of three experiments, each performed in duplicate. Error bars represent the standard deviation. (A) Data generated in the absence of BSA. (B) Data generated in the presence of BSA. △, ○, and □ represent propofol glucuronide formation in hepatic, renal, and intestinal microsomes, respectively.
In the absence of BSA propofol, Km was similar in HLMs and HKMs (107 versus 91 µM); however, the value for the intestine was greater (458 µM) (Table 2). Inclusion of 2% BSA caused reduction in Km for all tissues, which was more pronounced in the liver and kidney than in the intestine, with Km values of 5, 3, and 133 µM in the presence of BSA, respectively (Table 2). Similarly to data derived in the absence of BSA, Km values in the presence of 2% BSA were comparable in HLMs and HKMs. Under optimal conditions, inclusion of 2% BSA had no appreciable impact on Vmax in any of the tissues (Table 2). Scaled CLint,UGT values (ml/min per gram tissue) were highest in HKMs both in the presence and absence of BSA (Table 2). Upon inclusion of 2% BSA, CLint,UGT in HIMs increased by 4-fold in comparison with the large increases observed for HLMs (18-fold) and HKMs (23-fold) (Table 2).
Comparison of CLint Data Derived Using Different in Vitro Systems.
In the absence of BSA, propofol scaled CLint,UGT estimates derived from metabolite formation assays in HLMs and HKMs were lower than those from substrate depletion assays (0.6 versus 2.7 ml/min per gram tissue and 0.7 versus 1.2 ml/min per gram tissue, respectively) (Table 2). However, in the presence of 2% BSA, CLint,UGT data were comparable from both assays (Table 2). Consequently, the increase in propofol CLint,UGT upon inclusion of BSA in HLMs and HKMs was greater for the metabolite formation data in comparison with the substrate depletion data. For HIMs, CLint,UGT estimates obtained by metabolite formation were lower both in the presence (9-fold) and absence (22-fold) of BSA compared with those obtained by depletion (Table 2). Combined CLint,P450 and CLint,UGT data for hepatic microsomes (26 and 29 ml/min per gram tissue using the depletion or formation assays in the presence of BSA, respectively) gave higher total CLint per gram of liver than estimates obtained from hepatocytes (18 ml/min per gram tissue).
The in vitro fm,UGT for HLM CLint data derived using substrate depletion were 0.23 and 0.31 in the absence and presence of 2% BSA, respectively. When using HLM CLint,UGT data derived via metabolite formation in the presence of BSA, the resulting fm,UGT (0.37) was similar to using HLM CLint,UGT data obtained by substrate depletion. Use of metabolite formation CLint,UGT data obtained in the absence of BSA reduced the fm,UGT estimate. The fm,UGT determined in hepatocytes using the substrate depletion approach in the presence and absence of ABT (0.43) was similar to that determined from HLMs. The in vitro fm,UGT for HKMs and HIMs were 1.0 due to the lack of P450 clearance observed in these tissues.
Prediction of Propofol Clearance and Blood Concentration-Time Profiles Using Data from Different in Vitro Systems.
Blood concentration-time profiles and in vivo clearance were predicted using various combinations of in vitro CLint,UGT and CLint,P450 data from the different systems used; a summary of the prediction accuracy for each system is shown in Table 3 (full details for individual dose groups are presented in Supplemental Table 2). The predicted concentration-time profiles for all dose groups are shown in the Supplemental Figs. 4 and 5.
TABLE 3.
Accuracy of propofol in vivo clearance predicted using CLint,P450 and CLint,UGT data derived from different in vitro systems
Data are shown as mean (± S.D.) predicted CL/observed CL (%). Data for individual dose groups are detailed in Supplemental Table 2.
| In Vitro Method Used to Obtain Microsomal CLint,UGT | In Vitro Systems Used |
||
|---|---|---|---|
| Microsomes without BSA | Microsomes with BSA | Hepatocytes and HKMs and HIMs with BSA | |
| Substrate depletion | 16.8 (2.19) | 33.1 (4.78) | 28.3 (3.54) |
| Metabolite formation | 13.7 (1.75) | 34.7 (5.15) | 26.4 (3.59)a |
Substrate depletion approach used to determine hepatocyte CLint data.
Use of the PBPK model resulted in underestimation of clearance regardless of the source of the in vitro data. Predicted clearance did not exceed 35% of the observed values and recovery of the blood concentration-time profiles was poor when using CLint data from all in vitro systems (Table 3). As expected from comparison of the in vitro data, no difference was observed in the predicted in vivo clearance from microsomal CLint,UGT data derived by either the substrate depletion or metabolite formation methods in the presence of BSA (Table 3). The lower in vitro CLint obtained in hepatocytes or microsomes in the absence of BSA reduced the clearance prediction accuracy even further. Therefore, the microsomal CLint data obtained using the substrate depletion approach (in the presence of BSA) were used for further analysis and optimization of the PBPK model; a representative predicted blood concentration-time profile obtained using these data is shown in Fig. 3. When these in vitro data were used in the PBPK model, renal glucuronidation clearance contributed 11% to the predicted systemic clearance, which is much lower than the approximately 30% contribution that has been reported in vivo (Hiraoka et al., 2005; Takizawa et al., 2005a).
Fig. 3.

Predicted blood concentration-time profiles for propofol using optimized liver and kidney in vitro CLint,u data derived from microsomes in the presence of BSA. ○ represent mean ± S.D. observed blood concentration-time data for the 18 mg/kg dose level from Gepts et al. (1987). Black line represents predicted concentrations using in vitro CLint data from the substrate depletion microsomal assays (in the presence of BSA) without optimization; red line represents predicted concentrations using optimized kidney CLint,UGT data; and green line represents predicted concentrations using optimized CLint data for both liver and kidney.
With microsomal substrate depletion clearance data as inputs, the PBPK model was used to predict the extraction ratio due to metabolic clearance for the kidney and the liver at steady state. These data were compared with corresponding in vivo values detailed in a clinical study (Hiraoka et al., 2005). Using in vitro data derived in the presence of BSA, an underprediction of hepatic extraction was observed (predicted EH was 0.39, which represented 42% of the observed value). A more pronounced underprediction was found for ER, with the predicted value (0.07) being only 10% of the observed value.
Optimization of the Model for Prediction of Renal and Hepatic Metabolic Clearance.
Considering the underprediction observed, in vitro clearance parameters were optimized within the PBPK model by performing simultaneous fitting of the concentration-time data from intact and anhepatic patients (Table 4); in vitro data from microsomal substrate depletion assays in the presence of BSA were used as the initial estimates of CLint. Simultaneous optimization of in vitro hepatic and renal CLint in the PBPK model showed that renal glucuronidation clearance was underpredicted to a greater extent than hepatic clearance, resulting in an empirical scaling factor of 17 versus 9 required in the case of liver (Table 4). Coefficients of variation for the hepatic and renal CLint empirical scaling factors were 59 and 39%, respectively (Table 4). Use of the empirical scaling factors solely for in vitro kidney CLint,UGT data, in conjunction with the nonoptimized liver CLint, resulted in predicted clearance within 2-fold (59% on average) of observed values. Representative blood concentration-time profiles predicted using either nonoptimized in vitro data or optimized renal and hepatic clearance (in vitro hepatic and renal CLint data with the empirical scaling factors) are shown in Fig. 3. A high degree of correlation was observed for this dataset (R2 = 0.92, n = 94 data points) with 99% of the propofol blood concentrations predicted within 2-fold of the line of unity across the dose range (Fig. 4). Use of the optimized in vitro clearance data in the PBPK model predicted an overall fm,UGT value of 0.53, in agreement with in vivo estimates of approximately 0.6 (Favetta et al., 2002). In addition, predicted EH and ER at steady state based on the use of the optimized scalars for in vitro hepatic and renal CLint were within 20% of the observed values [91 and 82% of observed values, respectively; data from an additional independent study by Hiraoka et al. (2005)].
TABLE 4.
Empirical scaling factors used for prediction of Kp values and in vitro hepatic and renal CLint data determined by optimization of the PBPK model using simultaneous fitting of in vivo concentration-time data from intact and anhepatic patients
| Parameter | Initial Parameter Estimate | Model Fitted Parameter Estimatea | Model Fitted Scalara | CV% for Fitted Scalara |
|---|---|---|---|---|
| Kpb | — | — | 0.54 | 9.5 |
| Renal CLint,UGT (l/h per gram tissue) | 0.786c | 13.6 | 17.3 | 39 |
| Hepatic CLint (l/h per gram tissue)d | 1.57c | 14.5 | 9.21 | 59 |
CV, coefficient of variation.
Parameters estimated by simultaneous fitting of in vivo concentration-time data from intact and anhepatic patients, as detailed in the Materials and Methods.
Initial Kp values estimated using the Rodgers and Rowland (2006) method.
Initial CLint data taken from microsomal substrate depletion assays in the presence of BSA. Full details are presented in Table 2.
Combined CLint,P450 and CLint,UGT data.
Fig. 4.

Comparison of observed and predicted propofol blood concentrations using the whole-body PBPK model to simultaneously optimize liver and kidney CLint. Solid line represents unity; dashed line represents a 2-fold deviation from unity; ○ represents data from anhepatic patients (Veroli et al., 1992); ×, ◇, and □ represent data from the 6, 12, and 18 mg/kg dose levels in intact patients (Gepts et al., 1987), respectively.
Discussion
This is the first study to assess the prediction accuracy of renal versus hepatic clearance from CLint data determined in different in vitro systems by applying a PBPK modeling approach. Propofol was used as an example drug and clinical data reported in intact and anhepatic patients were used to refine the predictions of clearance from in vitro data and to optimize the PBPK model for prediction of propofol blood concentration-time profiles.
In Vitro Characterization of Propofol Metabolism in Microsomes.
No P450 clearance of propofol was detectable in HKMs, in agreement with studies reporting low P450 mRNA levels in the kidney (Nishimura and Naito, 2006; Bièche et al., 2007). The effect of albumin on CYP2B6 (the main enzyme for propofol P450 metabolism) has not been investigated to date; this enzyme is not reported to be involved in free fatty acid clearance and a marked impact of albumin is not anticipated, in agreement with the minimal increase (2-fold) in CLint,P450 observed in HLMs upon inclusion of BSA. Similar to previous reports, propofol liver CLint,P450 was greater than CLint,UGT (3-fold) in the absence of BSA with the extent of this difference being reduced upon inclusion of BSA (Al-Jahdari et al., 2006; Kilford et al., 2009).
This study shows that the trend of increased CLint,UGT upon inclusion of albumin is consistent in hepatic, renal, and intestinal microsomes; however, the extent differs between the tissues. Similarly to our findings, previous reports of the effect of albumin on UGT1A9 and 2B7 substrates in HLMs also showed a decrease in Km with minimal impact on Vmax (Rowland et al., 2007, 2008, 2009). This is considered to be due to the sequestration of inhibitory free fatty acids released during microsomal incubations, allowing estimation of the true Km (Rowland et al., 2007, 2008). Propofol kinetic parameters obtained in HLMs, HKMs, and HIMs were comparable with previously published data, where available (Supplemental Table 3 and Supplemental references). In the presence of BSA, HLM and HKM CLint,UGT values were similar from both metabolite formation and substrate depletion assays. In this study, a decrease in the propofol Vmax was observed at microsomal protein concentrations <0.1 mg/ml and in the presence of BSA, in agreement with a previous report (Walsky et al., 2012). Similar effects were observed at both 1 and 2% BSA, suggesting that the effect could not be simply rationalized by the differential concentration ratio of microsomal protein to BSA. Current findings are in contrast to an increase in Vmax reported for other UGT1A9 substrates in the presence of BSA and at low microsomal protein concentrations (Manevski et al., 2011, 2012). However, differences in assay conditions in the published studies prevent direct comparison with the findings herein; further investigation is required to confirm whether these effects on Vmax are directly associated with the use of BSA at very low protein concentrations. Current findings highlight the importance of careful selection of the assay conditions when determining glucuronidation kinetic parameters in the presence of BSA, particularly when using microsomal protein concentrations of <0.1 mg/ml.
The highest in vitro fm,UGT estimate was obtained from in-house hepatocyte data (0.43), whereas the fm,UGT calculated from HLM in vitro data derived in the presence of BSA was <0.4. These in vitro estimates ignore the contribution of extrahepatic metabolism and therefore it was not surprising that the fraction was lower than the value of 0.6 reported in vivo (Favetta et al., 2002). A mechanistic model that allows incorporation of metabolism in extrahepatic tissues is required to adequately assess the prediction of fm,UGT from in vitro CLint data derived in both hepatic and extrahepatic microsomes. However, inclusion of such data in the PBPK model still resulted in an underprediction of fm,UGT (0.39), with the predicted value being comparable with that derived from HLM data alone.
Prediction of Propofol Renal Glucuronidation and Hepatic Metabolism Using in Vitro Clearance Data from Different Systems and Optimization Using in Vivo Data.
This study represents the first report where a PBPK model has been used to predict propofol renal metabolism from in vitro clearance data, in contrast to previous propofol PBPK modeling efforts that have mainly focused on accommodating potential lung sequestration (Levitt and Schnider, 2005; Upton and Ludbrook, 2005). Inclusion of BSA in the microsomal assays improved the prediction of propofol in vivo clearance using the PBPK model, consistent with the trend reported previously for UGT1A9 substrates using static IVIVE methods (Rowland et al., 2008; Kilford et al., 2009; Gill et al., 2012). However, the underprediction trend was still apparent regardless of whether hepatocyte or microsomal data in the presence of 2% BSA were used as an input for hepatic CLint; predicted clearance was <35% of the observed value. Therefore, clinical data from three dose levels in intact patients and data reported for subjects during the anhepatic phase of liver transplantation were used to optimize the in vitro clearance parameters. Use of clinical data in conjunction with the developed PBPK model allowed differentiation between underprediction of hepatic and renal CLint and highlighted more pronounced underestimation of propofol renal CLint,UGT in comparison with hepatic clearance (17-fold versus 9-fold). Prior to optimization, predicted extraction ratios were 0.07 and 0.42 for kidney and liver, compared with 0.43–0.87 and 0.76–0.98 estimated in vivo, respectively (Hiraoka et al., 2005; Takizawa et al., 2005a, 2005b). Following optimization of renal in vitro glucuronidation data, the predicted in vivo clearance was within 2-fold of the observed values and the corresponding extraction ratios and fm,UGT values were in good agreement with the in vivo data (from independent studies than used for model development). The intestinal metabolism was not investigated in the PBPK model due to the anticipated low contribution of this tissue to its systemic clearance. This assumption is supported by UGT and P450 expression data for the intestine, showing low levels of UGT1A9 and CYP2B6 in comparison with the liver and kidney (Bièche et al., 2007; Court et al., 2012; Harbourt et al., 2012). Due to the lack of suitable in vivo data, we could not assess the prediction accuracy for in vitro CLint,UGT data for the intestine or potential for propofol sequestration in either lungs or other tissues, although this cannot be ruled out.
Reported UGT mRNA data showed regional differences in the kidney (Gaganis et al., 2007; Lash et al., 2008). In addition, glucuronidation capacity has been found to differ between the sections of the kidney in laboratory animals, with the highest activity observed in the proximal tubular cells of the cortex (Cojocel et al., 1983; Hjelle et al., 1986). However, data on regional differences in glucuronidation activity in human kidney tissue are very limited. Two studies have shown up to 3-fold higher glucuronidation clearance for naproxen and morphine (mainly cleared by UGT2B7) in cortical compared with medullary microsomes (Yue et al., 1988; Gaganis et al., 2007). In contrast, frusemide (primarily cleared by UGT1A9) glucuronidation clearance was comparable in microsomes from human kidney cortex and medulla (Kerdpin et al., 2008). Although these data may suggest that there are no regional differences in glucuronidation clearance via UGT1A9 in the kidney, these findings should be considered with caution because potential differences in microsomal recovery depending on the region of the kidney were not considered. The information on the proportion of the cortex and medulla used for the preparation of the microsomes employed in this study was not available; similarly, the region of the kidney used in the study reporting the kidney microsomal recovery (Al-Jahdari et al., 2006) is unknown, all of which may affect the glucuronidation activity and contribute to the particularly poor prediction of renal glucuronidation observed with the PBPK model prior to optimization of the in vitro data.
Although we have successfully developed a PBPK model for the prediction of propofol clearance and exposure from in vitro CLint data, we could not assess the use of our empirical scaling factors with other drugs. The availability of blood concentration-time data identifying drug metabolism across the kidney is extremely limited and hinders the assessment of this model with a wider range of compounds. In addition to the factors discussed above, poor prediction of renal metabolism may also suggest that well stirred assumptions, which are adequate for many tissues in PBPK modeling, are not appropriate for the kidney and more complex models may need to be considered. This will be particularly relevant for drugs that are also substrates of the renal transporters (Giacomini et al., 2010). Unlike the liver, the kidney is not a homogeneous tissue and consists of distinct regions with varying blood flows. However, in vivo and in vitro data required for development and validation of more complex kidney models, such as absolute abundance data for the UGTs and transporters in the various regions of the kidney, are currently lacking in the literature.
In conclusion, this study has shown consistent increases in propofol clearance estimates in hepatic, renal, and intestinal microsomes due to the “albumin effect” and highlighted the importance of careful selection of assay conditions when using BSA at very low microsomal protein concentrations. The analysis provides an example of the application of clinical data to refine the developed PBPK model and assess the predictive capacity of in vitro data for propofol renal and hepatic metabolic clearance in isolation. This study highlighted a more pronounced underprediction of renal glucuronidation than hepatic metabolism; further assessment of the contribution of extrahepatic clearance mechanisms and the adequacy of currently available scaling factors and models for renal metabolism is required.
Supplementary Material
Acknowledgments
The authors thank Sue Murby and Dr. David Hallifax (University of Manchester) for valuable assistance with the LC-MS/MS, and Dr. Peter Kilford for determination of the hepatocyte in vitro clearance data.
Abbreviations
- ABT
1-aminobenzotriazole
- BSA
bovine serum albumin
- CLint
unbound intrinsic clearance
- CLint,P450
unbound intrinsic clearance by cytochrome P450
- CLint,UGT
unbound intrinsic clearance by glucuronidation
- CLIV
intravenous clearance
- EH
hepatic extraction ratio
- ER
renal extraction ratio
- fm,UGT
fraction of metabolism due to glucuronidation
- fu,inc
fraction unbound from protein in the incubation
- HIM
human intestinal microsome
- HKM
human kidney microsome
- HLM
human liver microsome
- IVIVE
in vitro–in vivo extrapolation
- Kp
tissue to blood concentration ratio
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- P450
cytochrome P450
- PBPK
physiologically based pharmacokinetic
- UGT
uridine diphosphate glucuronosyltransferase
- Vss
volume of distribution at steady state
Authorship Contributions
Participated in research design: Gill, Gertz, Houston, Galetin.
Conducted experiments: Gill.
Performed data analysis: Gill.
Wrote or contributed to the writing of the manuscript: Gill, Gertz, Houston, Galetin.
Footnotes
This research was supported by a consortium of pharmaceutical companies within the Centre for Applied Pharmacokinetic Research at the University of Manchester; and a PhD studentship from the Biotechnology and Biological Sciences Research Council [Grant BB/F01760X/1] (to K.L.G.).
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Al-Jahdari WS, Yamamoto K, Hiraoka H, Nakamura K, Goto F, Horiuchi R. (2006) Prediction of total propofol clearance based on enzyme activities in microsomes from human kidney and liver. Eur J Clin Pharmacol 62:527–533 [DOI] [PubMed] [Google Scholar]
- Barter ZE, Bayliss MK, Beaune PH, Boobis AR, Carlile DJ, Edwards RJ, Houston JB, Lake BG, Lipscomb JC, Pelkonen OR, et al. (2007) Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr Drug Metab 8:33–45 [DOI] [PubMed] [Google Scholar]
- Bièche I, Narjoz C, Asselah T, Vacher S, Marcellin P, Lidereau R, Beaune P, de Waziers I. (2007) Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharmacogenet Genomics 17:731–742 [DOI] [PubMed] [Google Scholar]
- Brown HS, Griffin M, Houston JB. (2007) Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab Dispos 35:293–301 [DOI] [PubMed] [Google Scholar]
- Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13:407–484 [DOI] [PubMed] [Google Scholar]
- Cojocel C, Maita K, Pasino DA, Kuo C-H, Hook JB. (1983) Metabolic heterogeneity of the proximal and distal kidney tubules. Life Sci 33:855–861 [DOI] [PubMed] [Google Scholar]
- Court MH. (2005) Isoform-selective probe substrates for in vitro studies of human UDP-glucuronosyltransferases. Methods Enzymol 400:104–116 [DOI] [PubMed] [Google Scholar]
- Court MH. (2010) Interindividual variability in hepatic drug glucuronidation: studies into the role of age, sex, enzyme inducers, and genetic polymorphism using the human liver bank as a model system. Drug Metab Rev 42:209–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court MH, Duan SX, Hesse LM, Venkatakrishnan K, Greenblatt DJ. (2001) Cytochrome P-450 2B6 is responsible for interindividual variability of propofol hydroxylation by human liver microsomes. Anesthesiology 94:110–119 [DOI] [PubMed] [Google Scholar]
- Court MH, Zhang X, Ding X, Yee KK, Hesse LM, Finel M. (2012) Quantitative distribution of mRNAs encoding the 19 human UDP-glucuronosyltransferase enzymes in 26 adult and 3 fetal tissues. Xenobiotica 42:266–277 [DOI] [PubMed] [Google Scholar]
- Cubitt HE, Houston JB, Galetin A. (2009) Relative importance of intestinal and hepatic glucuronidation-impact on the prediction of drug clearance. Pharm Res 26:1073–1083 [DOI] [PubMed] [Google Scholar]
- Cubitt HE, Houston JB, Galetin A. (2011) Prediction of human drug clearance by multiple metabolic pathways: integration of hepatic and intestinal microsomal and cytosolic data. Drug Metab Dispos 39:864–873 [DOI] [PubMed] [Google Scholar]
- Dawidowicz AL, Fornal E, Mardarowicz M, Fijalkowska A. (2000) The role of human lungs in the biotransformation of propofol. Anesthesiology 93:992–997 [DOI] [PubMed] [Google Scholar]
- Doenicke AW, Roizen MF, Rau J, O’Connor M, Kugler J, Klotz U, Babl J. (1997) Pharmacokinetics and pharmacodynamics of propofol in a new solvent. Anesth Analg 85:1399–1403 [DOI] [PubMed] [Google Scholar]
- Favetta P, Degoute CS, Perdrix JP, Dufresne C, Boulieu R, Guitton J. (2002) Propofol metabolites in man following propofol induction and maintenance. Br J Anaesth 88:653–658 [DOI] [PubMed] [Google Scholar]
- Fisher MB, Campanale K, Ackermann BL, VandenBranden M, Wrighton SA. (2000) In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab Dispos 28:560–566 [PubMed] [Google Scholar]
- Fisher MB, Paine MF, Strelevitz TJ, Wrighton SA. (2001) The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism. Drug Metab Rev 33:273–297 [DOI] [PubMed] [Google Scholar]
- Gaganis P, Miners JO, Brennan JS, Thomas A, Knights KM. (2007) Human renal cortical and medullary UDP-glucuronosyltransferases (UGTs): immunohistochemical localization of UGT2B7 and UGT1A enzymes and kinetic characterization of S-naproxen glucuronidation. J Pharmacol Exp Ther 323:422–430 [DOI] [PubMed] [Google Scholar]
- Galetin A, Gertz M, Houston JB. (2008) Potential role of intestinal first-pass metabolism in the prediction of drug-drug interactions. Expert Opin Drug Metab Toxicol 4:909–922 [DOI] [PubMed] [Google Scholar]
- Gepts EM, Camu FM, Cockshott IDP, Douglas EJH. (1987) Disposition of propofol administered as constant rate intravenous infusions in humans. Anesth Analg 66:1256–1263 [PubMed] [Google Scholar]
- Gertz M, Harrison A, Houston JB, Galetin A. (2010) Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab Dispos 38:1147–1158 [DOI] [PubMed] [Google Scholar]
- Gertz M, Houston JB, Galetin A. (2011) Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab Dispos 39:1633–1642 [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill KL, Houston JB, Galetin A. (2012) Characterization of in vitro glucuronidation clearance of a range of drugs in human kidney microsomes: comparison with liver and intestinal glucuronidation and impact of albumin. Drug Metab Dispos 40:825–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grounds RM, Twigley AJ, Carli F, Whitwam JG, Morgan M. (1985) The haemodynamic effects of intravenous induction. Comparison of the effects of thiopentone and propofol. Anaesthesia 40:735–740 [DOI] [PubMed] [Google Scholar]
- Guitton J, Buronfosse T, Desage M, Flinois JP, Perdrix JP, Brazier JL, Beaune P. (1998) Possible involvement of multiple human cytochrome P450 isoforms in the liver metabolism of propofol. Br J Anaesth 80:788–795 [DOI] [PubMed] [Google Scholar]
- Harbourt DE, Fallon JK, Ito S, Baba T, Ritter JK, Glish GL, Smith PC. (2012) Quantification of human uridine-diphosphate glucuronosyl transferase 1A isoforms in liver, intestine, and kidney using nanobore liquid chromatography-tandem mass spectrometry. Anal Chem 84:98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y-L, Ueyama H, Tashiro C, Mashimo T, Yoshiya I. (2000) Pulmonary disposition of propofol in surgical patients. Anesthesiology 93:986–991 [DOI] [PubMed] [Google Scholar]
- Hiraoka H, Yamamoto K, Miyoshi S, Morita T, Nakamura K, Kadoi Y, Kunimoto F, Horiuchi R. (2005) Kidneys contribute to the extrahepatic clearance of propofol in humans, but not lungs and brain. Br J Clin Pharmacol 60:176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelle JT, Hazelton GA, Klaassen CD, Hjelle JJ. (1986) Glucuronidation and sulfation in rabbit kidney. J Pharmacol Exp Ther 236:150–156 [PubMed] [Google Scholar]
- Huang SM, Rowland M. (2012) The role of physiologically based pharmacokinetic modeling in regulatory review. Clin Pharmacol Ther 91:542–549 [DOI] [PubMed] [Google Scholar]
- ICRP (2002) Basic anatomical and physiological data for use in radiological protection: reference values. A report of age- and gender-related differences in the anatomical and physiological characteristics of reference individuals. ICRP Publication 89. Ann ICRP 32:5–265. [PubMed] [Google Scholar]
- Jones HM, Dickins M, Youdim K, Gosset JR, Attkins NJ, Hay TL, Gurrell IK, Logan YR, Bungay PJ, Jones BC, et al. (2012) Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica 42:94–106 [DOI] [PubMed] [Google Scholar]
- Kerdpin O, Knights KM, Elliot DJ, Miners JO. (2008) In vitro characterisation of human renal and hepatic frusemide glucuronidation and identification of the UDP-glucuronosyltransferase enzymes involved in this pathway. Biochem Pharmacol 76:249–257 [DOI] [PubMed] [Google Scholar]
- Kilford PJ, Stringer R, Sohal B, Houston JB, Galetin A. (2009) Prediction of drug clearance by glucuronidation from in vitro data: use of combined cytochrome P450 and UDP-glucuronosyltransferase cofactors in alamethicin-activated human liver microsomes. Drug Metab Dispos 37:82–89 [DOI] [PubMed] [Google Scholar]
- Kirkpatrick T, Cockshott ID, Douglas EJ, Nimmo WS. (1988) Pharmacokinetics of propofol (diprivan) in elderly patients. Br J Anaesth 60:146–150 [DOI] [PubMed] [Google Scholar]
- Lash LH, Putt DA, Cai H. (2008) Drug metabolism enzyme expression and activity in primary cultures of human proximal tubular cells. Toxicology 244:56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt DG, Schnider TW. (2005) Human physiologically based pharmacokinetic model for propofol. BMC Anesthesiol 5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevski N, Moreolo PS, Yli-Kauhaluoma J, Finel M. (2011) Bovine serum albumin decreases Km values of human UDP-glucuronosyltransferases 1A9 and 2B7 and increases Vmax values of UGT1A9. Drug Metab Dispos 39:2117–2129 [DOI] [PubMed] [Google Scholar]
- Manevski NM, Yli-Kauhaluoma J, Finel M. (2012) UDP-glucuronic acid binds first and the aglycone substrate binds second to form a ternary complex in UGT1A9-catalyzed reactions, in both the presence and absence of bovine serum albumin. Drug Metab Dispos 40:2192–2203 [DOI] [PubMed] [Google Scholar]
- Mazoit JX, Sandouk P, Scherrmann JM, Roche A. (1990) Extrahepatic metabolism of morphine occurs in humans. Clin Pharmacol Ther 48:613–618 [DOI] [PubMed] [Google Scholar]
- Nestorov I. (2003) Whole body pharmacokinetic models. Clin Pharmacokinet 42:883–908 [DOI] [PubMed] [Google Scholar]
- Nestorov I. (2007) Whole-body physiologically based pharmacokinetic models. Expert Opin Drug Metab Toxicol 3:235–249 [DOI] [PubMed] [Google Scholar]
- Nishimura M, Naito S. (2006) Tissue-specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab Pharmacokinet 21:357–374 [DOI] [PubMed] [Google Scholar]
- Oda Y, Hamaoka N, Hiroi T, Imaoka S, Hase I, Tanaka K, Funae Y, Ishizaki T, Asada A. (2001) Involvement of human liver cytochrome P4502B6 in the metabolism of propofol. Br J Clin Pharmacol 51:281–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KS, Rowland M. (1977) Hepatic clearance of drugs. I. Theoretical considerations of a “well-stirred” model and a “parallel tube” model. Influence of hepatic blood flow, plasma and blood cell binding, and the hepatocellular enzymatic activity on hepatic drug clearance. J Pharmacokinet Biopharm 5:625–653 [DOI] [PubMed] [Google Scholar]
- Pichette V, du Souich P. (1996) Role of the kidneys in the metabolism of furosemide: its inhibition by probenecid. J Am Soc Nephrol 7:345–349 [DOI] [PubMed] [Google Scholar]
- Poulin P, Jones RDO, Jones HM, Gibson CR, Rowland M, Chien JY, Ring BJ, Adkison KK, Ku MS, He H, et al. (2011) PHRMA CPCDC initiative on predictive models of human pharmacokinetics, part 5: Prediction of plasma concentration-time profiles in human by using the physiologically-based pharmacokinetic modeling approach. J Pharm Sci 100:4127–4157 [DOI] [PubMed] [Google Scholar]
- Price ML, Millar B, Grounds M, Cashman J. (1992) Changes in cardiac index and estimated systemic vascular resistance during induction of anaesthesia with thiopentone, methohexitone, propofol and etomidate. Br J Anaesth 69:172–176 [DOI] [PubMed] [Google Scholar]
- Reiner GN, Labuckas DO, García DA. (2009) Lipophilicity of some GABAergic phenols and related compounds determined by HPLC and partition coefficients in different systems. J Pharm Biomed Anal 49:686–691 [DOI] [PubMed] [Google Scholar]
- Rodgers T, Rowland M. (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257 [DOI] [PubMed] [Google Scholar]
- Rowland A, Gaganis P, Elliot DJ, Mackenzie PI, Knights KM, Miners JO. (2007) Binding of inhibitory fatty acids is responsible for the enhancement of UDP-glucuronosyltransferase 2B7 activity by albumin: implications for in vitro-in vivo extrapolation. J Pharmacol Exp Ther 321:137–147 [DOI] [PubMed] [Google Scholar]
- Rowland A, Knights KM, Mackenzie PI, Miners JO. (2008) The “albumin effect” and drug glucuronidation: bovine serum albumin and fatty acid-free human serum albumin enhance the glucuronidation of UDP-glucuronosyltransferase (UGT) 1A9 substrates but not UGT1A1 and UGT1A6 activities. Drug Metab Dispos 36:1056–1062 [DOI] [PubMed] [Google Scholar]
- Rowland A, Knights KM, Mackenzie PI, Miners JO. (2009) Characterization of the binding of drugs to human intestinal fatty acid binding protein (IFABP): potential role of IFABP as an alternative to albumin for in vitro-in vivo extrapolation of drug kinetic parameters. Drug Metab Dispos 37:1395–1403 [DOI] [PubMed] [Google Scholar]
- Rowland M, Peck C, Tucker G. (2011) Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol 51:45–73 [DOI] [PubMed] [Google Scholar]
- Servin F, Cockshott ID, Farinotti R, Haberer JP, Winckler C, Desmonts JM. (1990) Pharmacokinetics of propofol infusions in patients with cirrhosis. Br J Anaesth 65:177–183 [DOI] [PubMed] [Google Scholar]
- Servin F, Desmonts JM, Farinotti R, Haberer JP, Winckler C. (1988a) Pharmacokinetics of propofol administered by continuous infusion in patients with cirrhosis. Preliminary results. Anaesthesia 43 (Suppl):23–24 [DOI] [PubMed] [Google Scholar]
- Servin F, Desmonts JM, Haberer JP, Cockshott ID, Plummer GF, Farinotti R. (1988b) Pharmacokinetics and protein binding of propofol in patients with cirrhosis. Anesthesiology 69:887–891 [DOI] [PubMed] [Google Scholar]
- Simons PJ, Cockshott ID, Douglas EJ, Gordon EA, Hopkins K, Rowland M. (1988) Disposition in male volunteers of a subanaesthetic intravenous dose of an oil in water emulsion of 14C-propofol. Xenobiotica 18:429–440 [DOI] [PubMed] [Google Scholar]
- Soars MG, Burchell B, Riley RJ. (2002) In vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. J Pharmacol Exp Ther 301:382–390 [DOI] [PubMed] [Google Scholar]
- Takizawa D, Hiraoka H, Goto F, Yamamoto K, Horiuchi R. (2005a) Human kidneys play an important role in the elimination of propofol. Anesthesiology 102:327–330 [DOI] [PubMed] [Google Scholar]
- Takizawa D, Sato E, Hiraoka H, Tomioka A, Yamamoto K, Horiuchi R, Goto F. (2005b) Changes in apparent systemic clearance of propofol during transplantation of living related donor liver. Br J Anaesth 95:643–647 [DOI] [PubMed] [Google Scholar]
- Upton RN, Ludbrook G. (2005) A physiologically based, recirculatory model of the kinetics and dynamics of propofol in man. Anesthesiology 103:344–352 [DOI] [PubMed] [Google Scholar]
- Veroli P, O’Kelly B, Bertrand F, Trouvin JH, Farinotti R, Ecoffey C. (1992) Extrahepatic metabolism of propofol in man during the anhepatic phase of orthotopic liver transplantation. Br J Anaesth 68:183–186 [DOI] [PubMed] [Google Scholar]
- Vree TB, Baars AM, de Grood PMRM. (1987) High-performance liquid chromatographic determination and preliminary pharmacokinetics of propofol and its metabolites in human plasma and urine. J Chromatogr A 417:458–464 [DOI] [PubMed] [Google Scholar]
- Walsky RL, Bauman JN, Bourcier K, Giddens G, Lapham K, Negahban A, Ryder TF, Obach RS, Hyland R, Goosen TC. (2012) Optimized assays for human UDP-glucuronosyltransferase (UGT) activities: altered alamethicin concentration and utility to screen for UGT inhibitors. Drug Metab Dispos 40:1051–1065 [DOI] [PubMed] [Google Scholar]
- Yue QY, Odar-Cederlöf I, Svensson J-O, Säwe J. (1988) Glucuronidation of morphine in human kidney microsomes. Pharmacol Toxicol 63:337–341 [DOI] [PubMed] [Google Scholar]
- Zhao P, Zhang L, Grillo JA, Liu Q, Bullock JM, Moon YJ, Song P, Brar SS, Madabushi R, Wu TC, et al. (2011) Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther 89:259–267 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.