

Abstract
During infection of epithelial cells, the obligate intracellular pathogen Chlamydia trachomatis secretes the serine protease CPAF into the host cytosol to regulate a range of host cellular processes through targeted proteolysis. Here we report the development of an in vitro assay for the enzyme and the discovery of a cell-permeable CPAF zymogen-based peptide inhibitor with nanomolar inhibitory affinity. Treating C. trachomatis-infected HeLa cells with this inhibitor prevented CPAF cleavage of the intermediate filament vimentin, and led to loss of vimentin-cage surrounding the intracellular vacuole. Because Chlamydia is a genetically intractable organism, this inhibitor may serve as a tool to understand the role of CPAF in pathogenesis.
Keywords: Chlamydia trachomatis, inhibitor, pathogenesis, protease
The obligate intracellular pathogen, Chlamydia trachomatis is the causative agent of ocular and genital diseases of significant clinical and economic importance such as corneal scarring, pelvic inflammatory disease, ectopic pregnancy, and infertility.1 C. trachomatis is the leading cause of infectious blindness (trachoma) worldwide and the most common sexually transmitted pathogen in the developed world. C. trachomatis replicates within a parasitophorous vacuole, termed an inclusion, and secretes enzymes and proteins that efficiently modulate a wide range of host cellular processes such as lipid and membrane transport2-4, the actin cytoskeleton5, microtubule-based motors6, lysosomal recognition of the inclusion7, ERK/MEK signaling pathways8, and the onset of programmed cell death.9 Unfortunately, C. trachomatis is refractory to genetic manipulation10, thus the molecular mechanisms underlying chlamydial cooption of host cellular functions are poorly understood. In light of this limitation, enzymology and chemical biology approaches may provide insight into the function of specific enzymes important in C. trachomatis pathogenesis.
The serine protease CPAF (Chlamydia Protease-like Activity Factor) is secreted by C. trachomatis into the cytosol of host cells, where it cleaves several host proteins, including transcription factors (USF-1 and RFX5) required for MHC transcription11, and pro-apoptotic proteins (Bad, Puma, and Bid)12. In addition, CPAF mediates expansion of the inclusion by cleaving host intermediate filaments such as keratin 8 and vimentin.12-15 As such, mounting evidence points to CPAF as an important target for therapeutic intervention.
Beyond SDS-PAGE or Western blotting activity assays with impure enzyme preparations, detailed investigations on CPAF have been somewhat limited due to the lack of a robust in vitro assay suitable for obtaining activity and inhibition kinetic parameters. In response to this, we developed a facile assay for CPAF proteolysis that would support kinetic studies and serve as a platform for inhibitor discovery.
Few proteolysis sites have been mapped at specific residues since CPAF is known to proteolyze several different proteins and most of these substrates are degraded by multiple proteolytic events. However, Valdivia and coworkers determined the primary site of cleavage of human vimentin to be between Ser72 and Ser73 by N-terminal sequencing of immunoprecipitated vimentin cleaved by CPAF15. We envisioned that peptides derived from the vimentin primary sequence surrounding Ser72 might serve as a substrate for development of an in vitro assay for CPAF hydrolytic activity. Using solid-phase methods, we prepared a peptide substrate derived from the sequence of human vimentin (see Supplementary Materials). Peptide Abz-VRLRSSVPGV-NH2 (1) contained an N-terminal 2-anthranilic acid (Abz) moiety for fluorescence detection following HPLC separation of the products from CPAF-mediated proteolysis. Figure 1 depicts a typical chromatogram of the reaction. Mass spectrometry confirmed that CPAF proteolyzed this substrate into two peptide fragments at the anticipated scissile bond. Apparent kinetic parameters obtained for CPAF proteolysis of substrate 1 were 13.2 s-1, 0.88 mM, and 1.5 × 104 M-1s-1 for kcatapp, KMapp, and kcatapp/KMapp respectively.
Figure 1.
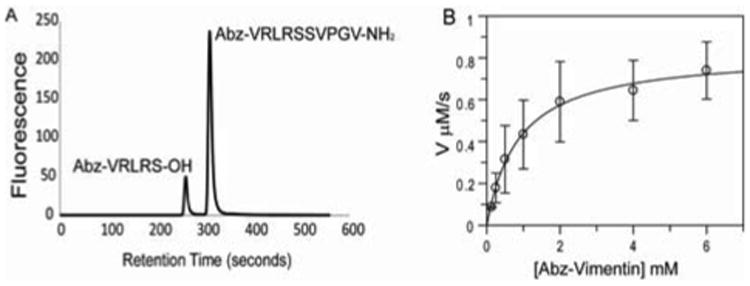
HPLC CPAF assay using Abz-VRLRSSVPGV-NH2: A. Representative HPLC trace showing 1 and the Abz containing hydrolysis product Abz-VRLRS-OH (2) as viewed by fluorescence detection at λem = 420 nm. B. Determination of apparent kinetic parameters for CPAF for 1. Assays were performed in a total volume of 100 μL containing Assay Buffer, CPAF (62.5 nM), and varying concentrations of Abz-VRLRSSVPGV-NH2 (0-6 mM).
CPAF is highly resistant to conventional broad-spectrum serine protease inhibitors such as fluorophosphonates and fluorosulfonates and has one only known inhibitor, the natural product lactacystin.16 Lactacystin acylates Ser499 within the Ser449-His105-Glu558 catalytic triad.16,17 To further confirm the utility of substrate 1 for inhibitor discovery, we examined the effect of lactacystin on CPAF activity ex vitro. Lactacystin exhibited CPAF an IC50 of 10.2 ± 2.3 μM, a value comparable to concentrations required to fully inhibit ex vivo.11
Indeed, the prospects of utilizing lactacystin as a probe of CPAF's function in chlamydial co-option of mammalian host cell function are improbable because this agent is an irreversible, potent inhibitor of the human proteasome. To design a CPAF-specific inhibitor we referred to the recent structural data on the zymogen and active forms of CPAF.17 CPAF is produced as a catalytically-inactive 609 amino acid zymogen that undergoes concentration-dependent autoproteolysis and self-activation by excision of an internal 40 amino acid inhibitory sequence (residues 243-283), releasing a 29 kDa C-terminal fragment and a 32 kDa N-terminal fragment that re-associate to form a stable heterodimer. Removal of the 40mer internal sequence allows the serine protease catalytic triad to assemble from residue contributions from both subunits, as was revealed from the recent crystal structure of recombinant CPAF (rCPAF) determined by Chai and coworkers.17 A mutated fragment of this 40-mer sequence fused to glutathione-S-transferase exhibited modest inhibitory activity against CPAF.17 Building upon these precedents, we hypothesized that binding of a 25 amino acid portion within the 243-283CPAF sequence that was predominantly alpha helical (residues 258-283) would likely bind within the mature CPAF heterodimer's active site (Figure 2). The enzyme-bound alpha helix may therefore compete for substrate binding to CPAF and emulate the inactive zymogen conformation by repositioning active site residues at the interface of the heterodimer. Since base strength amplification is the proposed mechanism of Ser499 nucleophilic activation,17 altering the relative positions of the three catalytic residues that were donated by two separate polypeptide chains may abrogate CPAF activity.
Figure 2.

CPAF inhibition assay comparison of lactacystin with inhibitors 3 and 4. A. HPLC-based assays were performed in a final volume of 100 μL containing Assay Buffer, CPAF (62.5 nM), substrate 1 (0.5 mM), and varying concentrations of inhibitor (0-240 μM). IC50 values for lactacystin, peptide 3, and peptide 4 were 10.2 ± 2.3 μM, 1.6 ± 0.6 μM, and 50 ± 7 nM respectively. B. Three-dimensional structure of CPAF with peptide 4 modeled into the active site of heterodimeric CPAF; C. Expansion view of the inhibitor 4 bound CPAF model showing predicted polyarginine binding to a region of negative electrostatic potential.
We subsequently prepared the peptide inhibitor 3 (H-SLFYSPMVPHFWAELRNHYATSGLK-NH2) that corresponded to 258-283CPAF by solid-phase peptide synthesis (See Supplementary Materials), and showed that this sequence was capable of inhibiting CPAF proteolysis of 1 in vitro using rCPAF. Peptide 3 exhibited an IC50 of 1.6 ± 0.6 μM, which was 6-fold more potent than lactacystin (Figure 2A).
Since C. trachomatis is an obligate intracellular pathogen, we designed a cell permeable variant of inhibitor 3 by introducing a nona-arginine C-terminal addition to this sequence, generating (H-SLFYSPMVPHFWAELRNHYATSGLK RRRRRRRRR-NH2, 4). When examined in vitro, peptide 4 yielded an IC50 of 50 ± 7 nM, a value ∼200-fold more potent than lactacystin and ∼30-fold better than inhibitor 3 (Figure 2A), making 4 the most potent CPAF inhibitor presently known. By analysis of the model of inhibitor 4 bound to mature CPAF (Figure 2B), we attribute the increase in inhibitory activity to enhanced binding due to favorable electrostatic interactions between the nona-arginine C-terminus and a large region of electronegative potential proximal to the active site where the helical 25mer is predicted to bind. We are currently performing structural studies of the peptide 4/rCPAF complex to confirm this hypothesis.
Valdivia and coworkers demonstrated that treating C. trachomatis LGV-L2 434-infected HeLa cells with lactacystin resulted in fiber oligermerization of vimentin in due to inhibition of CPAF-mediated proteolysis.15 To establish if CPAF was inhibited by peptide 4 during infection, vimentin cleavage was assessed in C. trachomatis LGV-L2 434 -infected HeLa cells.after being treated with a range of concentrations (2-10 M).15 Under similar conditions, infected HeLa cells were treated with a sequence scrambled control peptide that possessed no CPAF inhibitory activity in vitro (H-NFALSHFRLPLSTYKEMPYVSHWAGRRRRRRRRR-NH2, 5). Peptide 4, but not the scrambled peptide 5, markedly inhibited CPAF-mediated degradation of vimentin in a dose-dependent manner (Figure 3B). This result strongly suggested that peptide 4 not only penetrated the cell membrane, but also selectively targeted CPAF activity ex vivo. Permeability of these peptides is most likely modest with respect to the percentage of peptide being delivered, however the ability to inhibit still renders them useful.
Figure 3.
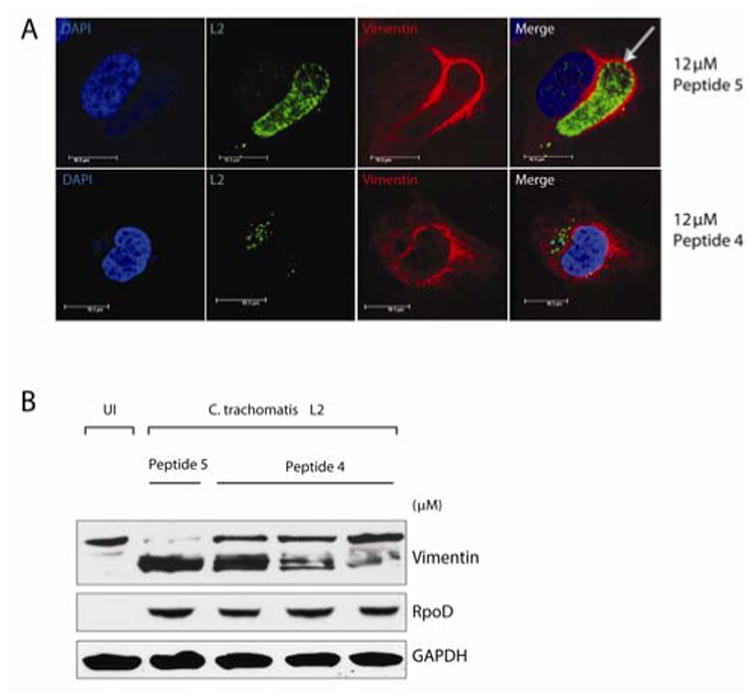
HeLa cells were infected with C. trachomatis L2, treated with control peptide 5 and peptide 4, and harvested at 30 hpi. A. Immunofluorescence microscopy by staining with an anti-vimentin antibody (red), and an anti-Chlamydia L2 antibody (green). Note loss of vimentin-cage (arrow) surrounding the inclusion in cells treated with peptide 4. B. Cells were lysed in the presence of protease inhibitors and resolved by SDS PAGE and subjected to immunblot analysis. Vimentin, RpoD (bacterial loading control) and GAPDH (host loading control) were visualized with substrate-specific antibodies.
Chlamydia remodels and recruits cytoskeletal components of the host cell such as F-actin and vimentin to form a dynamic scaffold or ‘cage’ that provides structural stability to the inclusion15. As the inclusion expands, secreted CPAF progressively modifies the intermediate filaments scaffold, presumably to increase the inclusion's flexibility and accommodate the increased bacterial load. In infected cells, CPAF processing of vimentin filaments occurs several hours after the hour post-infection (hpi) when CPAF is detectable in the cytosol.15 Treatment of C. trachomatis-infected HeLa cells with peptide 4, but not the control peptide 5, resulted in loss of vimentin processing (Figure 3B) and increased disorder in the position of the vimentin cage surrounding the inclusion (Figure 3A). These data suggest that peptide 4 selectively inhibits CPAF activity, which prevents vimentin cleavage and proper deposition of vimentin surrounding the intracellular vacuole (Figure 3A). Because intermediate filaments like vimentin are stable structures that provide mechanical support to maintain vacuole integrity in infected cells,15 it is likely that peptide 4 has altered the integrity of the vacuole, which may have a broader impact on bacterial survival within the host.
The development of an in vitro activity assay for CPAF coupled with the discovery of a cell-permeable nanomolar affinity inhibitor furthers our understanding of the role of CPAF in chlamydial pathogenesis. Since genetic manipulation of Chlamydia is not possible at this time, we hope that these enzymology and chemical biology tools will enable investigations into the kinetic and molecular mechanism of this unique enzyme, as well as analyses of the substrate specificity, small molecule inhibitor discovery, mechanism of action, and role in pathogenesis in vivo.
Supplementary Material
Acknowledgments
We thank Dr. Jijie Chai at the College of Biological Sciences, China Agricultural University, Beijing, China for the gift of the pET30a(CPAF) expression vector, and Dr. Charles Pemble IV of the Duke University Macromolecular X-ray Crystallography Shared Resource for assistance with computational modeling.
Funding Sources: This work was generously supported by Duke University, the National Institutes of Health awards AI081694 (R.H.V.), AI46611 (D.M.), the Burroughs Wellcome Trust Program in the Pathogenesis of Infectious Diseases (R.H.V.), and a CR Hauser Fellowship (M.B.).
Footnotes
Supporting information: Supplementary experimental methods and enlarged figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schachter J. Sex Transm Dis. 1999;26:279–280. doi: 10.1097/00007435-199905000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Carabeo RA, Mead DJ, Hackstadt T. Proc Natl Acad Sci U S A. 2003;100:6771–6776. doi: 10.1073/pnas.1131289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scidmore MA, Fischer ER, Hackstadt T. J Cell Biol. 1996;134:363–374. doi: 10.1083/jcb.134.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Ooij C, Kalman L, van I, Nishijima M, Hanada K, Mostov K, Engel JN. Cell Microbiol. 2000;2:627–637. doi: 10.1046/j.1462-5822.2000.00077.x. [DOI] [PubMed] [Google Scholar]
- 5.Carabeo RA, Grieshaber SS, Fischer E, Hackstadt T. Infect Immun. 2002;70:3793–3803. doi: 10.1128/IAI.70.7.3793-3803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grieshaber SS, Grieshaber NA, Hackstadt T. J Cell Sci. 2003;116:3793–3802. doi: 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- 7.Fields KA, Fischer E, Hackstadt T. Infect Immun. 2002;70:3816–3823. doi: 10.1128/IAI.70.7.3816-3823.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su H, McClarty G, Dong F, Hatch GM, Pan ZK, Zhong G. J Biol Chem. 2004;279:9409–9416. doi: 10.1074/jbc.M312008200. [DOI] [PubMed] [Google Scholar]
- 9.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. J Exp Med. 1998;187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomson NR, Clarke IN. Future Microbiology. 2010;5:555–561. doi: 10.2217/fmb.10.31. [DOI] [PubMed] [Google Scholar]
- 11.Zhong G, Fan P, Ji H, Dong F, Huang Y. J Exp Med. 2001;193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pirbhai M, Dong F, Zhong Y, Pan KZ, Zhong G. J Biol Chem. 2006;281:31495–31501. doi: 10.1074/jbc.M602796200. [DOI] [PubMed] [Google Scholar]
- 13.Savijoki K, Alvesalo J, Vuorela P, Leinonen M, Kalk-kinen N. FEMS Immunol Med Microbiol. 2008;54:375–384. doi: 10.1111/j.1574-695X.2008.00488.x. [DOI] [PubMed] [Google Scholar]
- 14.Dong F, Su H, Huang Y, Zhong Y, Zhong G. Infect Immun. 2004;72:3863–3868. doi: 10.1128/IAI.72.7.3863-3868.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar Y, Valdivia RH. Cell Host & Microbe. 2008;4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong G, Liu L, Fan T, Fan P, Ji H. J Exp Med. 2000;191:1525–1534. doi: 10.1084/jem.191.9.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Z, Feng Y, Chen D, Wu X, Huang S, Wang X, Xiao X, Li W, Huang N, Gu L, Zhong G, Chai J. Cell Host & Microbe. 2008;4:529–542. doi: 10.1016/j.chom.2008.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.