

Abstract
Despite reports that the PB1-F2 protein contributes to influenza virus pathogenicity in the mouse model, little is known about its significance in avian hosts. In our previous study the A/Vietnam/1203/04 (H5N1) wild-type virus (wtVN1203) was more lethal to mallard ducks than a reverse genetics (rg)–derived VN1203. In search of potential viral factors responsible for this discrepancy, we found that synonymous mutations (SMs) had been inadvertently introduced into 3 genes of the rgVN1203 (rgVN1203/SM-3). Of 11 SMs in the PB1 gene, 3 resided in the PB1-F2 open reading frame, caused amino acid (aa) substitutions in the PB1-F2 protein, and reduced virus lethality in mallard ducks. The wtVN1203 and recombinant viruses with repairs to these 3 aa’s (rgVN1203/R-PB1-F2) or with repairs to all 11 SMs (rgVN1203/R-PB1) were significantly more pathogenic than rgVN1203/SM-3. In cultured cells repairing 3 mutations in PB1-F2 increased viral polymerase activity and expression levels of viral RNA.
Keywords: A/Vietnam/1203/2004, PB1-F2, mallard ducks, H5N1, highly pathogenic influenza A virus
Introduction
Highly pathogenic avian influenza viruses of the H5 and H7 subtypes have usually been non-pathogenic in ducks. However, this pattern changed in 2002 when the H5N1 virus that had emerged in 1997 killed most of the exotic waterfowl in Hong Kong [6, 23]. Multiple segments of the viral genome, including those that encode the surface glycoproteins hemagglutinin (HA) and neuraminidase (NA) [25-27], the polymerase complex, and the nonstructural NS1 protein [10, 21, 22], have been reported to contribute to H5N1 pathogenicity in various animal models. The H5N1 strain A/Vietnam/1203/04 (VN1203) was shown to be highly pathogenic in mice, ferrets, and mallard ducks [14, 21]. Our previous comparison of the virulence of the wild-type virus (wtVN1203) and a reverse genetics–derived virus (rgVN1203) in mallard ducks showed wtVN1203 to be more lethal [14].
Here, we explored the possible causes of this discrepancy in virulence. Although the coding regions of 10 of the 11 proteins of wtVN1203 and rgVN1203 encoded identical proteins, we found that 11 synonymous mutations (SMs) had been inadvertently introduced into the PB1, 2 into the NP, and 1 into the NA gene of rgVN1203. Although SMs do not necessarily alter the aa sequence of the resulting protein, they do influence gene expression by affecting the mRNA folding [16] and the rate of protein translation [3]. Moreover, 3 of the 11 SMs, at nt positions 246, 261, and 354 of the PB1 open reading frame (ORF), resulted in aa substitutions in the N-terminal domain of PB1-F2, at positions 51, 56 and 87. PB1-F2, a recently described pro-apoptotic protein of influenza A viruses (IAVs), was discovered while searching for CD8+ epitopes in alternative reading frames of IAV genes and is encoded by an alternative (+1) ORF in the PB1 gene [2]. The C-terminal domain of PB1-F2 contains the mitochondrial signal and can trigger apoptosis in specific immune cells [2, 29]. Knockout of PB1-F2 diminished the pathogenicity of the H1N1 subtype in mice [28]. Recently, PB1-F2 has been shown to play an important role in promoting lung pathology in both primary viral infection and secondary bacterial infection in mice [18].
The facts that the PB1 subunit plays a central role in the catalytic activities of the viral RNA polymerase complex [1, 8, 9, 15, 24] and that the PB1-F2 protein contributes to viral pathogenicity [4, 5, 18, 28] led us to investigate the potential contributions of the 11 SMs in the PB1 gene, including those that encode 3 aa changes in the PB1-F2 protein, to the pathogenicity of the rgVN1203 virus in mallard ducks. Correction of the aa changes in the PB1-F2 protein and repair of the 11 SMs in the PB1 gene caused similar virus behavior, suggesting that PB1-F2 is a novel factor in VN1203 pathogenicity in this species.
Materials and methods
Generation of recombinant influenza viruses
The H5N1 IAV A/Vietnam/1203/04 (VN1203) sample was provided by the World Health Organization collaborating laboratories and stored in the repository of St. Jude Children’s Research Hospital. The virus was propagated in 10-day-old embryonated chicken eggs and handled at St. Jude in biosafety level 3+ facilities approved by the U.S. Department of Agriculture and Centers for Disease Control and Prevention. The 8 gene segments of the VN1203 virus had previously been cloned into a dual-promoter plasmid, pHW2000 [11, 21], resulting in 11 synonymous mutations in PB1, 2 in NP, and 1 in NA. The SMs in the VN1203 plasmids were repaired by using QuickChange site-directed mutagenesis kits (Stratagene) to generate a gene sequence identical to that of the wild-type (wt) VN1203 virus. Four reverse genetics (rg)-derived viruses were created by DNA transfection as described previously [12]: one with repair of all SMs in PB1, NP, and NA (VN1203/R-3), one with repair of all SMs in PB1 (VN1203/R-PB1), one with repair of the 3 SMs affecting PB1-F2 (VN1203/R-PB1-F2), and one with no repairs (VN1203/SM-3). Briefly, 293T and Madin-Darby canine kidney (MDCK) cells were co-cultured and transfected with 1 μg of each of the eight plasmids and 18 μl of transit LTI (Panvera) in a total volume of 1 ml of Opti-MEM I (Gibco, NY). The supernatant was removed 72 h post-transfection (p.t.), and 100 μl was injected into the allantoic cavity of 10-day-old embryonated chicken eggs. After 48 h, the allantoic fluid was harvested and total RNA was extracted by using an RNeasy kit (QIAGEN). Gene segments were amplified by reverse transcription-PCR, using the universal primer set for IAV [13]. Viral cDNAs were sequenced by the Hartwell Center for Biotechnology at St. Jude to confirm the identity of the virus.
Inoculation of mallard ducks
All animal experiments were approved by the Animal Care and Use Committee of St. Jude Children’s Research Hospital and performed in compliance with relevant institutional policies, the Association for the Accreditation of Laboratory Animal Care guidelines, the National Institutes of Health regulations, and local, state, and federal laws. Studies with mallard ducks were performed as described previously [14]. Briefly, groups of ten 4-week-old mallard ducks (Anas platyrhynchos) were inoculated with 106 50% egg infectious doses (EID50) of each stock virus in a 1-ml volume (0.3 ml each was applied to the trachea and the throat and 0.2 ml each to nares and eyes). All ducks were observed daily for 12 days. Ducks that exhibited severe disease signs were euthanized and recorded as having died on the following day. Tracheal and cloacal swabs were collected every other day starting on day 3 post-inoculation until virus was no longer isolated from cultured MDCK cells. The infectivity of positive samples was measured by determining the 50% tissue culture infectious dose (TCID50) after incubation at 37°C for 3 days. The limit of detection was 1 log10 TCID50/ml.
Cell lines and viral infection
DF-1 chicken fibroblast and 293T human embryonic kidney cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and antibiotics. Madin-Darby canine kidney (MDCK) cells were kept in minimal essential medium supplemented with 10% FCS and antibiotics. 293T and MDCK cells were maintained at 37°C, and DF-1 cells were incubated at 39°C. Cells were washed with phosphate-buffered saline (PBS) and incubated with virus at the indicated multiplicity of infection for 1 h. The inoculum was aspirated and cells were incubated with medium containing 4% bovine serum albumin (BSA) and antibiotics. Supernatants were collected at specific intervals for virus titration.
Dual luciferase assay of viral polymerase activity
Subconfluent monolayers of DF-1 chicken fibroblast cells or 293T human embryonic kidney cells were transfected by using transit LTI (Panvera) according to the manufacturer’s instructions with 1 μg of a firefly luciferase reporter plasmid flanked by chicken polymerase I (Pol I) promoter (pARO3-vluci) for DF-1 or human PolI promoter (pPolI A-luci) for 293T and murine terminator sites, which produces a viral RNA (vRNA)-like RNA transcript; 0.02 μg of Renilla luciferase-expressing plasmid; and a mixture of plasmids expressing PB2, PB1, PA, and NP in quantities of 1, 1, 1, and 2 μg, respectively. Cell extracts were prepared in 500 μl of passive lysis buffer (Promega) 24 h p.t. Luciferase activity was then assayed with a dual luciferase assay reagent kit (Promega) and read on a Veritas microplate luminometer (Turner BioSystems). Viral polymerase activity was expressed as the ratio of firefly to Renilla luciferase signal.
RNA isolation and analysis of viral vRNA, mRNA, and cRNA by primer extension assays
The different influenza virus RNA species produced during infection were quantified as described by Robb et al. [20]. Briefly, adherent DF-1 cells in 35-mm dishes were infected with recombinant virus at an MOI of 1. Total RNA was isolated 4, 6, 8, and 10 h p.i. by using the RNeasy kit. Total RNA (~ 1 μg) was mixed with an excess of DNA primers, labeled at the 5′ end with 30 μCi of [γ-32P]ATP and T4 polynucleotide kinase, and denatured by heating to 95°C for 3 min. The mixture was cooled to 50°C, and the primer extensions were performed after addition of 100 U of SuperScriptIII reverse transcriptase (Invitrogen) to the reaction buffer provided with the enzyme and incubation for 90 min at 50°C. Two PB1 gene-specific primers for A/VN/1203/04 were used in the separate reverse transcription reactions: 5′-TGATTTCGAGTCTGGAAGGA-3′ (to detect vRNA) and 5′-TCCATGGTGTATCCTGTCCC-3′ (to detect mRNA and cRNA). Another specific primer for detection of cellular 5s rRNA was included as an internal control for total RNA: 5′-TCCCAGGCGGTCTCCCATCC-3′. Transcription products were analyzed on 8% polyacrylamide gels containing 7 M urea in TBE buffer and were detected by autoradiography. Viral RNA bands were quantified with the PC-BAS software package (Fuji).
Statistical analysis
For the in vivo studies (Figs. 2 & 3), weight and temperature data were expressed as the percent change from day 0 for each duck. Temperature and weight changes caused by the different viruses were compared by regression with the individual duck included as a random effect to account for individual variations among the animals. Model simplification was performed and tested with parametric bootstrapping and likelihood ratio tests [7]. Because morbidity occurs in the days immediately following infection, regression analysis was restricted to time points prior to recovery. If significant effects were found, a simple effect of virus was investigated by ANOVA for each time point, followed by a Tukey-Kramer multiple comparison test, if appropriate. Kaplan-Meier analysis was performed for each survival curve. Virus titers were analyzed as the log10TCID50 values for each day. Analyses to determine the effects of various mutations on the morbidity and mortality of mallard ducks excluded the PBS control. All analysis was performed in R version 2.9.0 (www.R-project.org).
Figure. 2.
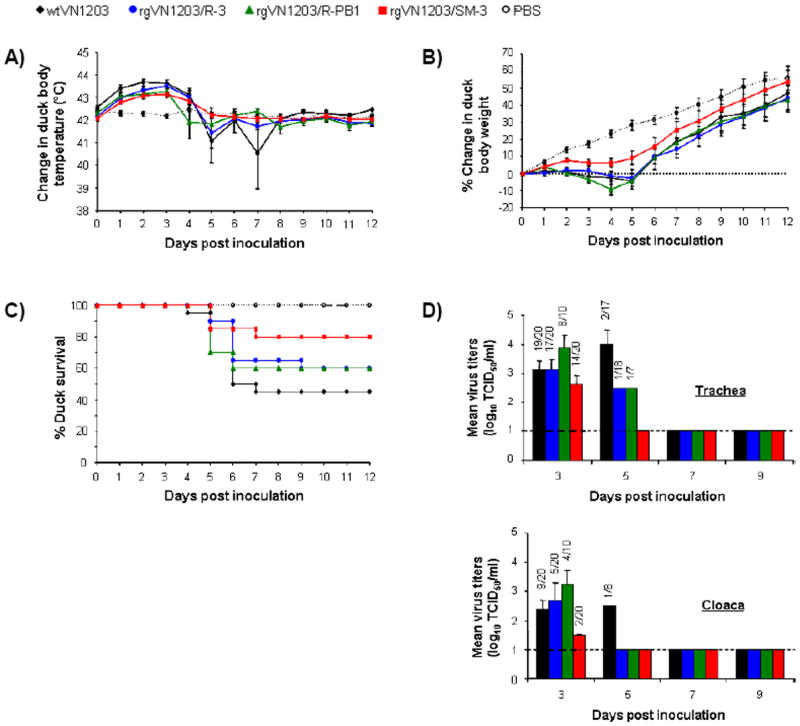
Effect of synonymous mutations on virus lethality in mallard ducks. (A) Mean ± SE body temperature change of mallard ducks a.i. (B) Mean ± SE percent weight change of mallard ducks a.i. (C) Survival rate of mallard ducks a.i. (D) Tracheal and cloacal mean ± SE virus titers of inoculated mallard ducks 3 and 5 days a.i. Dashed line indicates detection limit (1 log 10 TCID50/ml).
Figure. 3.
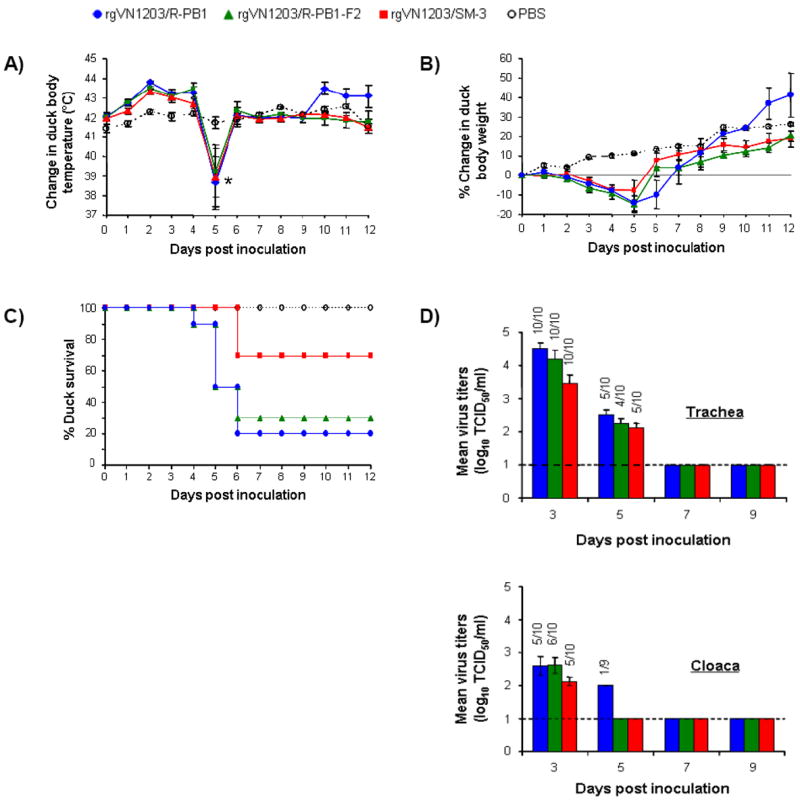
Effect of amino acid changes encoded within the PB1-F2 reading frame on virus lethality in mallard ducks. (A) Mean ± SE body temperature change of mallard ducks a.i. *The ducks showing a body temperature decrease on day 5 died the next day. (B) Mean ± SE percent weight change of ducks a.i. (C) Survival rate of ducks a.i. (D) Tracheal and cloacal mean ± SE virus titers of inoculated ducks 3 and 5 days a.i. Dashed line indicates detection limit (1 log 10 TCID50/ml).
Results
Sequence comparison of wtVN1203 and rgVN1203
To investigate the difference in pathogenicity between wtVN1203 and rgVN1203, we sequenced the entire genomes of both viruses. Although 10 of the 11 protein sequences were identically encoded by the two viruses, we found 11 SMs in the PB1 gene, 2 in the NP gene, and 1 in the NA gene of rgVN1203 (referred to hereafter as rgVN1203/SM-3) (Fig. 1A). Because most of the SMs were in PB1, a key component of the viral RNA-dependent RNA polymerase (RdRp), we first compared the codon triplet frequency at the affected nt positions in the PB1 genes of wtVN1203 and rgVN1203/SM-3. The VN1203 HA binds preferentially to α-(2,3)–linked sialic acid receptors, and therefore the codon triplets were evaluated by using PB1 of strictly avian influenza viruses (n = 1,088). The bioinformatics analysis showed significant differences in codon triplet frequency between the wtVN1203 and rgVN1203/SM-3 PB1 genes, with ratios as high as 1:112.1 (Table 1). A change at the third position of the codon triplets in PB1 of rgVN1203/SM-3 resulted in both rare and frequent codon usage. It is noteworthy that 3 of 11 SMs in PB1, at nt positions 246, 261, and 354 of the ORF, resulted in aa changes in the PB1-F2 protein: T51→M, V56→A, and E87→G, respectively (Fig. 1B).
Figure. 1.
Sequence comparison of wtVN1203 and rgVN1203. (A) The positions of synonymous mutations (SMs) in the PB1, NP, and NA genes of rgVN1203/SM-3 in comparison with the wtVN1203 nucleotide sequences. (B) Alignment and location of mutations in the PB1-F2 protein. SMs at positions 246, 261, and 354 of the rgVN1203/SM-3 PB1 open reading frame caused aa changes (red lettering) at positions 51, 56, and 87 of PB1-F2 (shown in red letters).
Table 1.
Comparison of codon usage bias at the positions of synonymous mutations in the PB1 gene of rgVN1203/SM-3 and wtVN1203 a.
| nt pos. | A/VN/1203/04 | Codon triplet | Frequency (out of 1,088) | Percent (%) | Ratio (wt : rg) |
|---|---|---|---|---|---|
| 63 | wt | ACC | 875 | 78.77 | 95.2 : 1 |
| rg | ACA | 9 | 0.83 | ||
| 246 | wt | TAC | 63 | 5.79 | 1 : 16.3 |
| rg | TAT | 1,025 | 94.21 | ||
| 261 | wt | TGT | 914 | 84.01 | 5.3 : 1 |
| rg | TGC | 174 | 15.99 | ||
| 354 | wt | AGA | 937 | 86.12 | 7.6 : 1 |
| rg | AGG | 123 | 11.31 | ||
| 516 | wt | GAG | 102 | 9.38 | 1 : 9.2 |
| rg | GAA | 931 | 85.57 | ||
| 1017 | wt | ATA | 9 | 0.83 | 1 : 112.1 |
| rg | ATT | 1,009 | 92.74 | ||
| 1809 | wt | AAC | 80 | 7.35 | 1 : 12.6 |
| rg | AAT | 1,007 | 92.56 | ||
| 1926 | wt | AAT | 1,046 | 96.14 | 58.1 : 1 |
| rg | AAC | 18 | 1.65 | ||
| 2094 | wt | AAA | 989 | 90.90 | 10.2 : 1 |
| rg | AAG | 97 | 8.92 | ||
| 2184 | wt | ATC | 17 | 1.56 | 1 : 62.9 |
| rg | ATT | 1,069 | 98.25 | ||
| 2223 | wt | GCC | 42 | 3.86 | 1 : 23.9 |
| rg | GCT | 1,005 | 92.37 |
Codon triplet frequency was evaluated by using PB1 of 1,088 strictly avian influenza viruses.
Repair of synonymous mutations in rgVN1203/SM-3 virus enhance its lethality in mallard ducks
SMs do not necessarily cause changes in the aa sequence of the resulting protein, but their presence can affect gene transcription and translation processes [3, 16]. To assess the effect of the changes in the PB1, NP, and NA genes on pathogenicity, we generated recombinant viruses with repairs in all 3 genes (rgVN1203/R-3), repairs in the PB1 gene only (rgVN1203/R-PB1), or no repairs (rgVN1203/SM-3) and compared their lethality in mallard ducks. The parental wtVN1203 virus was also included in this experiment. We first determined the pathogenicity index (PI) of each virus on the basis of the frequency of death and disease signs within 10 days after inoculation (a.i.) via natural routes with 106 50% egg infectious doses (EID50) of stock virus (Table 2). The wtVN1203 virus exhibited the highest PI (1.24), followed by rgVN1203/R-PB1 (1.01) and rgVN1203/R-3 (0.99). The PI of rgVN1203/SM-3 was the lowest (0.58) among the viruses. Severe disease signs (cloudy eyes, ataxia, and neurologic disorders) were more frequent in ducks inoculated with wtVN1203, rgVN1203/R-3, or rgVN1203/R-PB1 than in those inoculated with rgVN1203/SM-3 (Table 2).
Table 2.
Pathogenicity of wtVN1203, rgVN1203/R-3, rgVN1203/R-PB1, and rgVN1203/SM-3 in mallard ducks a.
| Virus | Pathogenicity index | No. of deaths/total no. | No. with clinical disease signs/total no. | |
|---|---|---|---|---|
| Cloudy eyes | Ruffled feathers, head twitching, ataxia, neurologic signs | |||
| wtVN1203 | 1.24 | 11/20 | 13/20 | 8/20 |
| rgVN1203/R-3 | 0.99 | 8/20 | 14/20 | 7/20 |
| rgVN1203/R-PB1 | 1.01 | 4/10 | 6/10 | 3/10 |
| rgVN1203/SM-3 | 0.58 | 4/20 | 10/20 | 3/20 |
Ducks were inoculated with 106 EID50 of virus via natural routes; n = 20, except for rgVN1203/R-PB1 (n = 10).
Inoculation with either virus caused a body temperature increase (as much as 1.5 °C greater than that of PBS-inoculated controls) within 3 days (Fig. 2A). However, after correction for the starting temperature in each group, we found no significant difference in body temperature within the first 4 days after inoculation (a.i.) (virus*time interaction, P=0.34). All ducks maintained their average body temperature from day 5 a.i. until the end of the experiment.
Body weight in the control group increased steadily during the 12 days of observation (Fig. 2B). Ducks inoculated with wtVN1203, rgVN1203/R-3, or rgVN1203/R-PB1 lost body weight during the first 5 days, whereas ducks inoculated with rgVN1203/SM-3 experienced only a brief plateau in weight gain (virus*time effect, P<0.0001). By day 4, ducks inoculated with rgVN1203/SM-3 were significantly heavier than those inoculated with rgVN1203/R-PB1 (simple effects ANOVA, P=0.03). Although survival in the inoculated groups did not differ significantly (Kaplan-Meier, P=0.08), higher mortality was observed after inoculation with wtVN1203, rgVN1203/R-3, or rgVN1203/R-PB1 than after inoculation with rgVN1203/SM-3 (Fig. 2C). The wtVN1203 virus caused the highest mortality (55%), followed by rgVN1203/R-3 and rgVN1203/R-PB1 (both 40%). The rgVN1203/SM-3 virus killed only 4 of 20 ducks (20%).
To investigate whether reduced virus replication explained the lower pathogenicity of rgVN1203/SM-3, we titrated virus shed from the trachea and cloaca (Fig. 2D). Ducks inoculated with wtVN1203, rgVN1203/R-3, or rgVN1203/R-PB1 viruses shed virus for 5 days; those inoculated with rgVN1203/SM-3 shed for only 3 days. The wtVN1203, rgVN1203/R-3, and rgVN1203/R-PB1 viruses were shed at higher levels from both the trachea and cloaca (ANOVA: trachea, P=0.39; cloaca, P=0.6). The highest titers were detected in the tracheas of ducks inoculated with rgVN1203/R-PB1 or wtVN1203 on days 3 and 5, respectively. Of the day-3 cloacal titers, that of rgVN1203/SM-3 was the lowest; on day 5, only wtVN1203 was detectable. These findings suggest that unrepaired PB1 reduced the lethality of rgVN1203/SM-3 in mallard ducks and that repair of NP and NA did not have a distinguishable effect on virus pathogenicity.
Amino acid changes encoded within the PB1-F2 reading frame of rgVN1203/SM-3 decrease lethality in mallard ducks
Because sequence analysis revealed 3 aa mutations encoded by SMs within the PB1-F2 reading frame of rgVN1203/SM-3, we investigated the role of these molecular changes in decreased lethality in mallard ducks. We generated a recombinant virus in which only these 3 SMs (at PB1 nt 246, 261, and 354) were repaired (rgVN1203/R-PB1-F2) and compared its pathogenicity with that of rgVN1203/R-PB1 and rgVN1203/SM-3. Although the PI values of all recombinant viruses were higher than those in the first experiment (Table 3), the PI of rgVN1203/SM-3 (0.78) was again the lowest. The rgVN1203/R-PB1 and rgVN1203/R-PB1-F2 viruses had PI values of 1.60 and 1.48, respectively. Inoculation with rgVN1203/R-PB1 or rgVN1203/R-PB1-F2 caused a markedly greater frequency of severe clinical signs than inoculation with rgVN1203/SM-3 (Table 3).
Table 3.
Pathogenicity of rgVN1203/R-PB1, rgVN1203/R-PB1-F2, and rgVN1203/SM-3 in mallard ducks a.
| Virus | Pathogenicity index | No. of deaths/total no. | No. with clinical disease signs /total no. | |
|---|---|---|---|---|
| Cloudy eyes | Ruffled feathers, head twitching, ataxia, neurologic signs | |||
| rgVN1203/R-PB1 | 1.6 | 8/10 | 9/10 | 5/10 |
| rgVN1203/R-PB1-F2 | 1.48 | 7/10 | 9/10 | 4/10 |
| rgVN1203/SM-3 | 0.78 | 3/10 | 4/10 | 1/10 |
Ducks were inoculated with 106 EID50 of virus via natural routes; n = 10.
All virus-inoculated ducks developed fever within 4 days a.i. (Fig. 3A), but no significant difference in body temperature was observed between the three virus groups (virus*time interaction, P=0.57). The body temperature of some ducks decreased dramatically (~3°C below the mean) on day 5, and they died on the subsequent day. Ducks in all virus groups lost body weight within 5 days a.i. (Fig. 3B), but the rate of weight loss did not differ (virus*time interaction, P=0.13). It is noteworthy that ducks inoculated with rgVN1203/SM-3 began to recover almost a full day earlier than the other ducks. The mortality rate was high in ducks inoculated with rgVN1203/R-PB1 (8/10) and rgVN1203/R-PB1-F2 (7/10) but considerably lower in ducks inoculated with rgVN1203/SM-3 (3/10) (Fig. 3C; Kaplan-Meier, P=0.02).
Tracheal swab titers on day 3 a.i. were higher in the rgVN1203/R-PB1 and rgVN1203/R-PB1-F2 groups than in the rgVN1203/SM-3 group (ANOVA, P<0.016; Fig. 3D), suggesting that the lower lethality of rgVN1203/SM-3 reflected reduced virus replication. The pattern of cloacal swab titers on day 3 was similar, although no significant difference was observed (ANOVA, P=0.24), and only rgVN1203/R-PB1 was detectable on day 5. Overall, these results suggest that the 3 aa changes in PB1-F2 contributed to the reduced lethality of rgVN1203/SM-3 in mallard ducks.
Mutations in PB1-F2 decrease viral polymerase activity
To characterize the effect of the 3 aa changes in PB1-F2 on transcription and translation of the virus genome, we compared levels of viral polymerase activity in the presence of repaired vs. unrepaired PB1-F2 in a minigenome system. IAV-like ribonucleoprotein (RNP) complexes were reconstituted in DF-1 chicken fibroblast cells or 293T human embryonic kidney cells by transfecting the cells with combinations of plasmids expressing vRNA-like RNA containing a firefly luciferase reporter gene under the control of an avian polymerase I (Pol I) promoter (for DF-1) or a human Pol I promoter (for 293T) and with other plasmids for expression of the RNP-associated viral proteins PB2, PB1, PA, and NP. Transfection efficiency was monitored in each sample by expression of the Renilla luciferase protein (dual-luciferase assay). The viral polymerase complex with unrepaired PB1-F2 had significantly less activity in DF-1 cells (20%-40% less; ANOVA, P=0.05; Fig. 4A) and 293T cells (35–50% less; ANOVA, P<0.0001; Fig. 4B) 24 h post-transfection (p.t.). The similar patterns of activity in the two cell lines showed that the activity was not cell type-dependent. These results indicate that the 3 aa changes in PB1-F2 directly or indirectly interfere with the function of the viral polymerase. These findings are similar to those observed in HEK293 cells by another group [17].
Figure. 4.
Viral polymerase activity in cultured cells. (A) DF-1 cells and (B) 293T cells were transfected with a plasmid containing an antisense firefly luciferase reporter gene flanked by a Pol I promoter and terminator sites, and with the Pol I/II-driven constructs expressing the viral polymerase proteins (PB2, PB1, PA, and NP) in the indicated combinations. Values represent the mean ± SE percentage of firefly to Renilla luciferase signal 24 h p.t. from 3 transfections. White: negative control without PB1; Red: unrepaired; Black-R: repaired PB1 or NP; Black-R/F2: corrected PB1-F2. * P≤0.05l, Dunnett’s test.
Mutations in PB1-F2 lower replication and transcription rates of viral RNAs
We next investigated whether the higher activity of RNP complexes containing corrected PB1 or corrected PB1-F2 would affect the accumulation of different species of viral RNA in virus-infected cells. DF-1 cells were infected at an MOI of 1 with rgVN1203/R-3, rgVN1203/R-PB1, rgVN1203/R-PB1-F2, or rgVN1203/SM-3. Cells were then lysed and total RNAs were isolated 4, 6, 8, and 10 h p.i. The presence of viral vRNA, cRNA, and mRNA was confirmed by primer extension assay using radioactively labeled PB1 gene-specific primers for detection of viral negative-strand and positive-strand RNAs derived from the PB1 gene segment. Small cellular 5s rRNA served as a loading control for total RNA. The 3 viral RNA species of all 4 viruses appeared to increase through 6 h p.i. (Fig. 5). The viral vRNA levels in cells infected with rgVN1203/SM-3 were lower than in those infected with the other 3 viruses (Tukey’s HSD, P<0.0001). Levels of cRNA and mRNA were lower in cells infected with rgVN1203/SM-3 than in cells infected with either rgVN1203/R-PB1 (cRNA: P<0.018; mRNA: P<0.0001) or rgVN1203/R-PB1-F2 (cRNA: P<0.044; mRNA: P<0.0001) at most time points. Viral mRNA dominated the viral RNA population and reached the highest levels 6 h after infection with rgVN1203/R-PB1 and rgVN1203/R-PB1-F2 (Tukey’s HSD: P<0.0001). Overall, viral cRNA was less abundant than vRNA and mRNA.
Figure. 5.
Viral RNA accumulation. DF-1 cells were infected with the indicated recombinant viruses at MOI = 1. Total RNA was isolated at the indicated time points and analyzed by Primer extension assays using PB1 gene-specific primers. Total RNA loading was monitored by measuring 5s rRNA expression.
Discussion
The H5N1 strain A/Vietnam/1203/04 (VN1203) belongs to the most lethal influenza A viruses tested in laboratories so far. Our findings show that a recombinant VN1203 virus containing 11 inadvertently introduced SMs in the PB1, NP, and NA genes showed decreased lethality in mallard ducks. We first sought to elucidate the role of these SMs in the reduced virulence. Mallard ducks inoculated with wtVN1203, rgVN1203/R-3, or rgVN1203/R-PB1 showed more severe disease signs, greater mortality, higher virus titers, and longer virus shedding from the trachea and cloaca than ducks inoculated with rgVN1203/SM-3. Interestingly, recombinant viruses in which all SMs in PB1 (rgVN1203/R-PB1) or only those affecting PB1-F2 (rgVN1203/R-PB1-F2) had been corrected were similarly highly lethal to the ducks. In contrast, ducks inoculated with rgVN1203/SM-3 showed less frequent disease signs and significantly less mortality. We therefore suggest that the differing lethality of wtVN1203 and rgVN1203/SM-3 was likely to be mediated by the 3 aa mutations in the PB1-F2 protein.
It remains unclear which aa substitution(s) (T51→M, V56→A, or E87→G) in PB1-F2 account for the lower pathogenicity of rgVN1203/SM-3. It is noteworthy that the majority of H5N1 viruses have M51, V56, and E87 in PB1-F2. Because E87→G is the only mutation that alters the charge of the aa (from negatively charged [acidic] to not charged [non-acidic]), the role of the aa change at this position warrants further investigation. In addition, it was recently shown that a single aa change (N66→S) in the PB1-F2 of the A/HK/156/97 (H5N1) and 1918 pandemic (A/Brevig Mission/18) influenza A viruses increases their pathogenicity in mice [5]. However, VN1203 is highly pathogenic to mice despite the presence of 66N in its PB1-F2 [21, 27]. This discrepancy suggests that the contribution of PB1-F2 to viral pathogenicity may be virus- and host-dependent.
In addition, we conducted functional assays in cultured cells in an effort to understand the molecular mechanisms involved in the biological function of PB1-F2. In chicken fibroblast DF-1 and human embryonic kidney 293T cell lines, significantly lower polymerase activity was observed in the RNP complexes containing the 3 uncorrected aa substitutions in PB1-F2. PB1-F2 is translated from an alternative reading frame of the PB1 gene segment and regulates viral polymerase activity by interaction with the PB1 protein [17]. Therefore, mutations in the PB1-F2 protein of rgVN1203/SM-3 may negatively affect the activity of the viral polymerase subunit PB1 and possibly of the entire RNP complex by impeding the interaction between PB1 and PB1-F2. The significantly lower replication rates observed in avian cells infected with rgVN1203/SM-3 multiple replication cycles is consistent with such a mechanism (data not shown). However, all recombinant viruses replicated to similar titers within 72 h after infection at a low MOI, suggesting that the 3 aa changes in PB1-F2 of rgVN1203/SM-3 did not completely attenuate, but rather only slowed, viral growth in cultured cells.
Greater polymerase activity would presumably lead to enhanced nuclear accumulation of viral RNAs in infected cells. We analyzed the levels of viral vRNA, mRNA, and cRNA in virus-infected cells by using a primer extension assay with PB1 gene-specific primers. Replication and transcription of viral RNA was increased when all SMs in the PB1-F2 reading frame or just those in PB1-F2 were corrected. Significantly less viral RNA accumulated in rgVN1203/SM-3-infected cells than in cells infected with rgVN1203/R-3, rgVN1203/R-PB1, or rgVN1203/R-PB1-F2. Therefore, the functional effects of PB1-F2 on viral polymerase activity play a role in viral RNA accumulation. McAuley et al. [19] recently reported that the effects of PB1-F2 expression on polymerase activity, PB1 accumulation, and viral replication were cell type- and virus strain-dependent. Therefore, our findings may be applicable only to the H5N1 virus strain used and to mallard ducks
Our study was limited by the absence of information about tissue tropism and systemic vs. non-systemic infection. In addition, all previous investigations of the role of PB1-F2 in virulence have used inbred mice [4, 5, 18, 28]. Because of the unavailability of inbred duck strains, the outcome of infection, particularly the mortality rate, differed between experiments. Significant differences were observed more frequently during the second experiment than during the first. However, despite this variability, rgVN1203/SM-3 was the least pathogenic of the recombinant viruses in both sets of experiments. Hence, our findings provide new insights into the significance of this viral protein in mallard ducks. Taken together, our findings identify the PB1-F2 protein of VN1203 as a novel viral factor in pathogenicity in mallard ducks.
Acknowledgments
This work was supported by the National Institutes of Health, U.S. Department of Health and Human Services, under Contract No. HHSN266200700005C, and by the American Lebanese Syrian Associated Charities (ALSAC).
We thank the World Health Organization Global Influenza Surveillance Network for providing the parental A/VN/1203/04 (H5N1) virus. We acknowledge Richard Webby for advice and suggestions; Scott Krauss, Patrick Seiler, Heather Forrest, David Carey, and Cedric Proctor for excellent technical assistance; and James Knowles for administrative assistance. We greatly appreciate the gift of pARO3-vluci plasmid by Hui-Ling Yen and of pPolI A-luci plasmid by Stephan Pleschka. Scientific editing was provided by David Galloway and Sharon Naron.
References
- 1.Biswas SK, Nayak DP. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J Virol. 1994;68:1819–1826. doi: 10.1128/jvi.68.3.1819-1826.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 3.Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S. Virus attenuation by genome-scale changes in codon pair bias. Science. 2008;320:1784–1787. doi: 10.1126/science.1155761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conenello GM, Palese P. Influenza A virus PB1-F2: a small protein with a big punch. Cell Host Microbe. 2007;2:207–209. doi: 10.1016/j.chom.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007;3:1414–1421. doi: 10.1371/journal.ppat.0030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis TM, Bousfield RB, Bissett LA, Dyrting KC, Luk GS, Tsim ST, Sturm-Ramirez K, Webster RG, Guan Y, Malik Peiris JS. Investigation of outbreaks of highly pathogenic H5N1 avian influenza in waterfowl and wild birds in Hong Kong in late 2002. Avian Pathol. 2004;33:492–505. doi: 10.1080/03079450400003601. [DOI] [PubMed] [Google Scholar]
- 7.Faraway JJ. Extending the Linear Model with R: generalized linear, mixed effects and nonparametric regression models. Chapman & Hall/CRC; Boca Raton, FL: 2006. p. 301. [Google Scholar]
- 8.Gonzalez S, Zurcher T, Ortin J. Identification of two separate domains in the influenza virus PB1 protein involved in the interaction with the PB2 and PA subunits: a model for the viral RNA polymerase structure. Nucleic Acids Res. 1996;24:4456–4463. doi: 10.1093/nar/24.22.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez S, Ortin J. Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. Embo J. 1999;18:3767–3775. doi: 10.1093/emboj/18.13.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatta M, Gao P, Halfmann P, Kawaoka Y. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science. 2001;293:1840–1842. doi: 10.1126/science.1062882. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann E, Neumann G, Hobom G, Webster RG, Kawaoka Y. “Ambisense” approach for the generation of influenza A virus: vRNA and mRNA synthesis from one template. Virology. 2000;267:310–317. doi: 10.1006/viro.1999.0140. [DOI] [PubMed] [Google Scholar]
- 12.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A. 2000;97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 14.Hulse-Post DJ, Franks J, Boyd K, Salomon R, Hoffmann E, Yen HL, Webby RJ, Walker D, Nguyen TD, Webster RG. Molecular changes in the polymerase genes (PA and PB1) associated with high pathogenicity of H5N1 influenza virus in mallard ducks. J Virol. 2007;81:8515–8524. doi: 10.1128/JVI.00435-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerry PS, Willsher N, Fodor E. A cluster of conserved basic amino acids near the C-terminus of the PB1 subunit of the influenza virus RNA polymerase is involved in the regulation of viral transcription. Virology. 2008;373:202–210. doi: 10.1016/j.virol.2007.11.030. [DOI] [PubMed] [Google Scholar]
- 16.Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazur I, Anhlan D, Mitzner D, Wixler L, Schubert U, Ludwig S. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell Microbiol. 2008;10:1140–1152. doi: 10.1111/j.1462-5822.2008.01116.x. [DOI] [PubMed] [Google Scholar]
- 18.McAuley JL, Hornung F, Boyd KL, Smith AM, McKeon R, Bennink J, Yewdell JW, McCullers JA. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe. 2007;2:240–249. doi: 10.1016/j.chom.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAuley JL, Zhang K, McCullers JA. The effects of influenza A virus PB1-F2 protein on polymerase activity are strain specific and do not impact pathogenesis. J Virol. 2009 doi: 10.1128/JVI.01785-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robb NC, Smith M, Vreede FT, Fodor E. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J Gen Virol. 2009;90:1398–1407. doi: 10.1099/vir.0.009639-0. [DOI] [PubMed] [Google Scholar]
- 21.Salomon R, Franks J, Govorkova EA, Ilyushina NA, Yen HL, Hulse-Post DJ, Humberd J, Trichet M, Rehg JE, Webby RJ, Webster RG, Hoffmann E. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. J Exp Med. 2006;203:689–697. doi: 10.1084/jem.20051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo SH, Hoffmann E, Webster RG. Lethal H5N1 influenza viruses escape host antiviral cytokine responses. Nat Med. 2002;8:950–954. doi: 10.1038/nm757. [DOI] [PubMed] [Google Scholar]
- 23.Sturm-Ramirez KM, Ellis T, Bousfield B, Bissett L, Dyrting K, Rehg JE, Poon L, Guan Y, Peiris M, Webster RG. Reemerging H5N1 influenza viruses in Hong Kong in 2002 are highly pathogenic to ducks. J Virol. 2004;78:4892–4901. doi: 10.1128/JVI.78.9.4892-4901.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torreira E, Schoehn G, Fernandez Y, Jorba N, Ruigrok RW, Cusack S, Ortin J, Llorca O. Three-dimensional model for the isolated recombinant influenza virus polymerase heterotrimer. Nucleic Acids Res. 2007;35:3774–3783. doi: 10.1093/nar/gkm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webster RG, Rott R. Influenza virus A pathogenicity: the pivotal role of hemagglutinin. Cell. 1987;50:665–666. doi: 10.1016/0092-8674(87)90321-7. [DOI] [PubMed] [Google Scholar]
- 26.Yamada S, Suzuki Y, Suzuki T, Le MQ, Nidom CA, Sakai-Tagawa Y, Muramoto Y, Ito M, Kiso M, Horimoto T, Shinya K, Sawada T, Kiso M, Usui T, Murata T, Lin Y, Hay A, Haire LF, Stevens DJ, Russell RJ, Gamblin SJ, Skehel JJ, Kawaoka Y. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature. 2006;444:378–382. doi: 10.1038/nature05264. [DOI] [PubMed] [Google Scholar]
- 27.Yen HL, Aldridge JR, Boon AC, Ilyushina NA, Salomon R, Hulse-Post DJ, Marjuki H, Franks J, Boltz DA, Bush D, Lipatov AS, Webby RJ, Rehg JE, Webster RG. Changes in H5N1 influenza virus hemagglutinin receptor binding domain affect systemic spread. Proc Natl Acad Sci U S A. 2009;106:286–291. doi: 10.1073/pnas.0811052106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol. 2006;80:7976–7983. doi: 10.1128/JVI.00415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zell R, Krumbholz A, Wutzler P. Influenza A virus PB1-F2 gene. Emerg Infect Dis. 2006;12:1607–1608. doi: 10.3201/eid1210.060511. [DOI] [PMC free article] [PubMed] [Google Scholar]