

Abstract
Phenethylisothiocyanate (PEITC), a naturally occurring isothiocyanate and potent cancer chemopreventive agent, works by multiple mechanisms, including the inhibition of cytochrome P450 (P450) enzymes, such as CYP2E1, that are involved in the bioactivation of carcinogens. PEITC has been reported to be a mechanism-based inactivator of some P450s. We describe here the possible mechanism for the inactivation of human CYP2E1 by PEITC, as well as the putative intermediate that might be involved in the bioactivation of PEITC. PEITC inactivated recombinant CYP2E1 with a partition ratio of 12, and the inactivation was not inhibited in the presence of glutathione (GSH) and not fully recovered by dialysis. The inactivation of CYP2E1 by PEITC is due to both heme destruction and protein modification, with the latter being the major pathway for inactivation. GSH-adducts of phenethyl isocyanate (PIC) and phenethylamine were detected during the metabolism by CYP2E1, indicating formation of PIC as a reactive intermediate following P450-catalyzed desulfurization of PEITC. Surprisingly, PIC bound covalently to CYP2E1 to form protein adducts but did not inactivate the enzyme. Liquid chromatography mass spectroscopy analysis of the inactivated CYP2E1 apo-protein suggests that a reactive sulfur atom generated during desulfurization of PEITC is involved in the inactivation of CYP2E1. Our data suggest that the metabolism of PEITC by CYP2E1 that results in the inactivation of CYP2E1 may occur by a mechanism similar to that observed with other sulfur-containing compounds, such as parathion. Digestion of the inactivated enzyme and analysis by SEQUEST showed that Cys 268 may be the residue modified by PIC.
Introduction
Isothiocyanates are chemopreventive compounds occurring naturally in cruciferous vegetables, such as cabbage, cauliflower, broccoli, and watercress (Keum et al., 2004; Zhang, 2004; Nakamura and Miyoshi, 2006). In these vegetables, isothiocyanates are stored as glucosinolates. Cutting or chewing the vegetables releases myrosinase, which hydrolyzes the glucosinolates, and the intermediates produced undergo rearrangement to form the isothiocyanates (Nakamura and Miyoshi, 2006). The chemopreventive effects of naturally occurring isothiocyanates have been characterized in a number of animal models and several clinical studies (London et al., 2000; Keum et al., 2004; Zhang, 2004; Nakamura and Miyoshi, 2006). Epidemiologic studies indicate an inverse relation between consumption of dietary isothiocyanates and the risk of developing lung, breast, and colon cancers (Zhao et al., 2001; Seow et al., 2002; Fowke et al., 2003). Evidence indicates that isothiocyanates exert their anti-carcinogenic effects by multiple mechanisms. The inhibition of cytochrome P450 (P450) enzymes, as well as the potent induction of a number of phase II metabolic and cellular defensive enzymes, has been implicated in the chemopreventive action of isothiocyanates (Keum et al., 2004; Zhang, 2004). Effects on apoptosis and cell cycle arrest are reported to be other mechanisms that may be responsible for the chemopreventive activity of isothiocyanates (Nakamura and Miyoshi, 2006). Isothiocyanates have also been suggested to suppress angiogenesis (Xiao and Singh, 2007) and metastasis of cancer cells (Hwang and Lee, 2006).
Phenethyl isothiocyanate (PEITC) is one of the most extensively studied isothiocyanates due to its promising chemopreventive effect, and it has been entered into phase I clinical trials as a preventive agent against lung cancer in smokers and ex-smokers (National Cancer Institute, 1996). The chemopreventive effect of PEITC has been demonstrated in animal models of nitrosamine-induced cancer. Administration of PEITC significantly inhibited carcinogenesis in animals treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (Morse et al., 1989), N-nitrosomethylbenzylamine (Solt et al., 2003), N-nitrosodiethylamine (Pereira, 1995), or N-nitrosodimethylamine (Chung et al., 1985). Cytochrome CYP2E1 plays a major role in the bioactivation of a number of nitrosamines (Yamazaki et al., 1992). PEITC has been shown to be a mechanism-based inactivator of rodent and human CYP2E1 (Moreno et al., 2001; Nakajima et al., 2001). The potency of PEITC in inhibiting CYP2E1 has been shown in vivo. In rats, a single dose of PEITC significantly suppressed the bioactivation of N-nitrosodimethylamine (Guo et al., 1992). Since this bioactivation is selectively catalyzed by CYP2E1, these results suggest that inhibition of P450s might contribute to the chemopreventive effect of PEITC. In addition to the animal studies, studies in humans have demonstrated that a single ingestion of watercress, one of the major sources of PEITC, inhibited the hydroxylation of chlorzoxazone, an in vivo probe for human CYP2E1 activity (Leclercq et al., 1998).
Although the mechanism for the inactivation of P450s by isothiocyanates is not yet fully understood, previous reports suggest that the inactivation of P450s by isothiocyanates could occur as a result of protein modification by a reactive intermediate of PEITC rather than by destruction of the heme moiety (Leclercq et al., 1998; Kent et al., 2001). Isothiocyanates require bioactivation to reactive intermediates, which then inactivate the P450s. Therefore, elucidation of the pathways for the P450-dependent oxidative metabolism of isothiocyanates is essential for understanding the mechanisms for the inactivation of P450s in humans. The major metabolic pathway for human metabolism of PEITC is believed to be a mercapturic acid pathway, which is independent of P450 and is initiated by conjugation of PEITC with glutathione (GSH) followed by successive cleavage of the γ-glutamyl residue, removal of the glycine residue, and finally N-acetylation to give the N-acetylcysteine conjugate (Zhang, 2004).
The primary objective of this study was to elucidate the mechanism of suicide inactivation of CYP2E1 by PEITC through characterizing the oxidative pathway for the metabolism of PEITC and identifying a reactive intermediate that might be involved in the inactivation. Reactive intermediates of isocyanates formed during their oxidative metabolism have previously been shown to bind to the P450 protein (Goosen et al., 2001; Moreno et al., 2001). Phenethyl isocyanate (PIC) has been suggested to be a putative reactive intermediate of PEITC. Electrospray ionization (ESI) liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was used to detect and identify GSH-adduct of PIC, and digestion of the inactivated P450 revealed a modified peptide that formed during the oxidative metabolism of PEITC by human CYP2E1. Although externally added PIC covalently bound to the CYP2E1 apoenzyme and there is evidence for modified peptides, the binding did not affect the activity of the protein. These data suggest that a reactive intermediate other than PIC may be responsible for the inactivation of human CYP2E1.
Materials and Methods
Materials.
Dilauroyl-l-α-phosphatidylcholine (DLPC), NADP, NADPH, catalase, GSH, PIC, phenethylamine, and bovine serum albumin were purchased from Sigma Chemical Co. (St. Louis, MO). PEITC was purchased from Trans World Chemical Co. (Rockville, MD), trypsin was purchased from Promega (Madison, WI), and 7-ethoxy-4-(trifluoromethyl)-coumarin (7-EFC) was obtained from Molecular Probes, Inc. (Eugene, OR). Chemical standards of the PEITC-GSH and PIC-GSH conjugates were prepared as described in the following sections. High-performance liquid chromatography (HPLC)-grade acetonitrile was from Fisher (Pittsburgh, PA), and trifluoroacetic acid (TFA) was from Pierce (Rockford, IL).
Enzymes.
The cDNA for human cytochrome CYP2E1 (generously provided by Dr. P. F. Guengerich, Vanderbilt University) was expressed in Escherichia coli MV1304 cells. Expression and purification of the protein was carried out according to published methods (Larson et al., 1991) with some modifications (Hanna et al., 1998). NADPH was purified after expression in E. coli Topp3 cells as previously described (Shen et al., 1989).
Effect of PEITC on the Catalytic Activity of Human CYP2E1.
The de-ethylation of 7-EFC to 7-hydroxy-4-(trifluoromethyl)coumarin (7-HFC) (Buters et al., 1993) was used to assess the inactivation of CYP2E1 by PEITC using a two-stage incubation protocol. CYP2E1 and reductase were reconstituted with lipid for 45 minutes at 4°C as previously described (Hanna et al., 1998). The primary reaction mixture contained varying concentrations of PEITC in methanol (the final concentration of methanol was 1%), 1 μM CYP2E1, 1 μM reductase, 100 μg/ml DLPC, and 2000 units of catalase in 50 mM potassium phosphate buffer (pH 7.4). Control samples contained methanol instead of PEITC. Reactions were initiated by adding NADPH to a final concentration of 1.2 mM and incubated at 30°C. The activity remaining at 0, 1, 2, 3, 4, and 5 minutes following the addition of NADPH was determined by measuring the activity for the O-de-ethylation of 7-EFC to 7-HFC spectrofluorometrically (Buters et al., 1993) on a Shimadzu model RF-5301PC spectrofluorometer (Shimadzu Scientific Instruments, Columbia, MD) with excitation at 410 nm and emission at 510 nm. For this assay, 15 μl aliquots of the primary mixture were transferred to secondary reaction mixtures containing 0.1 mM 7-EFC, 0.2 mM NADPH, and 40 μg/ml bovine serum albumin in 50 mM potassium phosphate buffer (pH 7.4) in a final volume of 0.6 ml. The reaction mixtures were incubated for 10 minutes, and the reactions were terminated by adding 0.35 ml of acetonitrile.
Determination of the Partition Ratio.
The partition ratio for the inactivation of CYP2E1 by PEITC was determined by incubating the enzyme with increasing concentrations of PEITC and allowing the inactivation to go to completion. The reconstitution was carried out as previously described. Primary reaction mixtures were incubated for 10 minutes at 30°C, and the activity remaining was determined by measuring the 7-EFC O-de-ethylation activity as previously described. The percent activity remaining was plotted as a function of the molar ratio of PEITC to CYP2E1 to determine the turnover number (Ghanbari et al., 2006).
Irreversibility of the Inactivation of CYP2E1 by PEITC.
CYP2E1 (1 μM) and reductase (1 μM) were reconstituted with lipid (100 μg/ml) and inactivated by incubation with 25 μM PEITC and 1.2 mM NADPH in the primary reaction mixture as previously described. Control samples were incubated without PEITC and NADPH, or with PEITC but no NADPH. After incubation for 10 minutes at 30°C, the samples were analyzed for 7-EFC O-de-ethylation activity as previously described. P450 heme content was determined using CO difference spectra and HPLC as described later in this section. The remainder of the samples was dialyzed overnight at 4°C in Slide-A-Lyzer cassettes (Thermo Scientific, Rockford, IL) against 1 liter of 50 mM potassium phosphate buffer (pH 7.4) containing 20% glycerol and 0.1 mM EDTA. After dialysis, the samples were subjected once again to quantitation of P450 and heme as described later. Portions of each sample were reconstituted with fresh lipid (final concentration of fresh lipid: 50 μg/ml), and an equal volume of the sample was reconstituted with fresh lipid and fresh reductase (final concentration of added reductase: 1 μM). These reconstituted samples were then assayed for 7-EFC activity as previously described.
Spectrophotometric Quantitation of P450 Heme.
The P450 contents of the aforementioned samples were analyzed before and after dialysis by measuring their reduced CO difference spectra. Aliquots (200 μl) of each sample were diluted with 400 μl of ice-cold 50 mM potassium phosphate buffer (pH 7.4). Dithionite and carbon monoxide were added and the reduced CO difference spectra were recorded between 400 and 500 nm on a UV spectrophotometer (Shimadzu Scientific Instruments) as previously described (Omura and Sato, 1964). The maximal absorbance at 450 nm was used to quantify P450 heme using an extinction coefficient of 91 mM cm−1.
HPLC Analysis of P450 Heme.
The inactivated samples were also subjected to HPLC analysis for heme before and after dialysis. Each sample was injected onto a 250-mm × 4.60-mm C4 reverse-phase HPLC column (Phenomenex, Torrance, CA) (solvent A, H2O and 0.1% TFA; solvent B, 100% acetonitrile and 0.1% TFA). The flow rate was 1 ml/min, and a linear gradient from 35% B to 95% B over 30 minutes was used. The elution of heme was monitored at 398 nm using diode array detection.
Effect of GSH on the Inactivation of Human CYP2E1 by PEITC.
Since isothiocyanates are known to react nonenzymatically with GSH (Jiao et al., 1994), we tested the effect of GSH on the ability of PEITC to inactivate human CYP2E1. Human CYP2E1 and reductase were reconstituted with lipid for 45 minutes at 4°C. The reconstituted sample contained 1 μM CYP2E1, 1 μM reductase, 100 μg/ml DLPC, and 2000 units of catalase in 50 mM potassium phosphate buffer (pH 7.4). The reconstitution mixture was then divided into three equal samples that were incubated with 1.2 mM NADPH or 1.2 mM NADPH together with 50 μM PEITC, or with 1.2 mM NADPH together with 50 μM PEITC and 1 mM GSH. Control samples received methanol instead of PEITC. Reactions were initiated by adding NADPH, and they were incubated at 30°C. PEITC was added 1 minute prior to adding NADPH to minimize the nonenzymatic binding of GSH to PEITC before starting the metabolic reaction. The activity remaining at 0, 2, 4, 6, and 12 minutes following the addition of NADPH was determined by measuring the O-de-ethylation activity for the conversion of 7-EFC to 7-HFC spectrofluorometrically as described earlier.
LC-MS/MS Analysis of Phenethylamine and the GSH Conjugates of PEITC and PIC.
For the analysis of the metabolites and GSH conjugates of PEITC formed by human CYP2E1, CYP2E1 (1 μM) and reductase (1 μM) were reconstituted with lipid (100 μg/ml) as previously described and preincubated with 2 mM GSH. After preincubation, PEITC (final 50 μM) and NADPH (final 1.2 mM) were added, and the reactions were incubated for 30 minutes at 30°C (total volume of reaction mixture: 600 μl). Control samples were incubated with PEITC in the absence of NADPH. After the incubation, the samples were applied to 1 ml AccuBond ODS-C18 solid phase extraction cartridges (Agilent Technologies, Palo Alto, CA). The cartridges were conditioned previously with 2 ml of methanol followed by 2 ml of water. After the samples were loaded, the cartridges were washed with 1 ml of water and then eluted with 2 ml of methanol, followed by 0.5 ml of acetonitrile. The eluted samples were dried under N2 gas and resuspended in 50% methanol. The samples were analyzed on a C18 reverse-phase column (Luna; 3 μm; 4.6 × 100 mm; Phenomenex) using a linear gradient of 20 to 30% B over 5 minutes followed by a linear gradient to 40% B over 15 minutes, and then to 90% B over 15 minutes at a flow rate of 0.3 ml/min (solvent A, water and 0.5% acetic acid adjusted to pH 4.0; solvent B, 100% acetonitrile and 0.5% acetic acid). The column effluent was directed into the ESI source of an liquid chromatography quadrupole (LCQ) mass spectrometer (Thermo Electron Corporation, Waltham, MA). The ESI conditions were: sheath gas flow rate, 90 arbitrary units; auxiliary gas, 30 arbitrary units; spray voltage, 4.5 kV; capillary temperature, 170°C; capillary voltage, 30 V; and tube lens offset, 25 V. Data were acquired in positive ion mode using Excalibur software (Thermo Electron Corporation) with one full scan followed by two data-dependent scans of the most intense and the second most intense ions.
For identification of their PEITC-GSH and PIC-GSH conjugates, the chemical standard mixtures were prepared just before use. For the PEITC-GSH standard mixture, 10 μl of PEITC in methanol (100 mM) was added to 1 ml of a GSH solution (1 mM) in 50 mM potassium phosphate buffer (pH 7.4) and stirred for 2 to 4 hours at room temperature (Jiao et al., 1994). The mixture was diluted 500-fold with methanol/water (50/50, v/v) and subjected to LC-MS/MS analysis. Formation of the PEITC-GSH conjugate was confirmed by mass and MS/MS spectra. For the PIC-GSH standard mixture, 10 μl of PIC in acetone (100 mM) was added to 1 ml of a GSH solution (0.5 mM) in acetonitrile/water (70/30, v/v) and stirred for 1 to 2 hours at room temperature (Jochheim et al., 2002). The mixture was diluted 250-fold with methanol/water (50/50, v/v) and subjected to LC-MS/MS analysis. Formation of PIC-GSH was confirmed by mass and MS/MS spectra.
We also analyzed the GSH conjugate of PIC produced in human liver microsomes. Frozen human liver samples were a gift from Dr. F. Peter Guengerich (Vanderbilt University, Nashville, TN). Microsomes were prepared as previously described with minor modifications (Guengerich, 1994; Teiber and Hollenberg, 2000). Human liver microsomes (0.5 mg protein/ml) in 50 mM potassium phosphate buffer (pH 7.4) were preincubated with 2 mM GSH for 5 minute . After preincubation, PEITC (final 50 μM) and NADPH (final 1.2 mM) were added, and the reactions were incubated for 30 minutes at 37°C (total volume of reaction mixture: 600 μl). Control samples were incubated with PEITC in the absence of NADPH. After the incubation, the samples were applied to 1 ml AccuBond ODS-C18 solid-phase extraction cartridges (Agilent Technologies) as previously mentioned. The extraction and LC-MS/MS analysis of the PIC-GSH conjugate were conducted using the same procedure described earlier in this section.
LC-MS/MS Analysis of the Protein-Adducts Formed by PEITC and PIC in the Absence of Metabolism.
CYP2E1(1 μM) was reconstituted only with lipid (100 μg/ml) and incubated with 25 μM PEITC or 10 μM PIC for 10 minutes at 30°C. After incubation, the samples (50 μl) were subjected to ESI LC-MS/MS analysis. The samples were analyzed on a C4 reverse-phase column (Jupiter; 5 μm; 2.0 × 150 mm; Phenomenex) equilibrated with 40% acetonitrile and 0.1% TFA at a flow rate of 0.3 ml/min. The acetonitrile concentration was increased linearly to 50% during the first 15 minutes, to 70% by 25 minutes, and to 90% by 30 minutes. The column effluent was diverted for the first 10 minutes. The flow was then directed into the LCQ mass spectrometer, and the mass spectra were recorded. The ESI source conditions were optimized using horse heart myoglobin. The ESI conditions were: sheath gas flow rate, 90 arbitrary units; auxiliary gas, 30 arbitrary units; spray voltage, 4.2 kV; capillary temperature, 230°C; capillary voltage, 30 V; and tube lens offset, 25 V. Data were acquired in the positive ion mode and analyzed using Excalibur software (Thermo Electron Corporation). The protein envelopes were deconvoluted using the Thermoquest Excalibur 1.0 SR1 Qual Browser (Thermo Fisher Scientific, Waltham, MA) to obtain the mass associated with each protein envelope.
LC-MS/MS Analysis of Protein-Adducts during the Metabolism of PEITC.
CYP2E1 (1 μM) and reductase (0.8 μM) were reconstituted with lipid (100 μg/ml) and the 2E1 inactivated by incubation with 25 μM PEITC in the primary reaction mixture as described earlier. Control samples were incubated with PEITC in the absence of NADPH. The samples were subjected to LC-MS/MS using a Vydac C18 reverse-phase column (protein and peptide C18; 5 μm; 2.0 × 150 mm; WR Grace & Co., Columbia, MD) equilibrated with 35% acetonitrile and 0.1% TFA at a flow rate of 0.3 ml/min. The acetonitrile concentration was increased linearly to 50% during the first 15 minuteS, to 70% by 25 minutes, and to 90% by 30 minutes. The column effluent was diverted for the first 10 minutes. The flow was then directed into the LCQ mass spectrometer, and spectra were obtained using the same conditions as described previously. The protein envelopes were deconvoluted using the Thermoquest Excalibur 1.0 SR1 Qual Browser to obtain the mass associated with each protein envelope.
Effect of PIC on the Enzyme Activity of Human CYP2E1.
CYP2E1 and reductase were reconstituted with lipid for 45 minutes at 4°C. The primary reaction mixture contained 1 μM CYP2E1, 1 μM reductase, 100 μg/ml DLPC, and 2000 units of catalase in 50 mM potassium phosphate buffer (pH 7.4). The primary reaction mixtures received varying concentrations of PIC in acetonitrile (final concentration of acetonitrile: 1%) and were incubated at 30°C. Control samples contained acetonitrile instead of PIC. The activity remaining after 10 minutes of incubation was determined by measuring the O-de-ethylation of 7-EFC to 7-HFC as described earlier.
Binding Spectra for the Interaction of PEITC or PIC with CYP2E1.
CYP2E1 (1 μM) was reconstituted with lipid (final concentration of 1 μg/ml) in 50 mM potassium phosphate buffer (pH 7.4) for 45 minutes at 4°C. The sample was divided equally into the reference and sample cuvettes. Samples were scanned from 350 to 500 nm on a UV spectrophotometer (Shimadzu Scientific Instruments). Scans were recorded following the addition of PEITC stock solutions dissolved in methanol or PIC stock solutions dissolved in acetonitrile (25 μM each; final concentration of 0.1% methanol or acetonitrile) to the sample cuvette and the same amount of solvent to the reference cuvette.
LC/MS/MS Analysis of Tryptic Digest and Identification of the Modified Site.
Inactivation of CYP2E1 was carried out as described earlier. The samples were then denatured by incubation in 8M urea at 55°C for 30 minutes followed by dilution with 50 mM ammonium bicarbonate (to reduce the urea concentration) at 4°C and dialyzed against 50 mM ammonium bicarbonate at 4°C. The samples were then reduced with 5 mM dithiothreitol at 60°C for 30 minutes and subsequently digested with trypsin at a 1:20 trypsin:protein ratio overnight at 37°C. The reactions were terminated with 1 µl of 10% trifluoroacetic acid. The samples were concentrated in a speed vac to a final volume of 30 µl and used for analysis as described later in this section.
High-pressure liquid chromatography separation was performed on a Shimadzu LC-10AD system (Shimadzu Scientific Instruments). The mobile phase consisted of 0.025% TFA, 0.025% formic acid in water (aqueous phase) and 0.025% TFA, and 0.025% formic acid in acetonitrile (organic phase). The flow rate was of 0.3 ml/min with an initial gradient from 10% B to 35% B in 25 minutes and then to 90% B in 60 minute, followed by elution with 90% B for 10 minutes and then re-equilibration to 10% B for 15 minutes. The LC-MS/MS system was a Shimadzu LCQ-Deca XP ion trap mass spectrometer with a Phenomenex Jupiter C18 column (150 × 2.00 mm, 5 µm). The instrument was operated in positive ESI mode with a capillary temperature of 210°C and capillary voltage of 2 V. The analysis was performed in the data-dependent acquisition mode using one full scan followed by two data-dependent scans of the first and the second most intense ions. The modified peptides from the PEITC inactivated CYP2E1 were identified based on a mass shift of the parent peptide equal to that of the metabolite, PIC (147 amu) or reactive sulfur species (180 amu, shown in Fig. 10) or desulfurization adduct (96 amu) using SEQUEST Bioworks software (Thermo Finnigan, San Jose, CA). Kyle et al. (2012) have shown that the mechanism of P450 inactivation by methyl parathion involves oxidative desulfuration and have identified an adduct that increased in mass by 96 amu. Peptides were identified if they met the proteolytic cleavage dependent cross-correlation scores of 2.0 for [M+H]1+, 2.2 for [M+H]2+, and 3.75 for [M+H]3+ and a matching tolerance of 2.0%. Sequence assignments were also based on selecting peptides that displayed minimum delta correlation values greater than 0.1 and SP values (preliminary score of the peptide) greater than 400 and 600 for the singly and doubly charged ions, respectively. Peptides that had a probability score less than 1.0 ×10−4 were discarded.
Fig. 10.

Possible pathway for the oxidative metabolism of PEITC by human CYP2E1. MW, molecular weight.
Results
Inactivation of Human CYP2E1 by PEITC.
The kinetics for the inactivation of human CYP2E1 by PEITC was studied by measuring the loss in the 7-EFC O-de-ethylation activity. As shown in Fig. 1, CYP2E1 in the reconstituted system was inactivated by PEITC in a time- and concentration-dependent manner. Linear regression analysis of the time course data were used to estimate the initial rate constants (kobs) for the inactivation of CYP2E1 by PEITC. From the double reciprocal plot (inset) of the values for kobs and the concentration of PEITC, the inhibition constant (KI), maximal inactivation rate constant (kinact), and half-life (t1/2) values for the inactivation of the 7-EFC catalytic activity of CYP2E1 by PEITC were determined to be 11 μM, 0.23 minute−1, and 3.0 minutes, respectively. Cytochrome b5 is not required for the inactivation of CYP2E1 by PEITC (unpublished data).
Fig. 1.

Time- and concentration-dependent inactivation of reconstituted human CYP2E1 by PEITC following incubation with NADPH. The incubation conditions and measurement of 7-EFC O-de-ethylation activity were described under Materials and Methods. The concentrations of PEITC that were used were (●) 0, (□) 1, (▴) 2.5, (◊) 5, (○) 10, (▪) 25 and (Δ) 50 μM. The data represent the mean and S.D. from six separate experiments done in duplicate. The inset shows the double-reciprocal plot of the rates of inactivation as a function of the PEITC concentration.
Partition Ratio.
The partition ratio (r), a measure of the efficiency of an inactivator, is defined as the number of product molecules produced per inactivation event (Ghanbari et al., 2006). As shown in Fig. 2, the turnover number (r + 1) was derived from the intercept between the line obtained by linear regression (lower PEITC concentrations) and the average straight line for % activity remaining at higher PEITC concentrations. The turnover number is approximately 13; this number includes the one molecule of inactivator that inactivates the enzyme, assuming a 1:1 stoichiometry for the inactivation, therefore the value for the partition ratio is 12.
Fig. 2.

Inactivation of the human CYP2E1 7-EFC O-de-ethylation activity as a function of the PEITC:CYP2E1 concentration ratio. Assay conditions were as described in Materials and Methods. The percent of human CYP2E1 7-EFC O-deethylation activity remaining after complete inactivation was plotted against the molar ratio of PEITC to CYP2E1. The data shown represent the mean of three separate experiments.
Irreversibility of Inactivation of CYP2E1 by PEITC.
The inactivation of CYP2E1 by PEITC was found to be irreversible by dialysis overnight (Table 1). PEITC-inactivated samples or control samples containing PEITC but no NADPH were dialyzed extensively as described in Materials and Methods. The samples were tested for 7-EFC O-de-ethylation activity, the amount of P450 was quantified using the reduced CO difference spectra, and the heme remaining was determined using HPLC. Table 1 shows the percent activity in samples prior to and after dialysis. A small decrease in activity (approximately 20%) was observed in control samples containing PEITC but no NADPH before and after 10 minutes of incubation (82 and 77%, respectively), and this small loss was still observed after dialysis even when fresh reductase was added. No significant loss in the CO-binding spectra or the heme content was observed in the control samples, indicating virtually no effect of PEITC on human CYP2E1 in the absence of NADPH. In contrast, samples inactivated with PEITC for 10 minutes lost almost 80% of their activity. After dialysis, a small percentage of the CYP2E1 activity (8%) was recovered, suggesting that some of the PEITC was carried over from the primary reaction mixture and competitively inhibited the 7EFC activity prior to dialysis. Reconstitution of the inactivated, dialyzed samples with fresh reductase did not result in a significant increase in catalytic activity, suggesting that the inactivation was confined to alterations of the P450. In contrast to the 80% loss in enzymatic activity, the PEITC-inactivated samples showed only a 30% loss in heme and a 56% loss in the P450 content as measured by the reduced-CO spectrum. This indicates that although some of the activity loss could be attributed to heme loss, the major loss in catalytic activity was due to modification of the protein. The P450 content measured by the reduced-CO spectrum after inactivation by PEITC was lower than the native heme content; this suggests that the covalent binding of a reactive intermediate of PEITC in the P450 active site resulted in an impairment of the heme-coordination, hindered access of CO to the heme, or decreased ability to reduce the heme iron. The difference between the enzymatic activity remaining (19%) and the P450 content (44%) further indicated modification of the protein by the PEITC. Both the losses in the P450 and in the heme content could not be recovered after removal of excess PEITC by dialysis. These results indicate that the inactivation of CYP2E1 by PEITC is not reversible and that modification of the reductase is not responsible for inactivation.
TABLE 1.
Effect of PEITC on the 7-EFC O-de-ethylation activity, P450 concentration, and the amount of heme remaining for human CYP2E1
Assay conditions were as described in Materials and Methods. Samples were first dialyzed before reconstitution with fresh reductase. The values shown represent the means ± S.D. from two experiments done in duplicate.
| % Activity Remaining | % P450Remaining | % Heme Remaining | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 min | 10 min | After Dialysis | FrFresh Reductase | 0 min | 10 min | After Dialysis | 0 Min | 10 Min | After Dialysis | |
| −PEITC, −NADPH | 100 | 91 ± 4 | 100 | 100 | 100 | 96 ± 8 | 100 | 100 | 100 ± 4 | 100 |
| +PEITC,−NADPH | 82 ± 4 | 77 ± 2 | 82 ± 4 | 85 ± 9 | 103 ± 8 | 93 ± 10 | 104 ± 3 | 100 ± 1 | ||
| +PEITC,+NADPH | 83 ± 5 | 19 ± 1 | 27 ± 4 | 26 ± 4 | 44 ± 3 | 42 ± 7 | 70 ± 2 | 64 ± 6 | ||
7-EFC, 7-ethoxy-4-(trifluoromethyl) coumarin; P450, cytochrome P450; PEITC, phenethyl isothiocyanate.
Effect of GSH on the PEITC-Dependent Inactivation of CYP2E1.
Isothiocyanates are known to react nonenzymatically with GSH exhibiting with first-order reaction kinetics (Jiao et al., 1994). The effect of GSH on the rate of inactivation of CYP2E1 by PEITC was investigated by incubating CYP2E1 with 50 μM PEITC together with 1 mM GSH. In this experiment, PEITC was added 1 minute prior to adding the NADPH to minimize the nonenzymatic reaction of GSH with PEITC before the initiation of the metabolic reaction. Figure 3 shows that GSH only slightly slowed the rate of inactivation of human CYP2E1. Significant inactivation of CYP2E1 by PEITC still occurred in a time-dependent manner, suggesting that the activation of PEITC and the subsequent inactivation of human CYP2E1 still occurred even in the presence of GSH. Presumably these two reactions are still possible due to the relatively slow kinetics for the reaction of PEITC with GSH and the rapid metabolism of PEITC by CYP2E1 leading to the inactivation of the enzyme.
Fig. 3.

Effect of GSH on the rate of inactivation of CYP2E1 by PEITC. CYP2E1 was incubated with (Δ) 50 μM PEITC, (▲) 50 μM PEITC and 1 mM GSH, or (●) without any additions, and then assayed for residual 7-EFC activity as described in Materials and Methods.
Formation of PEITC-GSH Conjugates Non-enzymatically.
We examined the metabolism of PEITC in the presence of GSH for two reasons. One was to use GSH to trap any reactive intermediates formed during the inactivation that escaped the active site and then to elucidate the structures of the GSH conjugates by LC-MS/MS. Second, we used GSH in the reaction mixtures because it more closely reflected the physiologic condition in human liver because PEITC can react directly with GSH nonenzymatically (Jiao et al., 1994). Therefore, GSH-adduct of PEITC might be formed nonenzymatically as well as in the reconstituted reaction system in the presence of GSH. As shown in Fig. 4A, LC-MS/MS analysis facilitated detection of GSH conjugates having MH+ ions at m/z (mass-to-charge ratio) 471 eluting at 16.7 minutes in reaction mixtures that were incubated either in the presence or absence of NADPH. This ion peak was identical to the peak that was observed when the PEITC-GSH standard was analyzed (Fig. 4B), and the molecular mass of this peak corresponds to the sum of the masses of PEITC (163 Da) plus GSH (307 Da). In addition, the MS/MS product ions of this peak were consistent with those of a PEITC-GSH conjugate (Fig. 4C). The characteristic fragment ions observed for samples incubated in the presence of GSH include neutral losses of 75 and 129 Da, corresponding to the glycine (Gly) and pyroglutamate (Glu) residues of GSH, respectively (Baillie and Davis, 1993). Fragment ions were observed at m/z 396, 342, and 239, which were derived from the loss of Gly, the loss of Glu, and the combined losses of Glu, Gly, and CO, respectively. Cleavage of the C–S bond within the GSH moiety of the GSH conjugate resulted in the fragment ions observed at m/z 308 and m/z 179 (Cys-Gly + H+) following further loss of Glu from GSH, as shown in Fig. 4D.
Fig. 4.

ESI LC-MS/MS analysis of GSH conjugates of PEITC with precursor ions at m/z 471 generated in the reconstituted system containing human CYP2E1. CYP2E1 was reconstituted with reductase and incubated with PEITC in the presence or absence of NADPH. GSH conjugates of PEITC were extracted and analyzed as described under Materials and Methods. (A) Extracted ion chromatogram at m/z 471 for the sample incubated with human CYP2E1 and PEITC in the presence or absence of NADPH (upper left panel), and a chemical standard mixture of PEITC and GSH in acetonitrile/water (bottom left panel). (B) LC-MS/MS spectrum for the ion with an m/z of 471 eluting at 16.7 minutes. (C) The proposed structure of the GSH conjugate of PEITC.
Profiling of the Oxidative Metabolites of PEITC Formed by Human CYP2E1.
We have identified two metabolites of PEITC formed in the presence of NADPH and GSH, a GSH conjugate of PIC (Fig. 5) and phenethylamine (Fig. 6). These two metabolites were only formed in the presence of NADPH, demonstrating that they are products of oxidative metabolism of PEITC. The data for the LC-MS/MS analysis of the GSH conjugate of PIC are presented in Fig. 5. GSH conjugates having an MH+ ion at m/z 455 were detected in the reaction mixture with NADPH (Fig. 5A). This ion was identical to the one found in the PIC-GSH standard (Fig. 5B), and the molecular mass of this ion corresponded to the sum of the mass of PIC (147 Da) plus GSH (307 Da). In addition, the MS/MS product ions shown in Fig. 5C further demonstrated that this ion is the PIC-GSH conjugate. The fragment ion at m/z 380 was derived from the loss of Gly, the ion at m/z 326 was from the loss of Glu, and the ion at m/z 223 was from the combined losses of Glu, Gly, and CO (Fig. 5C). The fragment ions at m/z 308 (GSH) and m/z 179 (Cys-Gly + H+) were also found in the product ions (Fig. 5D).
Fig. 5.

ESI LC-MS/MS analysis of GSH conjugates of PEITC and PIC with precursor ions at m/z 455 generated by in the reconstituted system containing human CYP2E1. CYP2E1 was reconstituted with reductase and incubated with PEITC or PIC in the presence or absence of NADPH. GSH conjugates of PEITC were extracted and analyzed as described under Materials and Methods. (A) Extracted ion chromatogram at m/z 455 in the sample incubated with human CYP2E1 in the presence and absence of NADPH (top panel), and a chemical standard mixture of PIC and GSH in acetonitrile/water (bottom panel). (B) LC-MS/MS pattern of the ion with an m/z of 455 eluting at 14.9 minutes. (C) The proposed structure of the GSH conjugate of PIC.
Fig. 6.

ESI LC-MS/MS analysis of phenethylamine with a precursor ion at m/z 122 formed by the metabolism of PEITC by human CYP2E1 in the reconstituted system. CYP2E1 was reconstituted with reductase and incubated with PEITC in the presence or absence of NADPH. Phenethylamine was extracted and analyzed as described under Materials and Methods. Upper panel, extracted ion chromatogram at m/z 122 of the sample incubated with human CYP2E1 and PEITC in the presence NADPH (left panel), and its MS/MS pattern (right panel). Lower panel, extracted ion chromatogram at m/z 122 for the chemical standard sample of phenethylamine (left panel) and its MS/MS pattern (right panel).
Figure 6 shows the extracted ion chromatogram for a metabolite of PEITC eluting at 12.7 minutes with an MH+ ion at m/z 122 (upper left panel) that was identified as phenethylamine using an authentic chemical standard (lower left panel). The MS/MS product ions having m/z values at 122 from both samples showed an ion with an m/z 105 (upper and lower right panels), which was generated by loss of the amine from the phenyl ethyl group. In the absence of NADPH, no formation of phenethylamine was observed.
Protein Adducts of PEITC and PIC with CYP2E1 Analyzed by ESI LC-MS/MS.
PEITC and PIC both react nonenzymatically with nucleophiles and form conjugates with GSH in the reaction mixture. Therefore, we investigated the reactivity of PIC and PEITC with the P450 apolipoprotein. CYP2E1 was incubated with 25 μM PEITC or 10 μM PIC in the absence of reductase for 10 minutes and was then analyzed using ESI LC-MS/MS. The mass of CYP2E1 incubated in the absence of PEITC or PIC (control) was 54,838 Da (Fig. 7, upper panel; Table 2). Figure 7 (middle panel) shows the mass obtained when CYP2E1 was incubated with 25 μM PEITC. The mass was identical to that of the control, indicating that PEITC does not covalently bind to the P450 apolipoprotein in the absence of metabolism. In contrast, the data obtained when CYP2E1 was incubated with 10 μM PIC showed two ion peaks with masses of 54,837 Da and 54,982 Da (Fig. 7, lower panel; Table 2) corresponding to the unmodified and covalently modified P450. The mass increase of 145 Da is the result of the formation of an adduct of PIC (molecular weight 147) with the protein, and these data clearly indicate that PIC can form a covalent adduct with the P450 apolipoprotein in the absence of metabolism.
Fig. 7.

ESI LC-MS/MS analysis of the protein adduct of PIC with CYP2E1. Incubations and separations were as described under Materials and Methods. Data represent the deconvoluted spectrum of the CYP2E1 sample containing methanol (control, upper panel), 25 μM PEITC (middle panel), and 10 μM PIC (lower panel). The control and the sample incubated with PEITC show only the nonadducted CYP2E1 (54,838 Da), and the sample incubated with PIC exhibits both nonadducted (54,837 Da) and PIC-adducted CYP2E1 (54,982 Da).
TABLE 2.
ESI LC-MS/MS analysis of CYP2E1 incubated with ad without reductase in the presence of PIC or PEITC
Incubations and separations were as described under Materials and Methods.
| Reaction system | Treatment | Observed Mass | Modifying Mass Unitd |
|---|---|---|---|
| P450 alone (control) | Methanol | 54838a | —– |
| P450 alone | 25 µM PEITC | 54838a | —– |
| P450 alone | 10 µM PIC | 54837a, 54982a | 145 |
| P450+reductase (control) methanol | 54843a | —– | |
| P450+reductase | 25 µM PEITC | 54842 ± 1b | —– |
| P450+reductase | 25µM PEITC+NADPH | 54843 ± 1c, 55018 ± 6c | 175 ± 6 |
ESI, electrospray ionization; LC-MS/MS, liquid chromatography-tandem mass spectrometry; PEITC, phenethyl isothiocyanate.
Data represent the mean of two injections in which the difference between the protein masses for the two injections was within three mass units.
b,cData represent the mean and standard deviations from three and four injections, respectively.
Difference in the protein mass between nonadducted and adducted peaks observed in each deconvoluted spectra.
ESI LC-MS/MS Analysis of Protein Adduct Formation during the Metabolism of PEITC.
The modified protein formed as a result of inactivation by PEITC was analyzed by LC-MS/MS, and the results are shown in Table 2. In this analysis, the human CYP2E1 and reductase eluted very close together under the liquid chromatography conditions used (retention times: CYP2E1 at 21.4 minutes and reductase at 22.0 minutes). The PEITC-inactivated CYP2E1 sample displayed two major peaks, and the mass of the PEITC-inactivated P450 (55,018 Da) had increased by 175 ± 6 mass units when compared with the unmodified P450 (54,843 Da) in the same sample (Table 2). This mass difference is larger than that of PIC-derived apolipoprotein adduct (147 Da; see Fig. 7; Table 2) and is consistent, within the experimental error of the LC-MS/MS measurement, with the mass of a PIC-derived protein adduct + 1 sulfur atom (147 ; 32 Da). Alternatively, this mass difference could also be accounted for by the addition of an intermediate that resulted from formation of a covalent adduct with oxidized PEITC (PEITC, 163 Da + one oxygen atom, 16 Da).
Effect of Covalent Binding of PIC to the CYP2E1 on Catalytic Activity.
The demonstration of the formation of a PIC-derived protein adduct by LC-MS (Fig. 7; Table 2) showed that PIC can form protein adducts with CYP2E1 and that PIC could be one of the reactive intermediates responsible for inactivation of CYP2E1. Therefore, the effect of covalent binding of PIC on the enzymatic activity of CYP2E1 was evaluated. CYP2E1 was incubated in the reconstituted system with reductase and PIC at 0- to 100-fold excesses of PIC over CYP2E1 for 10 minutes, and the effect on the catalytic activity of human CYP2E1 was investigated by measuring the loss in 7-EFC O-de-ethylation activity as described in the Materials and Methods. Surprisingly, the data in Fig. 8A show that PIC has relatively little effect on CYP2E1 activity, even at the highest concentration of PIC tested (10 μM) where formation of a P450 protein adduct is observed. To demonstrate that the externally added PIC was able to bind to the active site of CYP2E1, the binding spectra for PIC and PEITC (for positive control) with CYP2E1 were investigated. As shown in Fig. 8B, the binding of either PEITC or PIC to CYP2E1 resulted in typical type I difference spectra (Estabrook and Werringloer, 1978), demonstrating that PIC binds to the active site of CYP2E1 in a manner similar to that of PEITC. These data indicate that although the PIC formed by the oxidative metabolism of PEITC covalently binds to the CYP2E1 apolipoprotein, it is not responsible for the loss in the enzymatic activity of CYP2E1.
Fig. 8.

(A) Effect of PIC on the 7-EFC O-de-ethylation activity of human CYP2E1. Assay conditions were as described in Materials and Methods. The percent of human CYP2E1 7-EFC O-de-ethylation activity remaining 10 minutes after the addition of PIC was plotted against the molar ratio of PIC to CYP2E1. The data shown represent the mean of three separate experiments done in duplicate. (B) Difference binding spectra for the addition of 25 μM PEITC (blue) or PIC (red) to CYP2E1 (1 µM). The conditions for the binding spectra are described in Materials and Methods.
Formation of a PIC-GSH Conjugate in Human Liver Microsomes.
Although PIC does not appear to be involved in the inactivation of CYP2E1 by PEITC, the biologic effects of PIC on other cellular macromolecules is of interest because of its high reactivity with proteins. As described earlier, we have detected a GSH conjugate of PIC formed in the human CYP2E1-reconstituted system in the presence of NADPH (Fig. 5), and we analyzed the formation of the GSH conjugate in human liver microsomes to confirm the formation of the PIC adduct under more physiologic conditions. The data for the LC-MS/MS analysis of the GSH conjugate of PIC are presented in Fig. 9. A GSH conjugate having a MH+ ion at an m/z 455 was detected in the reaction mixture with NADPH. This ion was identical to the one found in the PIC-GSH standard (unpublished data). In addition, the MS/MS product ions at m/z 380, 326, 223, 308, and 179 were observed, which were same as those shown in Fig. 4B, demonstrating that this ion is a conjugate of PIC with GSH.
Fig. 9.

ESI LC-MS/MS analysis of GSH conjugates of PEITC generated by human liver microsomes. The incubations, extractions of the GSH conjugates of PITC, and analyses were performed as described in Materials and Methods. The data show the extracted ion chromatogram at m/z 455 in the sample incubated with human liver microsomes and PEITC in the presence (upper panel) or absence (bottom panel) of NADPH.
Identification of the Site of Modification of CYP2E1 by PEITC.
CYP2E1, which has been inactivated by PEITC, was digested by trypsin and analyzed by LC-MS/MS to detect and identify modified peptides. SEQUEST Bioworks 3.2 software was used to look for the expected mass shift of 180 amu for the reactive sulfur species or 147 amu for PIC or 96 amu for desulfurization species to identify the adducted peptides. The SEQUEST search identified the human 2E1 isozyme with sequence coverage of about 69%. Analysis of the tryptic digest resulted in the identification of a peptide that was modified by PIC, as can be seen by the mass increase of 147 amu. The peptide with MH+ at m/z 1556 was identified as the peptide modified by PIC. The modified peptide was determined to have the sequence DLTDCLLVEMEK on the basis of the MS/MS data, with the reactive metabolite, PIC (147 amu) being attached to the cysteine.
Although incubation with PIC was found not to inactivate the protein, the site at which it binds to the protein was identified using trypsin digested 2E1 that had been inactivated by incubation in the reconstituted system with PEITC and NADPH. Analysis of this tryptic peptide revealed a mass shift of +147 units at cysteine residue 268 of the peptide DLTDCLLVEMEK as demonstrated by the mass shift of 147 for the y8 ion. The MS/MS spectrum of the doubly charged [M+2H]2+ ion at m/z at 778.7 from the peptide modified by PIC is shown in Fig. 11. The identities of the b and y ions for the modified peptide were predicted using University of California-San Francisco’s Protein Prospector, and the spectrum indicates no increase in mass from the y4 to the y7 ions with an increase in the mass of the y8 ion by 147 and the mass of the b fragment ions showed no increase in mass up to the b4 ion with an increase from the b5 ion on. The mass increase of 147 Da between that expected for y8 and the observed y8 ion indicated that the modified residue is the cysteine at position 268. Several of the adducts containing fragment ions were identified, including those at m/z 862.7.0 (y7+), 1112.0 (y8+), 1227.0 (y9+), 1328.4 (y10+), as well as 695.6 (b5+), 808.8 (b6+), 1021.0 (b8+), 1150.0 (b9+), 1281.0 (b10+), and 1410.4 (b11+). The peptide was detected in its native unmodified form in the reconstituted CYP2E1 incubated with PEITC in the absence of NADPH, indicating that this metabolite is formed only in the presence of NADPH. Also, no adducts were detected with an increased mass of 180 or 96 amu.
Fig. 11.
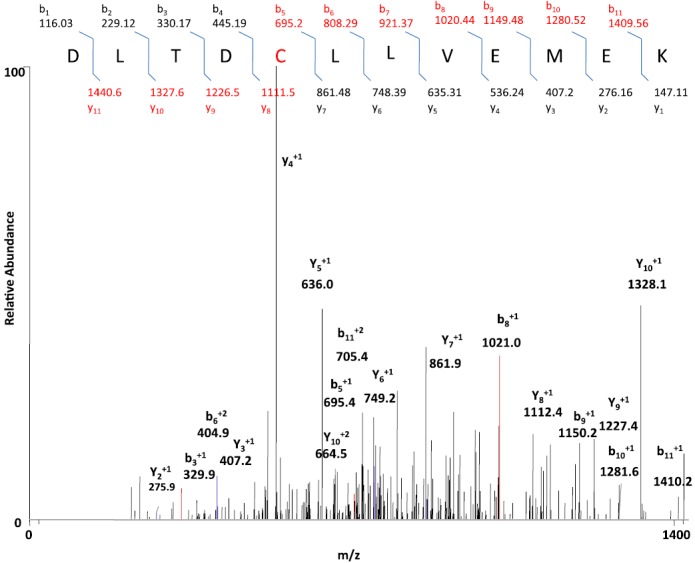
MS spectrum from the fragmentation of DLTDCLLVEMEK of the PIC-adducted peptide with a mass increment of 147 Da. CYP2E1 was reconstituted with reductase and PEITC in the presence and absence of NADPH. The samples were digested with trypsin and analyzed by LC-MS/MS and SEQUEST software as described in Materials and Methods. The figure shows the profile for the doubly charged ion with m/z 778.7 for the modified peptide 264DLTDCLLVEMEK275 with a mass increment of 147 amu and modification at the cysteine residue. The inset shows the b ion and y ions of the adducted peptide.
Discussion
The primary objectives of this study were to understand the mechanism of inactivation of CYP2E1 by PEITC through the oxidative pathway for the metabolism of PEITC and to identify the reactive intermediate that might be involved in the inactivation of PEITC. PEITC has previously been shown to nonenzymatically bind to GSH (Jiao et al., 1994). We found that the inactivation of CYP2E1 by PEITC required oxidative metabolism and occurred in the presence and absence of GSH. This is consistent with earlier studies demonstrating that rat liver CYP2E1 was inactivated by PEITC in vivo (Chung et al., 1985).
Since isocyanates can be formed by the oxidative metabolism of isothiocyanates, it has been suggested that the isocyanates may be the moieties that bind to and inactivate the P450 protein (Goosen et al., 2001; Moreno et al., 2001). A GSH-adduct of PIC, an isocyanate metabolite of PEITC formed during oxidative metabolism of recombinant human CYP2E1, is shown in Fig. 5. In addition, externally added PIC was shown to covalently bind to the CYP2E1 apolipoprotein (Fig. 7; Table 2). However, the covalent binding of PIC to the apolipoprotein did not inactivate the P450. These results suggest that another reactive intermediate may be involved in the inactivation. The bioactivation of PEITC to PIC by desulfurization may, in fact, be a major pathway for the oxidative metabolism of PEITC by human CYP2E1. Studies have shown that isothiocyanates are oxidized to isocyanates, which undergo hydrolysis nonenzymatically to yield their amine counterparts (Lee, 1996; Goosen et al., 2001; Moreno et al., 2001). The amine metabolites can then react with the remaining isothiocyanates or isocyanates to produce thiourea and urea (Lee, 1996). Previously, our laboratory investigated the oxidative metabolism of benzylisothiocyanate (BITC) by recombinant rabbit CYP2E1. Using [14C]-labeled BITC labeled at the α carbon, benzylamine and benzoic acid were detected as the major metabolites (Moreno et al., 2001). In addition, N,N′-di-benzylthiourea and N,N′-di-benzylurea were also shown to be formed by recombinant CYP2B1(Goosen et al., 2001). These results were obtained for incubations performed in the absence of GSH. In the current study, we detected the PIC-GSH conjugate and phenethylamine in the presence of GSH, and the possible scheme is depicted in Fig. 10. PIC is relatively unstable and therefore decomposes rapidly to give phenethylamine (Lee, 1996). In the presence of GSH, PIC may undergo conjugation with GSH to form a stable conjugation product, as shown in Fig. 10. P450s have been demonstrated to catalyze the desulfurization of parathion to produce paraoxon (Murray and Butler, 1994). A similar pathway involving the initial desulfurization of PEITC to form PIC could be envisioned that involves the initial formation of the S-oxide of PEITC, and then this rearranges to eliminate the sulfur to produce PIC (Fig. 10). The sulfur atom in its singlet state is electrophilic and could readily bind covalently to nucleophiles (Halpert et al., 1980). In addition to the desulfurization pathway for PEITC, we have previously explored the metabolic pathway responsible for the production of benzoic acid and phenyl acetic acid via hydroxylation of the α carbon of PEITC or further oxidation of phenethylamine, respectively (Moreno et al., 2001). We developed an analytical method that allowed us to detect benzoic acid and phenyl acetic acid using LC-MS/MS with detection in the negative ion mode. Using this highly sensitive method, we were unable to detect these metabolites in reaction mixtures from human CYP2E1 incubated with PEITC in the presence of NADPH (unpublished data). Although further studies are necessary, these results suggest that the formation of benzoic acid and phenyl acetic acid might not occur or is a relatively minor pathway catalyzed by human CYP2E1. Our results presented here also suggest that GSH adducts of PIC may be formed under physiologic conditions where GSH is present, and the peptide mass following digestion of the inactivated protein would increase by 147, based on the mass increase of the GSH adduct of PIC. The mass due to a peptide adduct could also increase by 180 amu if the reactive sulfur species binds to the protein (Fig. 10) or increase by 96 amu if an oxidative desulfurization of PEITC occurs as seen with methyl parathion (Kyle et al., 2012).
Thus, identification of the inactivating species and the modified residue was attempted by digesting the control and the PEITC-inactivated samples with trypsin and by analyzing the peptide digests by ESI LC-MS/MS and SEQUEST software. Peptides obtained from the control and the inactivated samples were compared with each other and with the theoretically expected digests. This led to the identification of one peptide in the inactivated sample with an m/z of 1556, and the same peptide without any modification with an m/z of 1409 (1556−147) in the control sample. The modified peptide was then analyzed by mass spectrometry sequencing to locate the modified amino acid. Interpretation of the MS/MS data led to the identification of the sequence DLTDCLLVEMEK, corresponding to residues 264–275 and residue Cys268 was demonstrated as the residue modified by PIC (Fig. 11). Although, our studies and previous studies by Konsue and Ioannides (2010) have shown that PIC is not the reactive metabolite responsible for the inactivation of the enzyme, it may be important to investigate the biologic consequences of the modification of CYP2E1 by PIC.
Our results show that under conditions where the PEITC-inactivated samples lost almost 80% of their catalytic activity, 30% of the heme and 56% of the P450 content as assayed by the CO-binding spectrum were lost. It is possible that 30% of the loss in catalytic activity may be due to heme destruction and that the remaining 50% loss in activity is due to protein modification. Because the magnitude of the decrease in the ability of the inactivated P450 to bind CO is greater than the heme loss, it is possible that modification of the P450 apolipoprotein in the active site by the reactive intermediate may lead to a partial impairment of the heme-coordination or hinder access of the CO to heme. Our results demonstrate that inactivation of human CYP2E1 by PEITC results from both heme-destruction and protein-modification, whereas the latter appears to be the predominant pathway for inactivation.
Previous studies (Goosen et al., 2001; Moreno et al., 2001) demonstrated that benzyl isocyanate is produced during the metabolism of BITC by P450-catalyzed oxidative de-sulfuration and it has been suggested that it then reacts with the P450 apoprotein leading to inactivation. However, our present study demonstrated that although PIC covalently binds to human CYP2E1, it does not inactivate it, indicating that one or more other intermediates maybe responsible for the inactivation. Similar studies by Konsue and Ioannides (2010) have shown that PIC is not the metabolite responsible for mechanism-based inhibition of P450 (Konsue and Ioannides, 2010). Therefore, the reactive sulfur species formed as a result of the desulfurization may be another potential reactive intermediate responsible for the loss in activity. Previously, a reactive sulfur species released as a consequence of desulfurization was shown to be responsible for the covalent binding and inactivation of P450s by S-containing compounds, such as parathion (Norman et al., 1974; Halpert et al., 1980; Kyle et al., 2012) and CS2 (DeMatteis, 1974). A similar mechanism for inactivation by PEITC is supported by the data for the formation of the CYP2E1 protein adduct as analyzed by LC-MS/MS. Table 2 shows that a protein adduct was observed in the PEITC-inactivated sample. The most plausible explanation for the observed mass difference (175 Da) involves reaction of the protein with PIC (147 Da) together with sulfur (32 Da), taking into account our observation that PIC can covalently bind to the apolipoprotein in a nonenzymatic process without causing inactivation.
This explanation is consistent with our hypothesis that the PEITC-dependent inactivation of human CYP2E1 occurs by a mechanism similar to that of other S-containing compounds, such as parathion, where the reactive sulfur species is responsible for inactivation. Alternatively, the observed mass difference for the inactivated sample could be accounted for by the addition of an intermediate resulting from oxidized PEITC (163+16 Da). However, additional data to allow us to distinguish between these two possibilities have not yet been obtained. Future studies will focus on the fate of the sulfur from [35S]-labeled PEITC by detecting radioactive peptide and analyzing by mass spectrometry.
The P450-catalyzed generation of PIC may lead to additional biologic effects because of the strong reactivity of PIC toward nucleophiles, including nucleophiles on proteins. Isocyanates are known to be highly reactive (Lee, 1992, 1996), and toxicity has been observed with some isocyanates, such as methyl isocyanate, which causes severe pulmonary toxicity (Nemery et al., 1985), and naphthyl isocyanate, which has been shown to be mutagenic (Tamura et al., 1990). Other drugs and compounds that are metabolized to form isocyanates include tolbutamide (Guan et al., 1999), N-methylformamide (NMF) (Mutlib et al., 2006), and anticancer agents, such as nitrosoureas (Rice et al., 2005) and diarylsulfonylureas (Jochheim et al., 2002). Arguments have been made for the role of isocyanates in some of the biologic effects of these compounds. Methyl isocyanate formed from NMF is thought to be related to the hepatotoxicity induced by NMF (Mutlib et al., 2006). Alkyl isocyanates produced by the oxidative metabolism of tolbutamide and N,N′-bis(2-chloroethyl)-N-nitrosourea (BCNU) are known to inhibit glutathione reductase (Guan et al., 1999; Rice et al., 2005). Isocyanates are unstable in water and quickly hydrolyze to form the corresponding amines, which would limit their biologic activities. However, isocyanates can readily form reversible glutathione conjugates (Jochheim et al., 2002) so that the GSH conjugate can act as a carrier for otherwise short-lived isocyanates in in vivo situations.
Taken together, this information suggests the possibility that PIC and PIC-GSH conjugates may have unanticipated biologic effects, and it would be of great interest to follow up on the mechanistic pathway of the present findings and the involvement of PIC and its GSH conjugate in the chemopreventive effect of PEITC.
Acknowledgments
The authors thank Dr. P. F. Guengerich for the human CYP2E1 plasmid and for the human liver microsomes. The authors also thank Hsia-lien Lin for purification of the reductase and for fruitful discussions.
Abbreviations
- BITC
benzylisothiocyanate
- DLPC
dilauroyl-l-α-phosphatidylcholine
- 7-EFC
7-ethoxy-4-(trifluoromethyl) coumarin
- ESI
electrospray ionization
- GSH
reduced glutathione
- 7-HFC
7-hydroxy-4-(trifluoromethyl)coumarin
- HPLC
high-performance liquid chromatography
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- LCQ
liquid chromatography quadrupole
- m/z
mass-to-charge ratio
- NMF
N-methylformamide
- P450
cytochrome P450
- PEITC
phenethyl isothiocyanate
- PIC
phenethyl isocyanate
- TFA
trifluoroacetic acid
Authorship Contributions
Participated in research design: Yoshigae, Kent, Sridar.
Conducted experiments: Yoshigae, Sridar.
Contributed new reagents or analytic tools: Yoshigae.
Performed data analysis: Yoshigae, Sridar.
Wrote or contributed to the writing of the manuscript: Yoshigae, Kent, Sridar, Hollenberg.
Footnotes
This work was supported in part by the National Institutes of Health [Grant CA-16954]; and by Daiichi Sankyo Co., Ltd., Tokyo, Japan.
References
- Baillie TA, Davis MR. (1993) Mass spectrometry in the analysis of glutathione conjugates. Biol Mass Spectrom 22:319–325 [DOI] [PubMed] [Google Scholar]
- Buters JT, Schiller CD, Chou RC. (1993) A highly sensitive tool for the assay of cytochrome P450 enzyme activity in rat, dog and man. Direct fluorescence monitoring of the deethylation of 7-ethoxy-4-trifluoromethylcoumarin. Biochem Pharmacol 46:1577–1584 [DOI] [PubMed] [Google Scholar]
- Chung FL, Wang MY, Hecht SS. (1985) Effects of dietary indoles and isothiocyanates on N-nitrosodimethylamine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone alpha-hydroxylation and DNA methylation in rat liver. Carcinogenesis 6:539–543 [DOI] [PubMed] [Google Scholar]
- DeMatteis F. (1974) Covalent binding of sulfur to microsomes and loss of cytochrome P450 during the oxidative desulfuration of several chemicals. Mol Pharmacol 10:849–854 [Google Scholar]
- Estabrook RW, Werringloer J. (1978) The measurement of difference spectra: application to the cytochromes of microsomes. Methods Enzymol 52:212–220 [DOI] [PubMed] [Google Scholar]
- Fowke JH, Chung FL, Jin F, Qi D, Cai Q, Conaway C, Cheng JR, Shu XO, Gao YT, Zheng W. (2003) Urinary isothiocyanate levels, brassica, and human breast cancer. Cancer Res 63:3980–3986 [PubMed] [Google Scholar]
- Ghanbari F, Rowland-Yeo K, Bloomer JC, Clarke SE, Lennard MS, Tucker GT, Rostami-Hodjegan A. (2006) A critical evaluation of the experimental design of studies of mechanism based enzyme inhibition, with implications for in vitro-in vivo extrapolation. Curr Drug Metab 7:315–334 [DOI] [PubMed] [Google Scholar]
- Goosen TC, Mills DE, Hollenberg PF. (2001) Effects of benzyl isothiocyanate on rat and human cytochromes P450: identification of metabolites formed by P450 2B1. J Pharmacol Exp Ther 296:198–206 [PubMed] [Google Scholar]
- Guan X, Davis MR, Tang C, Jochheim CM, Jin L, Baillie TA. (1999) Identification of S-(n-butylcarbamoyl)glutathione, a reactive carbamoylating metabolite of tolbutamide in the rat, and evaluation of its inhibitory effects on glutathione reductase in vitro. Chem Res Toxicol 12:1138–1143 [DOI] [PubMed] [Google Scholar]
- Guengerich FP. (1994) Analysis and characterization of enzymes, in Principles and Methods of Toxicology (Hayes AW. ed) pp 1259–1313, Raven Press, New York [Google Scholar]
- Guo Z, Smith TJ, Wang E, Sadrieh N, Ma Q, Thomas PE, Yang CS. (1992) Effects of phenethyl isothiocyanate, a carcinogenesis inhibitor, on xenobiotic-metabolizing enzymes and nitrosamine metabolism in rats. Carcinogenesis 13:2205–2210 [DOI] [PubMed] [Google Scholar]
- Halpert J, Hammond D, Neal RA. (1980) Inactivation of purified rat liver cytochrome P-450 during the metabolism of parathion (diethyl p-nitrophenyl phosphorothionate). J Biol Chem 255:1080–1089 [PubMed] [Google Scholar]
- Hanna IH, Teiber JF, Kokones KL, Hollenberg PF. (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch Biochem Biophys 350:324–332 [DOI] [PubMed] [Google Scholar]
- Hwang ES, Lee HJ. (2006) Phenylethyl isothiocyanate and its N-acetylcysteine conjugate suppress the metastasis of SK-Hep1 human hepatoma cells. J Nutr Biochem 17:837–846 [DOI] [PubMed] [Google Scholar]
- Jiao D, Eklind KI, Choi CI, Desai DH, Amin SG, Chung FL. (1994) Structure-activity relationships of isothiocyanates as mechanism-based inhibitors of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis in A/J mice. Cancer Res 54:4327–4333 [PubMed] [Google Scholar]
- Jochheim CM, Davis MR, Baillie KM, Ehlhardt WJ, Baillie TA. (2002) Glutathione-dependent metabolism of the antitumor agent sulofenur. Evidence for the formation of p-chlorophenyl isocyanate as a reactive intermediate. Chem Res Toxicol 15:240–248 [DOI] [PubMed] [Google Scholar]
- Kent UM, Juschyshyn MI, Hollenberg PF. (2001) Mechanism-based inactivators as probes of cytochrome P450 structure and function. Curr Drug Metab 2:215–243 [DOI] [PubMed] [Google Scholar]
- Keum YS, Jeong WS, Kong AN. (2004) Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat Res 555:191–202 [DOI] [PubMed] [Google Scholar]
- Konsue N, Ioannides C. (2010) Phenethyl isocyanate is not the metabolite of phenethyl isothiocyanate responsible for mechanism-based inhibition of cytochrome P450. Arch Toxicol 84:751–759 [DOI] [PubMed] [Google Scholar]
- Kyle PB, Smith SV, Baker RC, Kramer RE. (2012) Mass spectrometric detection of CYP450 adducts following oxidative desulfuration of methyl parathion. J Appl Toxicol.DOI: 10.1002/jat.1792. [DOI] [PubMed] [Google Scholar]
- Larson JR, Coon MJ, Porter TD. (1991) Alcohol-inducible cytochrome P-450IIE1 lacking the hydrophobic NH2-terminal segment retains catalytic activity and is membrane-bound when expressed in Escherichia coli. J Biol Chem 266:7321–7324 [PubMed] [Google Scholar]
- Leclercq I, Desager JP, Horsmans Y. (1998) Inhibition of chlorzoxazone metabolism, a clinical probe for CYP2E1, by a single ingestion of watercress. Clin Pharmacol Ther 64:144–149 [DOI] [PubMed] [Google Scholar]
- Lee MS. (1992) Oxidative conversion by rat liver microsomes of 2-naphthyl isothiocyanate to 2-naphthyl isocyanate, a genotoxicant. Chem Res Toxicol 5:791–796 [DOI] [PubMed] [Google Scholar]
- Lee MS. (1996) Enzyme induction and comparative oxidative desulfuration of isothiocyanates to isocyanates. Chem Res Toxicol 9:1072–1078 [DOI] [PubMed] [Google Scholar]
- London SJ, Yuan JM, Chung FL, Gao YT, Coetzee GA, Ross RK, Yu MC. (2000) Isothiocyanates, glutathione S-transferase M1 and T1 polymorphisms, and lung-cancer risk: a prospective study of men in Shanghai, China. Lancet 356:724–729 [DOI] [PubMed] [Google Scholar]
- Moreno RL, Goosen T, Kent UM, Chung FL, Hollenberg PF. (2001) Differential effects of naturally occurring isothiocyanates on the activities of cytochrome P450 2E1 and the mutant P450 2E1 T303A. Arch Biochem Biophys 391:99–110 [DOI] [PubMed] [Google Scholar]
- Morse MA, Wang CX, Stoner GD, Mandal S, Conran PB, Amin SG, Hecht SS, Chung FL. (1989) Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA adduct formation and tumorigenicity in the lung of F344 rats by dietary phenethyl isothiocyanate. Cancer Res 49:549–553 [PubMed] [Google Scholar]
- Murray M, Butler AM. (1994) Hepatic biotransformation of parathion: role of cytochrome P450 in NADPH- and NADH-mediated microsomal oxidation in vitro. Chem Res Toxicol 7:792–799 [DOI] [PubMed] [Google Scholar]
- Mutlib A, Jiang P, Atherton J, Obert L, Kostrubsky S, Madore S, Nelson S. (2006) Identification of potential genomic biomarkers of hepatotoxicity caused by reactive metabolites of N-methylformamide: Application of stable isotope labeled compounds in toxicogenomic studies. Chem Res Toxicol 19:1270–1283 [DOI] [PubMed] [Google Scholar]
- Nakajima M, Yoshida R, Shimada N, Yamazaki H, Yokoi T. (2001) Inhibition and inactivation of human cytochrome P450 isoforms by phenethyl isothiocyanate. Drug Metab Dispos 29:1110–1113 [PubMed] [Google Scholar]
- Nakamura Y, Miyoshi N. (2006) Cell death induction by isothiocyanates and their underlying molecular mechanisms. Biofactors 26:123–134 [DOI] [PubMed] [Google Scholar]
- National Cancer Institute (1996) Clinical development plan: phenethyl isothiocyanate. J Cell Biochem Suppl 26:149–157 [PubMed] [Google Scholar]
- Nemery B, Dinsdale D, Sparrow S, Ray DE. (1985) Effects of methyl isocyanate on the respiratory tract of rats. Br J Ind Med 42:799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman BJ, Poore RE, Neal RA. (1974) Studies of the binding of sulfur released in the mixed-function oxidase-catalyzed metabolism of diethyl p-nitrophenyl phosphorothionate(parathion) to diethyl p-nitrophenyl phosphate (paraoxon). Biochem Pharmacol 23:1733–1744 [DOI] [PubMed] [Google Scholar]
- Omura T, Sato R. (1964) The Carbon Monoxide-Binding Pigment of Liver Microsomes. Ii. Solubilization, Purification, and Properties. J Biol Chem 239:2379–2385 [PubMed] [Google Scholar]
- Pereira MA. (1995) Chemoprevention of diethylnitrosamine-induced liver foci and hepatocellular adenomas in C3H mice. Anticancer Res 15 (5B):1953–1956 [PubMed] [Google Scholar]
- Rice KP, Penketh PG, Shyam K, Sartorelli AC. (2005) Differential inhibition of cellular glutathione reductase activity by isocyanates generated from the antitumor prodrugs Cloretazine and BCNU. Biochem Pharmacol 69:1463–1472 [DOI] [PubMed] [Google Scholar]
- Seow A, Yuan JM, Sun CL, Van Den Berg D, Lee HP, Yu MC. (2002) Dietary isothiocyanates, glutathione S-transferase polymorphisms and colorectal cancer risk in the Singapore Chinese Health Study. Carcinogenesis 23:2055–2061 [DOI] [PubMed] [Google Scholar]
- Shen AL, Porter TD, Wilson TE, Kasper CB. (1989) Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem 264:7584–7589 [PubMed] [Google Scholar]
- Solt DB, Chang Kw, Helenowski I, Rademaker AW. (2003) Phenethyl isothiocyanate inhibits nitrosamine carcinogenesis in a model for study of oral cancer chemoprevention. Cancer Lett 202:147–152 [DOI] [PubMed] [Google Scholar]
- Tamura N, Aoki K, Lee MS. (1990) Characterization and genotoxicity of DNA adducts caused by 2-naphthyl isocyanate. Carcinogenesis 11:2009–2014 [DOI] [PubMed] [Google Scholar]
- Teiber JF, Hollenberg PF. (2000) Identification of the human liver microsomal cytochrome P450s involved in the metabolism of N-nitrosodi-n-propylamine. Carcinogenesis 21:1559–1566 [PubMed] [Google Scholar]
- Xiao D, Singh SV. (2007) Phenethyl isothiocyanate inhibits angiogenesis in vitro and ex vivo. Cancer Res 67:2239–2246 [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Inui Y, Yun CH, Guengerich FP, Shimada T. (1992) Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis 13:1789–1794 [DOI] [PubMed] [Google Scholar]
- Zhang Y. (2004) Cancer-preventive isothiocyanates: measurement of human exposure and mechanism of action. Mutat Res 555:173–190 [DOI] [PubMed] [Google Scholar]
- Zhao B, Seow A, Lee EJ, Poh WT, Teh M, Eng P, Wang YT, Tan WC, Yu MC, Lee HP. (2001) Dietary isothiocyanates, glutathione S-transferase -M1, -T1 polymorphisms and lung cancer risk among Chinese women in Singapore. Cancer Epidemiol Biomarkers Prev 10:1063–1067 [PubMed] [Google Scholar]