

Abstract
Since the discovery of vitamin C, the number of its known biological functions is continually expanding. Both the names ascorbic acid and vitamin C reflect its antiscorbutic properties due to its role in the synthesis of collagen in connective tissues. Ascorbate acts as an electron-donor keeping iron in the ferrous state thereby maintaining the full activity of collagen hydroxylases; parallel reactions with a variety of dioxygenases affect the expression of a wide array of genes, for example via the HIF system, as well as via the epigenetic landscape of cells and tissues. In fact, all known physiological and biochemical functions of ascorbate are due to its action as an electron donor. The ability to donate one or two electrons makes AscH− an excellent reducing agent and antioxidant. Ascorbate readily undergoes pH-dependent autoxidation producing hydrogen peroxide (H2O2). In the presence of catalytic metals this oxidation is accelerated. In this review, we show that the chemical and biochemical nature of ascorbate contribute to its antioxidant as well as its prooxidant properties. Recent pharmacokinetic data indicate that intravenous (i.v.) administration of ascorbate bypasses the tight control of the gut producing highly elevated plasma levels; ascorbate at very high levels can act as prodrug to deliver a significant flux of H2O2 to tumors. This new knowledge has rekindled interest and spurred new research into the clinical potential of pharmacological ascorbate. Knowledge and understanding of the mechanisms of action of pharmacological ascorbate bring a rationale to its use to treat disease especially the use of i.v. delivery of pharmacological ascorbate as an adjuvant in the treatment of cancer.
Keywords: Ascorbate, Oxidative stress, Hydrogen peroxide, Cancer, Pharmacology, Nutrition
1. Introduction
In the 80 years since the discovery of vitamin C (ascorbic acid, AscH2; ascorbate, AscH−) [1,2], the number of its known biological functions is continually expanding. Because of the ease of oxidation of ascorbate, gaining the first understanding of its role as the antiscorbutic vitamin was a major challenge. As a water-soluble reducing agent and donor antioxidant, AscH− can undergo two consecutive, one-electron oxidations resulting in the formation of ascorbate radical (Asc•−) and dehydroascorbic acid (DHA) (Fig. 1). Ascorbate can be regenerated from ascorbate radical and DHA, either enzymatically or non-enzymatically. Ascorbate readily undergoes pH-dependent autoxidation producing hydrogen peroxide [3]. In the presence of catalytic metals this oxidation is accelerated [4]. Ascorbate can also have pro-oxidant effects. In fact the combination of iron and ascorbate has long been used as an oxidizing system; the combination of these two reagents is referred to as the Udenfriend system and is used for the hydroxylation of alkanes, aromatics, and other oxidations [5,6]. In addition, ascorbate serves as a reducing cofactor for many enzymes, for example hydroxylases that belong to the Fe2+–2-oxoglutarate-dependent families of dioxygenases.
Fig. 1.
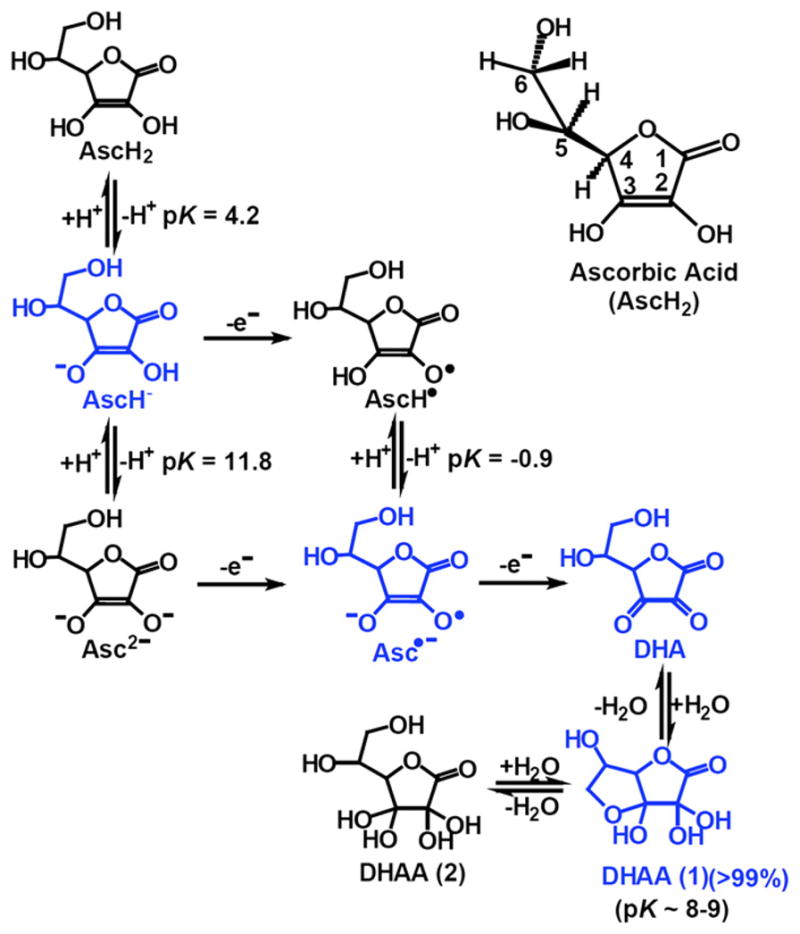
Structures of the chemical species associated with vitamin C. The structures in blue are those forms that dominate the biochemistry of vitamin C.
The uptake of ascorbate from the intestinal tract is very tightly controlled [7]. However, recent pharmacokinetic data indicate that intravenous administration of ascorbate can bypass this tight control resulting in highly elevated plasma levels [8]. Because ascorbate readily oxidizes to produce H2O2, pharmacological ascorbate has been proposed as a prodrug for the delivery of H2O2 to tumors [9–12]. This new knowledge brings insights into the controversial role of ascorbate in the treatment of cancer. In this review we present the fundamental chemistry and biochemistry of ascorbate that may have a role in the mechanisms of high dose, i.v. ascorbate in the treatment of cancer.
2. Chemistry and biochemistry
Ascorbic acid (AscH2, vitamin C) is a water-soluble ketolactone with two ionizable hydroxyl groups. It has two pKa’s, pK1 is 4.2 and pK2 is 11.6; thus, the ascorbate monoanion, AscH−, is the dominant form at physiological pH (Fig. 1). Ascorbate is an excellent reducing agent and readily undergoes two consecutive, one-electron oxidations to form ascorbate radical (Asc•−) and dehydroascorbic acid (DHA). The ascorbate radical is relatively unreactive due to resonance stabilization of the unpaired electron; it readily dismutes to ascorbate and DHA (kobs =2×105 M−1 s−1, pH 7.0) [4].
| (1) |
These properties make ascorbate an effective donor antioxidant [13].
Pure ascorbic acid is a white crystalline powder, extremely soluble in water making a colorless solution. Sodium ascorbate often contains significant quantities of oxidation products departing a yellow color. At pH values between 6 and 7.8, the purity and concentration of an ascorbate solution can be easily determined by its absorbance, ε265 =14,500 M−1 s−1 [14].
Ascorbate oxidizes readily. The rate of oxidation is dependent on pH and is accelerated by catalytic metals [14]. In the absence of catalytic metals, the spontaneous oxidation of ascorbate is quite slow at pH 7.0 [15]. This autoxidation, i.e. oxidation in the absence of catalytic metals, occurs via the ascorbate dianion, Asc2− [16]. At pH 7.0 the dominant species for vitamin C is AscH− (99.9%) with low concentrations of AscH2 (0.1%) and Asc2− (0.005%). The amount of Asc2− will increase by a factor of ten with a one unit increase in pH; this will increase the rate of autoxidation by a factor of 10. In an air-saturated phosphate buffer (pH 7.0), it has been estimated that the observed pseudo-first-order rate constant for the autoxidation of ascorbate is on the order of 10−7–10−6 s−1 at pH 7.0 [15], consistent with a rate constant of ≈300 M−1 s−1 for the true autoxidation of Asc2−, Reaction 2 [17].
| (2) |
Thus, true autoxidation is indeed very slow. In most laboratory settings, oxidation of ascorbate is catalyzed by adventitious catalytic metals in the buffers as well as metals that enter the buffer for laboratory equipment [15,18]. In near-neutral buffers with contaminating metals, the oxidation and subsequent loss of ascorbate can be very rapid.
3. Biosynthesis and uptake
Plants and most animals synthesize ascorbate from glucose. In primitive fish, amphibians and reptiles, ascorbate synthesis takes place in kidney, whereas liver is the site of synthesis in mammals. Humans, other primates, guinea-pigs and a few species of fruit-eating bats cannot synthesize ascorbate because the gene encoding L-gulonolactone oxidase (GLO), the enzyme required for the last step in ascorbate synthesis, is not functional [19]. Thus, dietary intake becomes vital, i.e. it is a vitamin.
The typical human diet contains both ascorbate and DHA; absorption occurs in the enterocytes of the small intestine. While ascorbate is accumulated in cells by Na+-dependent vitamin C transporters (SVCTs), DHA is absorbed via a Na+-independent facilitative glucose transporters (GLUTs) followed by intracellular reduction [20–24]. Under physiological conditions, DHA concentrations in plasma are very low, 2 μM or less, while plasma glucose levels are significantly higher (2–5 mM). Thus, high intracellular ascorbate concentrations are mostly due to the uptake of ascorbate by SVCT1 (SLC23A1) and SVCT2 (SLC23A2) [25,26]. Human SVCT1 (Km ≈65–240 μM) resides largely in brush-border surfaces of intestinal and renal tubular cells to mediate absorption and re-absorption [21]. Intestinal absorption and renal re-absorption determines the bio-availability of ascorbate. Because of the limited affinity of SVCT1 for ascorbate, plasma ascorbate concentrations are tightly controlled [8]. SVCT2 (Km ≈8–62 μM) is found in a range of neural, neuroendocrine, exocrine, and endothelial tissues as well as in osteoblasts [25,27]. Both isoforms of SVCT are sensitive to the changes in ascorbate levels [28,29]; it has been observed that high intracellular ascorbate lowered SVCTs in human platelets and in an intestinal epithelial cell line Caco-2TC7, while low levels up-regulated SVCTs. In addition, both SVCT1 and SVCT2 are remarkably specific for L-ascorbate; ascorbate-2-O-phosphate, DHA, glucose, and 2-deoxyglucose are not transported by SVCTs. The SVCTs have a greater affinity for L-ascorbate than D-isoascorbate; they depend on sodium to create the electrochemical gradient across the plasma membrane, thereby providing the energy required for uptake of ascorbate [27]. Recently an ascorbate analog 6-bromo-6-deoxy-L-ascorbic acid has been identified as completely specific for SVCTs but not GLUT1 or -3, the authors demonstrated that 6-bromo-6-deoxy-L-ascorbic acid was taken up through the SVCTs of human skin fibroblasts but not by neutrophils, which accumulate DHA through GLUTs [30]. It has been hypothesized that the SVCT pathway maintains intracellular levels of ascorbate under normal physiologic conditions, whereas the GLUT-mediated DHA uptake is restricted to specialized oxidizing conditions, such as those found in the surrounding milieu of the activated neutrophils [30]. Given that cancer development and progression is considered to have an inflammatory component [31], it is highly possible that ascorbate, is oxidized in the extracellular environment, and then taken up by tumor as well as stromal cells [32].
DHA is taken up with lower affinity (Km =0.8 mM) than ascorbate (Km =0.2 mM), but the maximal rates of uptake by adult human jejunum are similar for DHA and ascorbate (Vmax 28 vs. 35 pmol s−1 mg-protein−1) when glucose is absent [33]. Although the structure of DHA has similarities to glucose, the influence of glucose on DHA uptake varies between cell types. In adipocytes, neutrophils, osteoblasts and smooth muscle cells, the uptake of DHA is largely inhibited by physiological concentrations of glucose, which occurs to a lesser extent in astrocytes, enterocytes, and renal tubular cells [27]. Mammalian facilitative glucose transporters GLUT1 and GLUT3 mediate DHA transport with a similar efficiency to that of glucose. GLUT4 transports DHA with a higher affinity than 2-deoxyglucose (Km =0.98 vs. 5.2 mM) but a much lower Vmax [34]. Given that: (1) cellular uptake of ascorbate is regulated by both glucose and insulin [35,36]; and (2) ascorbate infusion has been shown to enhance glucose uptake and whole body glucose disposal in healthy subjects and diabetics [37], it is clear that more research is needed on the effects of ascorbate infusion and tissue uptake.
Human erythrocytes express a high number of GLUT1, but have no SVCT proteins [38]. The ascorbate level in erythrocytes is similar to the plasma from which they were taken [39,40]. It is speculated that erythrocytes recycle and release ascorbate to help maintain plasma levels of ascorbate. With the exception of erythrocytes, intracellular ascorbate concentrations are higher than extracellular fluids. Studies have shown that ascorbate accumulates to millimolar concentrations in neutrophils, lymphocytes, monocytes, and platelets (Table 1). In circulating lymphocytes, 4 mM can be achieved in healthy young women with oral ascorbate supplementation [41]. Intracellular ascorbate not only contributes to the intracellular reducing environment, it can also quench extracellular oxidants by transferring electrons across the plasma membrane [42]. In addition, there appears to be regulated ascorbate efflux from astrocytes to neurons, thereby bolstering antioxidant defense [43]. Furthermore, the release of ascorbate from cells may also reduce non-transferrin-bound iron, thereby stimulating the uptake of iron by cells [44].
Table 1.
Ascorbate content of human tissues.
| Tissue | Ascorbate (mmol/kg wet tissue) | Ascorbate (mM) | Ref. |
|---|---|---|---|
| Adrenal glands | 1.7–2.3 | [45] | |
| Pituitary gland | 2.3–2.8 | [45] | |
| Liver | 0.8–1 | [45] | |
| Spleen | 0.8–1 | [45] | |
| Pancreas | 0.8–1 | [45] | |
| Kidneys | 0.28–0.85 | [45] | |
| Skeletal muscle | 0.17–0.23 | [45] | |
| Brain | 0.74–0.85 | [45,226,227,234] | |
| Placenta | 0.23–0.72 | [48] | |
| Plasma (healthy) | 0.04–0.08 | [41,45,51] | |
| Red blood cell | 0.04–0.06 | [39] | |
| Neutrophil | 1.2 | [41] | |
| Lymphocyte | 4.0 | [41] | |
| Monocyte | 3.2 | [41] | |
| Platelet | 3.7 | [41,56] | |
| Cerebral spinal fluid | 0.15–0.25a | [235–237] | |
| Neuron | 10 | [238] | |
| Glial cells | 1 | [238] | |
| Lens | 2.5–3.4 | [46] | |
| Corneal epithelium | 12.5 | [49] | |
| Aqueous humor | 0.4–1.1 | [46] | |
| Alveolar macrophage | 0.32 | [50] | |
| Bronchoalveolar lavage | 0.04–0.06 | [51] | |
| Saliva | 0.04–0.05 | [45] |
Ascorbate levels in cerebral spinal fluid are approximately three to four times that of plasma.
In human and animal tissues, the highest concentrations of ascorbate are in the adrenal and pituitary glands (Table 1) [45–48]. Ascorbate is a physiological reductant in the dopamine β-hydroxylase-catalyzed reaction that converts dopamine to norepinephrine in the chromaffin granules of adrenal medulla. Likewise, peptidyl glycine α-amidating monooxygenase in the pituitary glands also requires high concentrations of ascorbate to catalyze the amidation reaction of carboxyl-terminal glycine residue of a number of peptides. In addition, millimolar levels of ascorbate also found in eye tissues [46,49]; this may protect these tissues from potential damage caused by solar radiation. Since tissues from lungs are constantly exposed to relatively high concentrations of oxygen and ROS, antioxidant enzymes and low-molecular-weight antioxidants, such as ascorbate, serve to protect cells and tissues from oxidative damage. Ascorbate levels in respiratory tract lining fluids are similar or lower than those in plasma [50]. However, there appears to be increased ascorbate content in the epithelial lining fluid and alveolar macrophage of smokers who have a relatively high dietary intake of vitamin C [51]. This increased content of ascorbate suggests that there is an adaptive defense mechanism against the ROS derived from cigarette smoke.
Human cell culture experiments usually lack ascorbate, as these cells are incapable of synthesizing this vitamin; in addition, there is no source of vitamin C in typical cell culture media formulations. However, cultured cells take up ascorbate, ascorbate-2-phosphate, as well as DHA when supplemented in the media. DHA is rapidly transported into cells by glucose transporters over a period of minutes while ascorbate and its phosphate require SVCTs and can take several hours [52]. Ascorbate 2-phosphate is most likely hydrolyzed by membrane esterases with the resulting ascorbate being transported into cells [53]. Compared to the same concentration of ascorbate, cells accumulate intracellular ascorbate via ascorbate 2-phosphate at a somewhat slower rate to reach the same intracellular concentration [54]. However, ascorbate 2-phosphate does not oxidize in the media and thus does not produce H2O2 as does ascorbate. Thus, the use of ascorbate 2-phosphate is preferred when studying the biology of vitamin C in cell culture. Millimolar concentrations of ascorbate can be achieved intracellularly by carrier-mediated mechanisms (Table 2). There is no detectable level of DHA in cells as intracellular DHA is readily reduced by an array of biological reductants and enzyme systems [55,56].
Table 2.
Ascorbate in cultured cells.
| Cell culture | Treatment | Intracellular ascorbate (mM) | Ref |
|---|---|---|---|
| A431 (human epidermoid carcinoma) | 1 mM ascorbatea, 18 h | 1 | [239] |
| Adj.PC-5 mouse plasmacytoma | 200 μM Ascb, 12 h | 7 | [240] |
| Alveolar macrophage (rat) | 0.1 mM Asc, 30 min | 3.2 | [241] |
| Alveolar type II epithelial cells (rat) | 0.1 mM Asc, 30 min | 3.2 | [241] |
| Astrocytes (rat) | DHAc, 100 μM, 10 min | 1.5 | [248] |
| B lymphocytes | 250 μM Asc, 4 h | 0.7–1.5 | [242] |
| EA.hy926 endothelial cells | 0.3 mM Asc, 60 min | 3–4 | [244] |
| HepG2 hepatic epithelial cells | 32 μM 14C-Asc, 3 min | 1.1 | [245] |
| HL-60 | 0.4–3 mM ascorbic phosphate | 0.5 | [246] |
| HUVEC | 1 mM ascorbate, 18 h | 3.5 | |
| INS-1 (rat pancreatic β-cell) | 0.4 mM Asc, 16–18 h | 2.4 | [247] |
| K562 (human erythroleukemia) | 500 μM DHA, 30 min | 3 | [248] |
| L6 skeletal myocytes (rat) | 100 μM DHA, 10 min | 12.5 | [242] |
| MIA PaCa-2 pancreatic cancer | 50 μM Asc, 4 h | 6 | d |
| 1 mM Asc, 4 h | 8 | ||
| 15 mM Asc, 4 h | 18 | ||
| Murine microvascular endothelial cells | 0.5 mM Asc, 12 h | 5 | [249] |
| Osteoblasts (mouse) | 1 mM Asc, 24 h | 8 | |
| Primary hepatocytes | 32 μM 14C-Asc, 3 min | 0.41 | [245] |
| SH-SY5Y neuroblastoma | 2 mM Asc, 16–18 h | 6 | [250] |
| Skin fibroblasts | 1 mM ascorbate, 18 h | 0.4 | [239] |
| U937 (human histocytic leukemia) | 50 μM Asc or Asc-2-phosphate, 24 h | 4 | [54] |
| U937 | 1 mM Asc or Asc-2-phosphate, 24 h | 7 | [54] |
Ascorbate.
L-Ascorbic acid.
Dehydroascorbate.
Thomas J van’t Erve, personal communication.
At physiological conditions DHA rapidly hydrolyzes to 2,3-L-diketogulonate (2,3-DKG), which is quite unstable [57]. 2,3-DKG subsequently decarboxylates yielding L-xylonate and L-lyxonate; these compounds can enter the pentose phosphate pathways [58]. Alternatively, L-erythrulose and oxalate can be produced from 2,3-DKG degradation. It has been proposed that highly reactive L-erythrulose rapidly glycates and crosslinks proteins. In fact, ascorbate-dependent modification of proteins was proposed to occur in human lens during diabetic and cataract formation. Oxalate monoalkylamide, one of the advanced glycation end products (AGEs) is a major product induced by incubation of human lens protein with DKG and other degradation product of ascorbate [59]. Oxalate is one of the major end products of ascorbate breakdown in humans, and it has the potential to crystallize as calcium oxalate in the urinary space in susceptible people [60,61]. However, in a study involving 16 cancer patients who had normal renal function, intravenous administration of ascorbate ranging from 0.2 to 1.5 g kg−1 body weight, resulted in approximately 30 to 80 mg of oxalic acid being excreted during the 6 h after the infusion [62]. While in primary hyperoxaluria, oxalic acid excretion ranged between 100 mg d−1 and 400 mg d−1. Oxalate nephrocalcinosis and calcium oxalate stones develop over months to years [63]. Overall, these studies suggest that patients with normal renal functions and a time-limited course of ascorbate infusions would not have an immediate risk of oxalate stone formation.
4. Biological functions
4.1. Ascorbate as enzyme cofactors of hydroxylases
In addition to the monooxygenases mentioned above, ascorbate is involved in many physiological and biochemical processes involving enzymatic reactions that are catalyzed by members of the Fe2+–2-oxoglutarate-dependent family of dioxygenases. These dioxygenases use 2-oxoglutarate as a co-substrate and as a source for two of the four electrons needed for the reduction of O2; they transfer one oxygen atom from O2 to the succinate product and one atom to the protein substrate. Ascorbate is required for maintaining iron in the ferrous state, thereby maintaining full activity of this class of enzymes. Reaction 3 [64,65].
| (3) |
Evidence that the Fe2+–2-oxoglutarate-dependent dioxygenases can modify proteins was gathered from the studies of hydroxylases in collagen metabolism. Located within the lumen of the endoplasmic reticulum, collagen prolyl-4-hydroxylase and prolyl-3-hydroxylase are responsible for the insertion of the hydroxyl groups onto prolyl residues, which is essential for the proper assembly of collagen [66]. There are at least three isozymes of collagen prolyl hydroxylase in humans each with distinct α subunits; but they all have protein disulfide isomerases (PDIs) to serve as β subunits, which catalyze the formation of intra- and inter-chain disulfide bonds during collagen synthesis [67]. Collagens are major constituents of the extracellular matrix (ECM), constituting ≈30% of total protein mass. Type I collagen is the major structural protein in the interstitial ECM, while type IV is prevalent in basement membrane [68]. Collagens in the extracellular matrix are important components of the physical barrier against invasion and metastasis of cancer cells [88,89]. Ascorbate has been shown to stimulate the production of types I and III collagen in human skin fibroblasts [70]. Cameron and Pauling proposed that mega doses of ascorbate would inhibit cancer growth by preventing cancer cell invasion [71]. More recently, it has been shown that a type IV collagen domain, produced as a recombinant protein, inhibits the proliferation of capillary endothelial cells and blood vessel formation, resulting in the suppression of tumor growth [72]. However, it is not known if very high levels of ascorbate will influence cancer growth by mechanisms related to collagen synthesis.
4.2. Ascorbate as a cofactor in the HIF system
As novel members of the dioxygenase family, cytoplasmic prolyl hydroxylases have been shown to regulate hypoxia-inducible transcription factor (HIF) [73]. These enzymes have no PDI subunits but have higher values of Km for oxygen than collagen prolyl hydroxylases. HIF protein is composed of an oxygen-sensitive HIF-α subunit and an oxygen-insensitive HIF-β subunit. In hypoxia, HIF-α is induced, enters the nucleus, and forms a heterodimer with HIF-β, which is constitutively expressed. In normoxia, HIF-α is efficiently hydroxylated by prolyl hydroxylases. Hydroxylated HIF-α then binds to the von Hippel–Lindau (pVHL) protein, which is part of an ubiquitin ligase complex, leading to ubiquitination and proteasomal degradation of the protein [74]. All three of the HIF prolyl hydroxylases (PHD1, PHD2, and PHD3) recognize the C-terminal hydroxylation site with a higher affinity than the N-terminal hydroxylation site [75]. In addition, an asparaginyl hydroxylase inhibits HIF through the hydroxylation of a C-terminal asparagine residue, blocking coactivator recruitment. Given that there are more than 800 direct HIF target genes yielding more than 60 gene products that are regulated by HIF [76–78], it is likely that the availability of ascorbate will have a profound effect on many cell functions such as angiogenesis, cell survival, glucose uptake, glycolysis and iron homeostasis.
As members of the Fe2+–2-oxoglutarate-dependent dioxygenase families, HIF prolyl- and asparaginyl hydroxylases require ascorbate as a reducing agent to maintain iron in the ferrous state. Values of Km for ascorbate with the HIF prolyl hydroxylases range from 140 to 300 μM [65], well below the millimolar concentrations of ascorbate in most of cells in healthy individuals [55]. Supplementation of ascorbate in a lymphoma cell model in SCID mice has been shown to diminish HIF levels and the rate of tumor growth [79]. Lu et al. demonstrated that the antitumorigenic effect of ascorbate is dependent on the activity of HIF hydroxylases [80]. By analyzing endometrial tumor tissues for ascorbate and three of its targets GLUT-1, BNIP3 and VEGF, Kuiper et al. [81] found that HIF-1α and its targets were all up-regulated in human tumor tissue. Tumors with higher ascorbate levels had low HIF-1 activation, while tumors with high HIF-1 activation and aggressive characteristics had lower ascorbate levels. These important findings indicate that having high tissue levels of ascorbate may protect against HIF-1 activation and aggressive tumor behavior.
As a downstream factor produced upon activation of HIF, VEGF plays an important role in tumor angiogenesis. In addition to direct cytotoxicity effects, pharmacological ascorbate has been shown to suppress capillary-like tube formation on cells grown on Matrigel [82]. However, it is still unknown whether the suppression of angiogenesis by pharmacological ascorbate is due to the production of H2O2 [83] or through the inhibition of HIF [84].
4.3. Ascorbate as a cofactor in histone demethylation
The newly discovered histone demethylases containing Jumonji catalytic domains, which can demethylate trimethylated lysines, also belong to the Fe2+–2-oxoglutarate dioxygenase family [85]. As with other members of this family, the Jumonji proteins have the ability to bind Fe2+, use 2-oxoglutarate as substrate, and require ascorbate for full catalytic activity. Furthermore, ascorbate supplementation also leads to increased histone acetylation as well as increased expression of HIF histone demethylases JMJD 1 and 2 in human embryonic stem cells [86]. Thus, we can speculate that epigenetic status of tissue may also depend on ascorbate to keep Fe2+–2-oxoglutarate dioxygenases functional [87]. Indeed, Esteban et al. [88] also postulated that ascorbate supplementation may enhance the function of epigenetic modifiers during reprogramming of induced pluripotent stem cells (iPSCs), as many of these modifiers are ascorbate-dependent dioxygenases. Considering that cancer cells have shared features of normal stem cells [89] and DNA hypermethylation is associated with cancer development, ascorbate and other nutritional supplements could prevent cancer by modulating the components of the systems that set the epigenetic code [90].
The necessity of ascorbate as a reducing agent to maintain full functioning of an array of enzymes shows the importance of vitamin C in maintaining the biochemical machinery of cells and tissues. These functions are often overlooked as many researchers focus only on the antioxidant abilities of ascorbate.
4.4. Ascorbate and cancer prevention
Epidemiologic evidence suggests that vitamin C-rich foods play a protective role against development of cancer [91–94], plasma concentrations of ascorbate have been shown to be inversely associated with cancer risk [95,96]; however, large scale randomized intervention trials comparing antioxidants (vitamins A, C, E and β-carotene) supplements given alone or in combination have not shown protective effects [97–99]. One of the drawbacks of randomized control trials (RCT) for nutrients is that most nutrients such as vitamins have threshold behavior [100]. For example, scurvy only develops when serum ascorbate levels are on the order of 8 μM or less [8]. The recommended dietary allowance (RDA) for vitamin C is 75–125 mg daily; consistent intake of 60 mg d−1 will in general prevent scurvy for 30–45 days once vitamin C intake ceased [101,102]. The fact that ascorbate is required to maintain full function of an array of enzymes indicates optimizing intake would optimize metabolism as well as prevent cancer and other degenerative disease [103,104].
5. Recycling of ascorbate
5.1. Reduction of ascorbate free radical
There are several enzymatic systems involved in the recycling of ascorbate radical (Asc•−) and DHA. The reduction of ascorbate radical by NADH-cytochrome b5 reductase has been shown in rat liver [105], where most of the reductase activity is localized in the outer mitochondria membrane. As a membrane-bound protein, NADH-cytochrome b5 reductase is also associated with the endoplasmic reticulum and the plasma membrane [106]. Cytochrome b5 donates an electron to ascorbate radical, which in turn, draws electrons from NADH. On the other hand, the transmembrane ascorbate radical reductase in erythrocytes can use electrons from NADH or intracellular ascorbate to regenerate extracellular ascorbate from ascorbate radical [107]. The plasma membrane ascorbate radical reductase reduces ascorbate radical generated in blood during oxidative stress as well as the ascorbate radical generated by reduction of α-tocoperoxyl radical during lipid oxidation. Reduction of ascorbate radical by thioredoxin reductase (TrxR) purified from rat liver suggests that TrxR can also function as a cytosolic ascorbate radical reductase, which may complement cellular ascorbate recycling. Thus, there are several biochemical routes for the one-electron reduction of ascorbate radical to regenerate ascorbate.
5.2. Reduction of dehydroascorbate
DHA is unstable at physiological pH with a half-life is 5–15 min at 37 °C [108–110]. It is estimated that the concentration of DHA is less than 2 μM in healthy humans; many reported measurements may have overestimated DHA levels due to the ease of oxidation of AscH− during sample processing. Extracellular DHA can be imported into cells using glucose transporters present in cells, where DHA can be reduced to ascorbate by glutathione (GSH) directly [111] or more efficiently by glutaredoxin in the cytoplasm [112]. Purified rat liver thioredoxin reductase also catalyzes NADPH-dependent reduction of DHA [113]. Thus, electrons from NADPH can find their way to DHA via thiols.
Purified bovine liver PDI has been found to react directly with DHA and GSH to catalyze the reduction of DHA to ascorbate [112]. The finding that PDI has DHA reductase activity is interesting since human collagen prolyl 4-hydroxylase has PDI as a β subunit [69]. It is possible that the β subunit also serves as DHA reductase to generate ascorbate from DHA since collagen hydroxylases require ascorbate to maintain iron in its ferrous state.
3α-Hydroxysteroid dehydrogenase belongs to the family of oxido-reductases that catalyze NADPH-dependent oxidoreduction of a variety of substrates. Del Bello et al. demonstrated that the NADPH-dependent DHA reductase from rat liver cytosol is identical to 3α-hydroxysteroid dehydrogenase [114]. The enzyme has a low affinity for DHA (Km = 4.6 mM) but a high affinity for NADPH (Km < 5 μM). Under pathological conditions, decreases in GSH and increases in DHA could activate 3α-hydroxysteroid dehydrogenase.
Thus, DHA has many routes for its two-electron reduction to regenerate ascorbate. Because of the relative instability of DHA, rapid reduction to ascorbate also prevents loss of vitamin C. Ascorbate is involved in a variety of biochemical functions that are fundamental to the functioning of a wide variety of enzyme systems; thus efficient recycling maintains the pool of ascorbate in cells and tissues.
The regeneration of ascorbate from ascorbate radical and DHA will allow many cycles of ascorbate oxidation, thus one molecule of ascorbate can produce many molecules of H2O2. This mechanism is an important one when considering pharmacological ascorbate in cancer treatment that will be discussed later.
6. Ascorbate as an antioxidant
The typical concentration of ascorbate in plasma from healthy humans is about 40–80 μM (Table 1). At these levels ascorbate functions as an endogenous antioxidant; for example, it serves as a co-antioxidant with vitamin E to protect LDL from detectable oxidative damage induced by aqueous peroxyl radicals [115]. Thermodynamically ascorbate readily donates an electron to potentially damaging oxidizing radicals such as hydroxyl radical (HO•), alkoxyl radical (RO•), peroxyl radical (LOO•), thiol radical (GS•), and tocopheroxyl radicals (TO•) [13]; this one-electron oxidation of AscH− results in the production of the ascorbate radical (Asc•−). Ascorbate radical is relatively unreactive, and can be reduced back to ascorbate by NADH- and NADPH-dependent reductases [116]; Asc•− can also undergo a pH-dependent disproportionation reaction, resulting in the formation ascorbate and DHA, Reaction 1 [117,118].
Physiological concentrations of ascorbate have been shown to inhibit LDL oxidation induced by endothelial cells, macrophages, copper ions, and AAPH (2,2′-azo-bis (2-amidinoproprane) dihydrochloride) [119,120]. An important feature of the action of ascorbate is its synergistic interaction with vitamin E [121]. Vitamin E is lipid-soluble; it is a primary antioxidant in LDL and lipid membrane oxidation [122]. Its one-electron oxidation product, the α-tocoperoxyl radical, can be reduced by ascorbate. The two-electron oxidation product, tocopherol quinone, undergoes ring breakage and thus vitamin E is lost [123]. In this context, ascorbate is important for maintaining vitamin E and inhibiting lipid oxidation. It has been shown that doubling plasma ascorbate concentrations by supplementation results in decreased rates of disappearance of vitamin E in smokers [124]. This is the first in vivo demonstration that ascorbate maintains vitamin E, most likely through “recycling”.
| (4) |
| (5) |
Ascorbate is clearly a co-antioxidant protecting lipid oxidation even in iron-loaded human plasma [125]. In human plasma lipid peroxides (cholesteryl ester hydroperoxides) were detectable only when serum ascorbate was depleted; iron-induced lipid peroxidation was inhibited by ascorbate in a concentration dependent manner [126]. Guinea pigs with iron overload had greater oxidative lipid damage that is suppressed with oral supplementation of ascorbate [127]. Thus, in the presence of catalytic iron, robust nutritional levels of ascorbate protect from oxidative damage.
7. Pro-oxidant effects of ascorbate
In the presence of catalytic metal ions, ascorbate can also exert pro-oxidant effects [14,128,129]. Ascorbate is an excellent one-electron reducing agent that can reduce ferric (Fe3+) to ferrous (Fe2+) iron, while being oxidized to ascorbate radical (Reaction 6). Depending on co-ordination environment, Fe2+ can readily react with O2, reducing it to superoxide radical (Reaction 7), which in turn dismutes to H2O2 and O2 (Reaction 8).
| (6) |
| (7) |
| (8) |
In a classic Fenton reaction, Fe2+ reacts with H2O2 to generate Fe3+ and the very oxidizing hydroxyl radical, Reaction 9. The presence of ascorbate can allow the recycling of Fe3+ back to Fe2+, which in turn will catalyze the formation of highly reactive oxidants from H2O2.
| (9) |
Depending on concentrations, the effects of ascorbate on models of lipid peroxidation can be pro- or antioxidant [14,130]. There is considerable variability in the literature; this variability appears to be a result of the different concentrations and form of transition metal ions in the experiments and the media [131].
The prooxidant effects of ascorbate may be important in vivo depending on the availability of catalytic metal ions. In healthy individuals, iron is largely sequestered by iron binding proteins such as transferrin and ferritin [132,133]. Transferrin is a glycoprotein that is synthesized in the liver. It is the major circulating iron binding protein with a high affinity, but low capacity for iron; this iron is essentially redox inactive [134]. Transferrin avidly binds to the transferrin receptor on the cell surface. The transferrin–transferrin receptor complex is internalized to an endosome, which releases iron in acidic conditions. The ferric iron released is reduced and transported to the cytoplasm where it is either utilized for synthesis of iron-containing proteins or bound in ferritin, an iron storage protein. Ferritin is capable of sequestering up to 4500 atoms of iron, but is normally only 20% saturated. Iron stored in ferritin can be released by appropriate reductants in the presence of a chelator or by degradation of ferritin in the lysosome.
Iron can be released from ferritin by biological reductants such as thiols, ascorbate and reduced flavins [135]. The released iron enables cells to synthesize cytochromes and iron-containing enzymes. However, uncontrolled release of iron from ferritin has the potential to form HO•, which can damage critical cellular components [136–138].
In pathological situations, such as thalassaemia or hemotochromatosis, non-transferrin-bound iron is present. Thus, supplemental ascorbate without administration of an iron chelator can lead to deleterious effects [14]. Tissue damage resulting from ischemia/reperfusion is another example of increased availability of catalytic metal occurring in vivo [139]. Intravenous ascorbate prior to vascular surgery increased concentrations of ascorbate radical and lipid hydroperoxides suggesting that catalytic iron released into the circulation during the ischemic phase of the surgery with ascorbate may promote iron-induced lipid peroxidation [140,141].
Elevated levels of catalytic metal ions have also been demonstrated in chronic inflammatory diseases [142]. There is an increased deposition of iron proteins in the synovial membranes in rheumatoid arthritis. Ascorbate radical has been detected in synovial fluid from patients with synovitis disease indicating that catalytic iron is in part responsible for the decreased levels of ascorbate and increased levels of DHA [141]. In addition, ascorbate concentrations were decreased while levels of catalytic iron increased in patients with sepsis, compared to healthy subjects [143].
Although there is no direct evidence that catalytic iron is increased in tumors, many patients with malignant disease have elevated serum or tissue ferritin concentrations [144–146]. Raised levels of circulating ferritin are found in childhood Hodgkins lymphoma and are associated with poor survival [147]; serum ferritin levels have been shown to be related to the stage of the disease and tumor volume in cervical cancer [148]. Furthermore, studies by Feng et al. [149] have shown that the peritoneal and subcutaneous microvessels of normal mice were largely impermeable to circulating ferritin, but in mice bearing solid or ascites tumors, circulating ferritin was found in the basal lamina and extravascular space, suggesting that tumor vessels are hyperpermeable to circulating macromolecules such as ferritin. In fact, ferritin staining was detected in stroma and histiocytes surrounding neoplastic cells of breast carcinoma tissue of patients [150]. Extracellular metal-containing proteins have been proposed to be essential for the pro-oxidant effects of ascorbate [10,129], iron-saturated ferritin could be a potential candidate as source of catalytic iron. A recent study by Deubzer et al. [151] demonstrated that ferritin released by neuroblastoma cells enhanced pharmacologic ascorbate induced-cytotoxicity, indicating that ferritin with high iron-saturation could be a source of catalytic iron [152]. Consistent with this, ascorbate has also been shown to be capable of releasing iron from cellular ferritin [158]. Ferritin is only one candidate as a source of catalytic iron; extracellular iron chelates are present in tissue and seem to be increased under pathological conditions [159]. It is clear that only low levels of catalytic metals are needed to substantially increase the rate of oxidation of ascorbate [15,18]. These many observations provide insights on the mechanism by which pharmacologic concentrations of ascorbate have potential in treating certain types of cancer.
8. Ascorbate and cancer treatment
The use of high-dose ascorbate in treating cancer patients began in the 1970s. These early studies demonstrated beneficial effects of high-dose ascorbate [155–158]. However, two double-blinded, randomized clinical trials at the Mayo Clinic did not show any benefit [169,160]. Subsequently, use of ascorbate for cancer treatment was considered ineffective and dismissed by the research and medical communities. However, a marked difference existed in these studies. Cameron’s group gave patients ascorbate intravenously as well as orally, while patients in the Mayo Clinic trials received only oral ascorbate. Some years later, clinical data were generated that demonstrated that when ascorbate is given orally, plasma concentrations are tightly controlled [101]. At oral doses of 200 mg, the steady-state plasma concentrations are ≈80 μM. As doses exceed 200 mg, relative absorption decreases, urine excretion increases and the fraction of bioavailable ascorbate is reduced [7]. Peak plasma values do not exceed ≈220 μM even after maximum oral dose of 3 g 6 times daily [8]. In contrast, when ascorbate is administered intravenously, millimolar concentrations can be achieved. In fact, infusion of 10 g of ascorbate in cancer patients results in plasma concentrations of 1 to 5 mM [161,162]. Thus, only intravenous administration of ascorbate can yield high plasma levels, i.e. pharmacological levels.
8.1. The role of hydrogen peroxide generation and removal
In vitro, the toxicity of ascorbate centers on the generation of H2O2 by ascorbate upon its oxidation [9,10,12,163,164]. The true autoxidation of ascorbate, i.e. in the absence of catalytic metals, via reaction 2 will generate considerable amounts of H2O2 when ascorbate is at millimolar levels. For example, in an aqueous solution containing 20 mM ascorbate at pH 7.4, the concentration of ascorbate dianion, Asc2−, will be on the order of 1 μM. This will result in a flux of H2O2 on the order of 10 nM s−1, in a typical cell culture experiment.
Catalytic iron (and perhaps copper) in the media and serum will increase the rate of ascorbate oxidation and associated generation of H2O2. Clement et al. found that Dulbecco’s modification of Eagle’s MEM (DMEM) generates more H2O2 than RPMI 1640 during a 6-hour incubation with increasing concentrations of ascorbate [165]. In addition to adventitious iron, DMEM has 0.25 μM Fe (NO3)3 in its formulation, which could contribute to this greater production of H2O2 in DMEM than that from RPMI. Serum usually contains total iron at a range of 10 to 50 μM, which could also be another factor that causes the variable toxicity of ascorbate in different media; however, some of this iron will be redox inactive as it will be sequestered in transferrin [134]; there will undoubtedly be a highly variable amount of iron present that has nonspecific peroxidase activity [166]. This iron will actively catalyze the oxidation of ascorbate. Thus, the amount of catalytic iron present in typical cell culture media is highly variable. Even with careful attention to detail, this can lead to considerable variability in experimental results, even in the same set of experiments.
α-Keto acids such as pyruvate and α-ketoglutarate directly react with H2O2 when present in cell culture media [172];
| (10) |
this will result in an apparent lowering of the toxicity of ascorbate [168]. Thus, the presence of α-keto acids in media must be taken into consideration in the design and interpretation of data from experiments where the oxidation of ascorbate and associated production of H2O2 are central to the study.
Because cells rapidly and efficiently remove extracellular H2O2, observed toxicities or cell killing induced by ascorbate is an inverse function of cell density [151,153,169]. There is an array of intracellular antioxidant enzymes involved in the removal of H2O2, such as catalase, glutathione peroxidase, and the peroxiredoxins. Although modest catalase activity has been detected in heart mitochondria [170], catalase is predominantly located in peroxisomes of mammalian cells. Concomitant administration of the catalase inhibitor amino-1,2,4-triazole has been shown to enhance ascorbate toxicity to Ehrlich ascites cells in vitro [171]. Using adenovirus constructs containing human catalase cDNA, Du et al. [12] demonstrated that overexpressing intracellular catalase protected cells from ascorbate-induced cytotoxicity. These results provide evidence supporting a fundamental role for H2O2 in ascorbate-induced toxicity.
Cytosolic glutathione peroxidase (GPx1), another antioxidant enzyme that removes peroxide, is widely distributed in tissues. GPx1 catalyzes GSH-dependent reduction of H2O2 to water. Gaetani et al. [172,173] have shown that the intracellular concentration of H2O2 in human erythrocytes is inversely proportional to the activity of catalase and GPx-1. GPx1 is responsible for eliminating low concentrations of H2O2; however, at high fluxes of H2O2 recycling of GPx1 by GSH becomes rate-limiting [174]; at high fluxes of H2O2 catalase is the enzyme that is principally responsible for removal of H2O2 [175].
Another antioxidant system that contributes to removal of H2O2 within cells is the family of peroxiredoxins, in particular, peroxiredoxin 2 (Prx2). Prx2 is the third most abundant protein in erythrocytes [176]. The rate constant of human Prx2 reacting with H2O2 is 1.3×107 M−1 s−1 [177] which is similar to that of catalase (1.7× 107 M−1 s−1 [178]). These complementary enzymes systems for the removal of intracellular H2O2 in RBCs will also make these cells efficient sinks for H2O2.
8.2. Ascorbate-induced cytotoxicity in vitro
Chen et al. [9,11] have demonstrated that some cancer cells have increased sensitivity to ascorbate-induced cytotoxicity compared to normal cells. In a complementary study, Du et al. [12] demonstrated that pancreatic cancer cells are more sensitive to pharmacological concentrations of ascorbate than their normal cell counterparts. The difference in sensitivity between normal and cancer cells towards ascorbate may be due to low levels of antioxidant enzymes and high endogenous levels of ROS in cancer cells [179–181]. The relative lower activities of catalase, glutathione peroxidase, and peroxiredoxins in cancer cells could potentially contribute to less efficient removal of H2O2 and increased sensitivity to ascorbate-induced cytotoxicity.
The rate of oxidation of ascorbate is typically a function of the level of catalytically active iron and copper in solution. Iron in cell culture media contributes significantly to the rate of H2O2 generation. Deferoxamine (Desferal® or DFO) is an iron-chelating agent that renders iron catalytically inactive with respect to ascorbate oxidation [15]. Although hydrophilic, DFO is cell-permeable; short-term exposure to DFO is not effective in accessing cytosolic labile iron pool (LIP) of cells from different origins [182]. However, short-term pre-incubation of cells to DFO appeared to protect cells from ascorbate-induced toxicity [184]. These observations indicate that DFO either is effective in accessing endosomal iron [182] or iron associated with cellular membranes [183]. Other cell culture studies have demonstrated that Fe2+ in the media can actually protect cells from the oxidative damage resulting from exposure to extracellular H2O2 [184,185]. Although hydroxyl radicals are produced via the Fenton reaction, most will not induce cell damage, but rather disappear by reactions with components of the media. The key is that H2O2 is removed. Thus, interpretation of data from experiments with iron, ascorbate, H2O2 or some combination is not always straightforward.
Oxidative stress has been shown to increase the levels of catalytic iron in tissues. A series of electron paramagnetic resonance (EPR) studies demonstrated that ultraviolet light increased the levels of labile iron in skin [186]; heat stress-induced oxidative events increased the level of labile iron in liver [187]; and ischemia reperfusion increased the level of catalytic iron in the heart [188]. In addition, ionizing radiation and some chemotherapeutic drugs have been shown to increase catalytic free iron levels [189–191]. Since it takes only very low concentrations of catalytic metals to bring about the rapid oxidation of ascorbate [192,193], approaches that increase catalytic iron could potentially enhance the cytotoxicity of pharmacologic ascorbate in vivo [194].
Porphyrin-based SOD mimics have a redox-active metal (Mn, Fe, and Cu) center and a stable porphyrin complex. The dismutation of O2 •− by Mn porphyrin complexes involves two steps in which the Mn center cycles between Mn(III) and Mn(II): the first step is the reduction of Mn(III) by O2 • − to yield Mn(II) and O2 and the second step the oxidation of Mn(II) by O2 • − to yield H2O2 and return the manganese to its resting state as Mn(III) porphyrin [195]. However, in the presence of a reductant such as ascorbate, Mn porphyrins function as superoxide reductases rather than dismutases; Mn(III) can be reduced to Mn(II) by ascorbate while Mn(II) can react with O2 forming O2 • −, which subsequently forms H2O2 and O2, Reaction 8. The prooxidant effects of Mn porphyrin and ascorbate have been studied by several groups. Gardner et al. [196] have demonstrated that MnTM-4-PyP5+ catalyzes the oxidation of ascorbate in vitro. Zhong et al. [197] have shown synergistic killing of human prostate cancer cell RWPE-2 by MnTM-4-PyP5+ and ascorbate. Most recently, Tian et al. [198] have shown that MnTM-4-PyP5+ synergizes with ascorbate inhibiting the growth of prostatic, pancreatic, and hepatic cancer cells. Furthermore, the cytotoxic effects of two Mn(III) alkylpyridylporphyrins (MnTE-2-PyP5+ and MnTnHex-2-PyP5+) and ascorbate have been demonstrated in Caco-2, HeLa, HCT116, and 4T1 cells [199,200]. The more lipophilic MnTnHex-2-PyP5+ appeared much more effective. Given that several Mn porphyrins have already been tested in vivo as SOD mimetics, and by themselves have shown low toxicities at micromolar levels [195], there is great potential for using Mn porphyrins as an adjuvant to enhance the efficacy of pharmacologic ascorbate.
8.3. High dose ascorbate in animal studies
As mentioned above, the uptake of oral ascorbate in humans is tightly controlled by the gut and kidney filtration [7]. Rats receiving ascorbate by gavage (0.5 mg g−1) increased blood and extracellular concentration to peak levels <150 μM [10]. By contrast, concentrations reached peak levels of nearly 3 mM in rats receiving intraperitoneal injections, while intravenous injection increased peak levels to >8 mM. In a similar study, mice receiving bolus intravenous injections of ascorbate (1 g kg−1), resulted in plasma concentrations of 15 mM [164]. Supplementation of ascorbate in drinking water at 1 g kg−1 only increased plasma concentrations to <50 μM. These results clearly indicate that pharmacologic concentrations of ascorbate cannot be obtained by oral administration.
In addition to the finding that millimolar levels of ascorbate were achieved by parenteral administration, Chen and colleagues [10] demonstrated that ascorbate radical was formed in extracellular fluid but was not detectable in whole blood. Moreover, H2O2 was detected in extracellular fluid, but not in the blood; H2O2 correlated with ascorbate radical concentration. These results indicate that plasma membrane associated ascorbate radical reductase and high levels of catalase, glutathione peroxidase, and peroxiredoxin enable erythrocytes to act as a sink for extracellular ascorbate radical and H2O2 [176,107]. On the other hand, ascorbate could be oxidized by catalytic iron associated with damaged proteins in the extracellular space [201–203, 209]. Considering that the permeability of tumor vessels is much higher compared to normal endothelium, tumor endothelium may permit the outflow of macromolecules, such as albumin, to interstitial fluid [169,204]. The fluid that accumulated in the peritoneal cavities of ascites tumor-bearing mice had a protein concentration several-fold greater than that of normal peritoneal fluid, with approximately 85% of the protein level of plasma, including albumin, in a proportion similar to that found in plasma [205,206]. Human albumin is the most abundant protein in the plasma; and has the capacity to bind to metal ions such as Cu2+ [207]. The first four amino acids of the N-terminus of human albumin Asp-Ala-His-Lys forms a tight-binding site for Cu2+ [207]. In fact, almost thirty years ago, Linus Pauling and his colleagues [194] designed a copper:glycylglycylhistidine complex that mimics the binding of albumin to copper; it enhanced the antitumor activity of ascorbate against Ehrlich ascites tumor cells. In addition, extracellular fluids contain little or no catalase activity, and low levels of SOD and GPx [208]. Thus, the H2O2 generated by ascorbate oxidation in the extracellular space could accumulate to a concentration greater than the intracellular level resulting in net rate of diffusion across the cell membrane into cells, resulting in toxicity to cancer cells [10].
Pharmacological ascorbate inhibits tumor growth in mice. In mice bearing tumor xenografts of pancreatic cancer, treatment with 4 g kg−1 ascorbate (i.p., twice daily) significantly decreased the rate of tumor growth [11,12,164] (Table 4). Upon cessation of treatment with pharmacological ascorbate, the rate of tumor growth increased. This rate was similar to the rate observed in the control group, i.e. no ascorbate (Du et al., unpublished results). In this tumor model pharmacologic ascorbate as a single agent was cytostatic, not cytotoxic. Obviously, the use of intravenous high dose ascorbate as a single agent for cancer was not curative. To test the combination effect of standard chemotherapy with ascorbate, Espey et al. [209] have shown that synergistic cytotoxic effects can be achieved with gemcitabine and ascorbate in pancreatic cancer both in vitro and in a nude-mouse model. These data provide support for investigating the use of pharmacologic ascorbate as an adjuvant for conventional cancer chemotherapies.
Table 4.
Pro-oxidant effects of ascorbate in animal models.
| Species | Cell type | Treatment | Effects | Ref |
|---|---|---|---|---|
| Balb/c nude mice | HT29 colon cancer | Asc 15 mg, 100, 1000 mg/kg, i.p., daily. 4 weeks. | 7/7 mice survived at 1000 mg/kg. No carcinogenic invasion. Tumor volumes decrease. ARSs and EiFs genes down regulated. | [262] |
| Balb/c nude mice | K562 leukemia | Vit K3 10 mg/kg, i.p.; Asc 1 g/kg, i.p. | Asc+vit K3 decrease tumor growth. | [263] |
| Ncr nude mice | Ovcar5 ovarian cancer | Asc 4 g/kg, i.p., twice daily. 30 days. | No adverse effects. Decrease tumor growth. | [11] |
| Ncr nude mice | Pan02 pancreatic cancer | Asc 4 g/kg, i.p., daily. 14 days. | No adverse effects. Decrease tumor growth. | [11] |
| Ncr nude mice | 9 L rat glioblastoma | Asc 4 g/kg, i.p., twice daily. 12 days. | No adverse effects. Decrease tumor growth. Prevent metastases. |
[11] |
| Ncr nude mice | Pan02 pancreatic cancer | Asc 4 g/kg, i.p., daily; Gemcitabine 30, 60 mg/kg, i.p., every 4 days. 21 days. | No side effects except osmotic stress. Asc+Gem significantly decrease tumor growth. |
[209] |
| Ncr nude mice | PANC-1 pancreatic cancer | Asc 4 g/kg, i.p., daily; Gemcitabine 30, 60 mg/kg, every 4 days. 33 days. | No side effects except osmotic stress. Asc+Gem significantly decrease tumor growth. |
[209] |
| Nude mice | MIA PaCa-2 pancreatic cancer | Asc 4 g/kg, i.p., twice daily. 14 days. | No adverse effects. Decrease tumor growth. | [12] |
| NMRI mice | TLT Murine hepatoma | Asc 1 g/kg, i.p., daily. 30 days. | Decrease tumor growth. | [164] |
| Nude mice | H322 non-small cell lung cancer | Asc 250 mg/kg, i.p.; 101 F6 nanoparticle i.v. | Asc synergistic with 101 F6 decrease tumor growth. | [264] |
| SCID mice | EHMS-10 mesothelioma cells | Asc 0.88, 8.8 g/mouse, i.v. single dose. | Decrease tumor growth. | [265] |
| Balb/c mice | Murine sarcoma S180 cells | Asc 5.5, 30 mg/mouse, i.p., every two days. | Decrease tumor growth; inhibit bFGF, VEGF and MMP2 genes. | [266] |
| Balb/c mice | Murine sarcoma S180 cells | Asc 1.5 mg/g, i.p., every three days. | Inhibit tumor establishment; RKIP and annexin A5 levels increase. | [267] |
8.4. Clinical studies
Although the two clinical trials using oral ascorbate at the Mayo Clinic failed to show any benefit, numerous case reports have provided encouraging results with intravenous ascorbate therapy [162,210,211]. However, most of the cases were reported without sufficient detail or follow-up for evaluation. Padayatty et al. [212] examined three well-documented cases in accordance with National Cancer Institute (NCI) Best Case Series guidelines. These cases suggest that high-dose, intravenous ascorbate therapy prolonged the survival of the patients with advanced cancer.
Intravenous ascorbate therapy is likely to be safe in most patients except under certain conditions [213]. Because the removal of H2O2 generated in blood requires glutathione, which in turn requires NADPH generated by G6PD, the rate-limiting enzyme of the pentose phosphate pathway [214], high-dose ascorbate can induce intravascular hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency [215–217]. One end-product of ascorbate oxidation is oxalic acid, which has the potential to crystallize as calcium oxalate in the urinary space in patients with pre-existing renal insufficiency [218]. In addition, rare cases of massive tumor hemorrhage have been reported in patients with advanced cancer following high-dose intravenous ascorbate [155,219]. Thus, as always, caution is needed, but there are clear indications for a potential role for ascorbate in cancer treatment.
Three phase I clinical trials of intravenous ascorbate in patients with advanced cancer measured plasma concentrations of ascorbate and evaluated clinical consequences [162,220,221]. In the study by Riordan and colleagues, patients were given continuous infusion of 0.15 to 0.7 g kg−1 day−1 for up to eight weeks; in the Hoffer study, ascorbate was administrated three times per week at fixed doses of 0.4, 0.6, 0.9 and 1.5 g kg−1; and in the most recent study by Monti et al. [221], patients received 50, 75 and 100 g per infusion (three infusions per week) for 8 weeks with concomitant i.v. gemcitabine and oral erlotinib. In the Riordan study, ascorbate concentrations in serum during therapy ranged from 0.28 to 3.8 mM. In the Hoffer study, peak plasma ascorbate concentrations ranged from 2.4 to 26 mM. When 1.5 g kg−1 was administered, plasma ascorbate concentrations exceeded 10 mM for ≈4.5 h [220], levels that have been shown to induce cell killing in a variety of cancer cells [9,11,12,164]. Finally in the Monti study, patients that received 100 g of ascorbate achieved peak plasma concentrations between 25 and 32 mM. All three trials demonstrated that high-dose intravenous ascorbate was well tolerated in cancer patients with normal renal function; the combination of ascorbate infusion with standard of care chemotherapies did not increase toxicity.
8.5. Ascorbate and conventional cancer therapy
The cytotoxicity of high-dose intravenous ascorbate is dependent on the ascorbate-induced production of H2O2. Hydrogen peroxide can deplete intracellular GSH, causing oxidative stress. In high doses ascorbate acts as a pro-oxidant to kill cancer cells. Initial concerns that ascorbate might reduce the effectiveness of standard chemotherapy and/or radiation therapy have not been demonstrated. Numerous reports suggest that pharmacological ascorbate may actually increase the efficacy of several chemotherapeutic drugs and radiation in vitro [164,222–224] (Table 3). Espey et al. [209] demonstrated that pharmacologic concentrations of ascorbate with gemcitabine resulted in a synergistic cytotoxic response in pancreatic tumor cell lines; gemcitabine–ascorbate combinations administrated to mice bearing pancreatic tumor xenografts consistently enhanced inhibition of growth compared to gemcitabine alone. Case reports [161,212] indicate that intravenous ascorbate can be safely used with chemotherapy and radiotherapy and could potentially enhance the effects of conventional cancer therapy. Furthermore, intravenous ascorbate has been shown to improve the quality of life in breast cancer patients during chemo-/radiotherapy and aftercare [225]. Clinical trials using combinations of chemotherapy and intravenous ascorbate are underway at several institutions (www.clinicaltrials.gov).
Table 3.
Pro-oxidant effects of ascorbate in cultured cells.
| Cell line | Treatment | Effects | Ref |
|---|---|---|---|
| Primary human diploid fibroblasts GM5399 | 20–500 μM Asc | Inhibit growth, DNA damage | [251] |
| Normal human skin fibroblast CCD-25 SK | Sodium ascorbate, 0.56–2.8 mM | No effect | [210] |
| Normal human colon fibroblast CCD-18 Co | Sodium ascorbate, 0.1–0.4 mM | No effect | [210] |
| Human embryonic fibroblast | Sodium ascorbate, 0.2 mM | Inhibit growth | [252] |
| HRE human normal renal epithelial cells | Asc 1.2 mM | Promote growth | [253] |
| Human normal myeloid cells | L-Ascorbic acid, 0.3 mM | No effect | [254] |
| Primary chicken embryo fibroblast | Sodium ascorbate, 0.2 mM | Inhibit growth | [252] |
| AN3 CA endometrial adenocarcinoma | Sodium ascorbate, 0.56–2.8 mM | Inhibit growth | [210] |
| Ehrlich ascites carcinoma | Ascorbate+3-amino-triazole | Synergistic killing | [171] |
| 5637 human bladder cancer | Asc EC50 < 5 mM | Inhibit growth | [11] |
| T24 human bladder cancer | Asc+vitK3 | Synergistic, Autoschizis, Necrosis, apoptosis | [255] |
| MB231 human breast cancer | 5 mM Asc | Decrease clonogenic survival | [9] |
| MCF-7 human breast cancer | 5 mM Asc | Decrease clonogenic survival | [9] |
| MCF-7 human breast cancer | Asc+porphyrin | Apoptosis, cell cycle disruption | [256] |
| Hs587t human breast cancer | 5 mM Asc | Decrease clonogenic survival | [9] |
| HeLa human cervical cancer | Sodium ascorbate, 0.25 mM; Asc EC50 >5 mM | No effect | [11,257] |
| HeLa human cervical cancer | Asc+MGd (Motexafin gadolinium) | Apoptosis, Lysosomal rupture | [257] |
| Normal human colon fibroblast CCD-18-Co | Sodium ascorbate, 0.1–0.8 mM | No effect | [210] |
| Human colon carcinoma cells | Sodium ascorbate, 0.1–3.2 mM | Inhibit growth | [210] |
| HEp-2 human epidermoid larynx carcinoma | Asc 500 μM+25 μM vitB12b | Apoptosis, DNA damage | [258] |
| A549 human lung cancer | Asc EC50 < 5 mM | Inhibit growth | [11] |
| HepG2 human hepatocellular carcinoma | Asc, Asc+MnTMPyP | Decrease clonogenic survival, mitochondria damage. | [198] |
| DU 145 human prostate cancer cells | Sodium ascorbate, 1–10 mM; Asc+MnTMPyP | Inhibit growth | [198] |
| LNCaP human prostate cancer cells | Sodium ascorbate, 1–10 mM; Asc+MnTMPyP | Inhibit growth | [198] |
| PC-3 human prostate cancer | Sodium ascorbate, 0.56–2.3 mM; Asc+MnTMPyP | Inhibit growth | [198] |
| MIA PaCa-2 human pancreatic carcinoma | Sodium ascorbate, 0.56–2.8 mM; | Inhibit growth | [210] |
| MIA PaCa-2 human pancreatic carcinoma | Asc 1–5 mM | Cytotoxic, autophagy | [12] |
| PANC-1 | Asc, Asc+MnTMPyP | Decrease clonogenic survival | [198] |
| Pan02 mouse pancreatic cancer | Asc 2.5–10 mM | cytotoxic | [11] |
| Human acute leukemic cells | L-Ascorbic acid, 0.3 mM | Inhibit growth | [254] |
| U937 human histocytic leukemia cells | L-Ascorbic acid, 50–300 μM | Inhibit growth | [163] |
| Human chronic lymphocytic leukemia | Asc 0.3–5 mM; Asc+ATO (Arsenic trioxide) | Cytotoxic | [259] |
| Kelly human neuroblastoma | 0.6–5 mM | Cytotoxic | [151] |
| SK-N-SH human neuroblastoma | 0.6–5 mM | Cytotoxic | [151] |
| Mouse neuroblastoma | Sodium ascorbate+5-FU Sodium ascorbate+X-irradiation |
Inhibit growth | [222] |
| 9 L rat glioblastoma | Asc 2.5–10 mM | Cytotoxic | [11] |
| LN18 human glioblastoma cell lines, human primary glioblastoma cells, GL261 mouse astrocytoma cell line | Asc+γ-irradiation | Double-stranded DNA breaks, G2/M arrest block | [224] |
| B16F10 murine melanoma | Asc 0.05–0.2 mM | Cell cycle arrest | [260] |
| Chinese hamster ovary (CHO) cells | Ascorbate+misonidazole | Inhibit growth | [261] |
9. Perspectives
The inhibition effects of pharmacologic ascorbate on tumor growth have been confirmed in many laboratories (Table 4). In murine models elevated ascorbate levels in plasma were verified following i.p. administration [11,164]. It is still unknown whether cells and tissues levels are also elevated [226,227]. Human platelets supplemented with 500 μM Asc for 30 min showed a decrease in SVCT2 levels, while in Asc depleted platelets SVCT2 expression was higher than native ones [29]. Thus in vivo studies for possible changes in SVCT expression levels following ascorbate infusion are needed. Another issue is a possible transient withdrawal effect upon cessation of treatment with pharmacologic ascorbate [228]. Male guinea pigs that received ascorbate 1 g kg−1 body weight per day by i.p. for 4 weeks had elevated plasma and urinary levels; however, in the weeks after the treatment withdrawn, mean plasma and urinary ascorbate were significantly lower than normal [229]. It is unknown if a similar rebound effect would occur in human subjects following high doses i.v. ascorbate. F2-isoprostane is a biomarker for lipid peroxidation in vivo, and its quantification in plasma and urine has emerged as the most reliable method to assess systemic oxidative stress in humans and animals [230–232]. For healthy young women taking oral doses of ascorbate 30–2500 mg daily, plasma and urine F2-isoprostanes were unchanged [41]. It is unclear whether plasma F2-isoprostanes will change following pharmacologic ascorbate infusion in cancer patients. Furthermore, since pharmacological ascorbate is a prodrug for H2O2 generation, there should be many applications where delivery of H2O2 via pharmacological ascorbate may have clinical benefit, such as infectious diseases caused by viruses, bacteria, and other pathogens [10,233].
10. Summary
More than eighty years since its discovery, our understanding of the functions of ascorbic acid has evolved from the prevention of scurvy to its potential use as a therapeutic drug for cancer treatment. Ascorbate maintains Fe2+ of collagen hydroxylases in an active state; therefore it plays a pivotal role in collagen synthesis; parallel reactions with a variety of dioxygenases affect the expression of a wide array of genes, for example via the HIF system, and possibly the epigenetic landscape of cells and tissues. The ability to donate one or two electrons makes ascorbate an excellent reducing agent and antioxidant. However, in the presence of catalytic metals, ascorbate also has pro-oxidant effects, where the redox-active metal is reduced by ascorbate and then in turn reacts with oxygen, producing superoxide that subsequently dismutes to produce H2O2.
Apart from its biochemical functions that are met by healthy nutritional levels, recent pharmacokinetic data indicate that intravenous administration of ascorbate bypasses the tight control of the gut and renal excretion; thus, intravenous administration of ascorbate will produce highly elevated plasma levels; this ascorbate will autoxidize resulting in a high flux of extracellular H2O2. This H2O2 will readily diffuse into cells challenging the intracellular peroxide-removal system, initiating oxidative cascades. These high fluxes of H2O2 appear to have little effect on normal cells but can be detrimental to certain tumor cells. Knowledge and understanding of these mechanisms brings a rationale to the use of high-dose ascorbate to treat disease and thereby is reviving interest in the use of i.v. ascorbate in cancer treatment. The full potential of pharmacological ascorbate in cancer treatment will only be realized with greater understanding of its basic mechanism of action in conjunction with the appropriate design of clinical trials.
Abbreviations
- Asc
a general abbreviation to include all forms, i.e. AscH2 +AscH−+Asc2−+DHA
- AscH2
ascorbic acid, vitamin C
- AscH−
ascorbate monoanion
- Asc2−
ascorbate dianion
- Asc•−
semidehydroascorbate, i.e. ascorbate radical
- DHA
dehydroascorbic acid
- HIF
hypoxia inducible factor
- PDI
protein disulfide isomerase
- SVCT
sodium-dependent vitamin C transporters
Footnotes
Supported by NIH grants CA137230 and 1R01GM073929, the Medical Research Service, Department of Veterans Affairs, and the Susan L. Bader Foundation of Hope.
References
- 1.Svirbely JL, Szent-Györgyi A. The chemical nature of vitamin C. Biochem J. 1933;27:279–285. [PMC free article] [PubMed] [Google Scholar]
- 2.Buettner GR, Schafer FQ. Albert Szent-Györgyi: vitamin C identification. Biochemist. 2006;28:31–33. [Google Scholar]
- 3.Calcutt G. The formation of hydrogen peroxide during the autoxidation of ascorbic acid. Experientia. 1951;7:26. doi: 10.1007/BF02165477. [DOI] [PubMed] [Google Scholar]
- 4.Bielski BHJ. Chemistry of ascorbic acid radicals. In: Seib PA, Tolbert BM, editors. Ascorbic Acid: Chemistry, Metabolism, and Uses. Vol. 200. American Chemical Society; Washington D.C: 1982. pp. 81–100. [Google Scholar]
- 5.Khan MM, Martell AE. Metal ion and metal chelate catalyzed oxidation of ascorbic acid by molecular oxygen. II. Cupric and ferric chelate catalyzed oxidation. J Am Chem Soc. 1967;89:7104–7111. doi: 10.1021/ja01002a046. [DOI] [PubMed] [Google Scholar]
- 6.Udenfriend S, Clark CT, Axelrod J, Brodie BB. Ascorbic acid in aromatic hydroxylation. I. A model system for aromatic hydroxylation. J Biol Chem. 1954;208:731–739. [PubMed] [Google Scholar]
- 7.Graumlich JF, Ludden TM, Conry-Cantilena C, Cantilena LRJ, Wang Y, Levine M. Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm Res. 1997;14:1133–1139. doi: 10.1023/a:1012186203165. [DOI] [PubMed] [Google Scholar]
- 8.Padayatty SJ, Sun H, Wang Y, Riordan HD, Hewitt SM, Katz A, Wesley RA, Levine M. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004;140:533–537. doi: 10.7326/0003-4819-140-7-200404060-00010. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, Shacter E, Levine M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102:13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Espey MG, Sun A, Lee J, Krishna MC, Shacter E, Choyke PL, Pooput C, Kirk KL, Buettner GR, Levine M. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104:8749–8754. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, Khosh DB, Drisko J, Levine M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A. 2008;105:11105–11109. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, Taghiyev AF, Du C, Knudson CM, Cullen JJ. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res. 2010;16:509–520. doi: 10.1158/1078-0432.CCR-09-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, [alpha]-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 14.Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996;145:532–541. [PubMed] [Google Scholar]
- 15.Buettner GR. In the absence of catalytic metals ascorbate does not autoxidize at pH 7: ascorbate as a test for catalytic metals. J Biochem Biophys Methods. 1988;16:27–40. doi: 10.1016/0165-022x(88)90100-5. [DOI] [PubMed] [Google Scholar]
- 16.Song Y, Buettner GR. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radic Biol Med. 2010;49:919–962. doi: 10.1016/j.freeradbiomed.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams NH, Yandell JK. Outer-sphere electron-transfer reactions of ascorbate anions. Aust J Chem. 1982;35:1133–1144. [Google Scholar]
- 18.Buettner GR. Ascorbate oxidation: UV absorbance of ascorbate and ESR spectroscopy of the ascorbyl radical as assays for iron. Free Radic Res Commun. 1990;10:5–9. doi: 10.3109/10715769009145927. [DOI] [PubMed] [Google Scholar]
- 19.Nishikimi M, Fukuyama R, Minoshima S, Shimizu N, Yagi K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J Biol Chem. 1994;269:13685–13688. [PubMed] [Google Scholar]
- 20.Vera JC, Rivas CI, Velasquez FV, Zhang RH, Concha II, Golde DW. Resolution of the facilitated transport of dehydroascorbic acid from its intracellular accumulation as ascorbic acid. J Biol Chem. 1995;270:23706–23712. doi: 10.1074/jbc.270.40.23706. [DOI] [PubMed] [Google Scholar]
- 21.Savini I, Rossi A, Pierro C, Avigliano L, Catani MV. SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids. 2008;34:347–355. doi: 10.1007/s00726-007-0555-7. [DOI] [PubMed] [Google Scholar]
- 22.Welch RW, Wang Y, Crossman AJ, Park JB, Kirk KL, Levine M. Accumulation of vitamin C (ascorbate) and its oxidized metabolite dehydroascorbic acid occurs by separate mechanisms. J Biol Chem. 1995;270:12584–12592. doi: 10.1074/jbc.270.21.12584. [DOI] [PubMed] [Google Scholar]
- 23.Diliberto EJ, Heckman GD, Daniels AJ. Characterization of ascorbic acid transport by adrenomedullary chromaffin cells. Evidence for Na+-dependent co-transport. J Biol Chem. 1983;258:12886–12894. [PubMed] [Google Scholar]
- 24.Welch RW, Bergsten P, Butler JD, Levine M. Ascorbic acid accumulation and transport in human fibroblasts. Biochem J. 1993;294:505–510. doi: 10.1042/bj2940505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang Y, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- 26.Corpe CP, Tu H, Eck P, Wang J, Faulhaber-Walter R, Schnermann J, Margolis S, Padayatty S, Sun H, Wang Y, Nussbaum RL, Espey MG, Levine M. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J Clin Invest. 2010;120:1069–1083. doi: 10.1172/JCI39191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson JX. Regulation of vitamin C transport. Annu Rev Nutr. 2005;25:105–125. doi: 10.1146/annurev.nutr.25.050304.092647. [DOI] [PubMed] [Google Scholar]
- 28.Savini I, Catani MV, Arnone R, Rossi A, Frega G, Del Principe D, Avigliano L. Translational control of the ascorbic acid transporter SVCT2 in human platelets. Free Radic Biol Med. 2007;42:608–616. doi: 10.1016/j.freeradbiomed.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 29.MacDonald L, Thumser AE, Sharp P. Decreased expression of the vitamin C transporter SVCT1 by ascorbic acid in a human intestinal epithelial cell line. Br J Nutr. 2002;87:97–100. doi: 10.1079/BJN2001492. [DOI] [PubMed] [Google Scholar]
- 30.Corpe CP, Lee JH, Kwon O, Eck P, Narayanan J, Kirk KL, Levine M. 6-Bromo-6-deoxy-L-ascorbic acid: an ascorbate analog specific for Na+-dependent vitamin C transporter but not glucose transporter pathways. J Biol Chem. 2005;280:5211–5220. doi: 10.1074/jbc.M412925200. [DOI] [PubMed] [Google Scholar]
- 31.Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–638. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 32.Agus DB, Vera JC, Golde DW. Stromal cell oxidation: a mechanism by which tumors obtain vitamin C. Cancer Res. 1999;59:4555–4558. [PubMed] [Google Scholar]
- 33.Malo C, Wilson JX. Glucose modulates vitamin C transport in adult human small intestinal brush border membrane vesicles. J Nutr. 2000;130:63–69. doi: 10.1093/jn/130.1.63. [DOI] [PubMed] [Google Scholar]
- 34.Rumsey SC, Daruwala R, Al-Hasani H, Zarnowski MJ, Simpson IA, Levine M. Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J Biol Chem. 2000;275:28246–28253. doi: 10.1074/jbc.M000988200. [DOI] [PubMed] [Google Scholar]
- 35.Cunningham JJ. The glucose/insulin system and vitamin C: implications in insulin-dependent diabetes mellitus. J Am Coll Nutr. 1998;17:105–108. doi: 10.1080/07315724.1998.10718734. [DOI] [PubMed] [Google Scholar]
- 36.Sherry S, Ralli EP. Further studies of the effects of insulin on the metabolism of vitamin C. J Clin Invest. 1948;27:217–225. doi: 10.1172/JCI101936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paolisso G, D’Amore A, Balbi V, Volpe C, Galzerano D, Giugliano D, Sgambato S, Varricchio M, D’Onofrio F. Plasma vitamin C affects glucose homeostasis in healthy subjects and in non-insulin-dependent diabetics. Am J Physiol Endocrinol Metab. 1994;266:E261–E268. doi: 10.1152/ajpendo.1994.266.2.E261. [DOI] [PubMed] [Google Scholar]
- 38.May JM, Qu ZC, Qiao H, Kouryab MJ. Maturational loss of the vitamin C transporter in erythrocytes. Biochem Biophys Res Commun. 2007;360:295–298. doi: 10.1016/j.bbrc.2007.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evans RM, Currie L, Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br J Nutr. 1982;47:473–482. doi: 10.1079/bjn19820059. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Tu H, Wang Y, Levine M. Vitamin C in mouse and human red blood cells: an HPLC assay. Anal Biochem. 2012;426:109–117. doi: 10.1016/j.ab.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci U S A. 2001;98:9842–9846. doi: 10.1073/pnas.171318198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lane DJR, Lawen A. Ascorbate and plasma membrane electron transport —enzymes vs efflux. Free Radic Biol Med. 2009;47:485–495. doi: 10.1016/j.freeradbiomed.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- 44.Han O, Failla ML, Hill AD, Morris ER, Smith JCJ. Reduction of Fe(III) is required for uptake of nonheme iron by Caco-2 cells. J Nutr. 1995;125:1291–1299. doi: 10.1093/jn/125.5.1291. [DOI] [PubMed] [Google Scholar]
- 45.Hornig D. Distribution of ascorbic acid, metabolites and analogues in man and animals. Ann N Y Acad Sci. 1975;258:103–118. doi: 10.1111/j.1749-6632.1975.tb29271.x. [DOI] [PubMed] [Google Scholar]
- 46.Kern HL, Zolot SL. Transport of vitamin C in the lens. Curr Eye Res. 1987;6:885–896. doi: 10.3109/02713688709034857. [DOI] [PubMed] [Google Scholar]
- 47.Rumsey SC, Levine M. Absorption, transport, and disposition of ascorbic acid in humans. J Nutr Biochem. 1998;9:116–130. [Google Scholar]
- 48.Streeter ML, Rosso P. Transport mechanisms for ascorbic acid in the human placenta. Am J Clin Nutr. 1981;34:1706–1711. doi: 10.1093/ajcn/34.9.1706. [DOI] [PubMed] [Google Scholar]
- 49.Brubaker RF, Bourne WM, Bachman LA, McLaren JW. Ascorbic acid content of human corneal epithelium. Invest Ophthalmol Vis Sci. 2000;41:1681–1683. [PubMed] [Google Scholar]
- 50.Van der Vliet A, O’Neill CA, Cross CE, Koostra JM, Valz WG, Halliwell B, Louie S. Determination of low-molecular-mass antioxidant concentrations in human respiratory tract lining fluids. Am J Physiol Lung Cell Mol Physiol. 1999;276:L289–L296. doi: 10.1152/ajplung.1999.276.2.L289. [DOI] [PubMed] [Google Scholar]
- 51.Bui MH, Sauty A, Collet F, Leuenberger P. Dietary vitamin C intake and concentrations in the body fluids and cells of male smokers and nonsmokers. J Nutr. 1992;122:312–316. doi: 10.1093/jn/122.2.312. [DOI] [PubMed] [Google Scholar]
- 52.May JM, Qu Z, Li X. Ascorbic acid blunts oxidant stress due to menadione in endothelial cells. Arch Biochem Biophys. 2003;411:136–144. doi: 10.1016/s0003-9861(02)00715-4. [DOI] [PubMed] [Google Scholar]
- 53.Chepda T, Cadau M, Girin PH, Frey J, Chamson A. Monitoring of ascorbate at a constant rate in cell culture: effect on cell growth. In Vitro Cell Dev Biol Anim. 2001;37:26–30. doi: 10.1290/1071-2690(2001)037<0026:MOAAAC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 54.Vislisel JM, Schafer FQ, Buettner GR. A simple and sensitive assay for ascorbate using a plate reader. Anal Biochem. 2007;365:31–39. doi: 10.1016/j.ab.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Washko PW, Wang Y, Levine M. Ascorbic acid recycling in human neutrophils. J Biol Chem. 1993;268:15531–15535. [PubMed] [Google Scholar]
- 56.Lloyd JV, Davis PS, Emery H, Lander H. Platelet ascorbic acid levels in normal subjects and disease. J Clin Pathol. 1972;25:478–483. doi: 10.1136/jcp.25.6.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simpson GLW, Ortwerth BJ. The non-oxidative degradiation of ascorbic acid at physiological conditions. Biochim Biophys Acta. 2000;1501:12–24. doi: 10.1016/s0925-4439(00)00009-0. [DOI] [PubMed] [Google Scholar]
- 58.Bánhegyi G, Braun L, Csala M, Puskás F, Mandl J. Ascorbate metabolism and its regulation in animals. Free Radic Biol Med. 1997;23:793–803. doi: 10.1016/s0891-5849(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 59.Nagaraj RH, Shamsi FA, Huber B, Pischetsrieder M. Immunochemical detection of oxalate monokylamide, an ascorbate-derived Maillard reaction product in the human lens. FEBS J. 1999;453:327–330. doi: 10.1016/s0014-5793(99)00571-2. [DOI] [PubMed] [Google Scholar]
- 60.Chai W, Liebman M, Kynast-Gales S, Massey L. Oxalate absorption and endogenous oxalate synthesis from ascorbate in calcium oxalate stone formers and non-stone formers. Am J Kidney Dis. 2004;44:1060–1069. doi: 10.1053/j.ajkd.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 61.Traxer O, Huet B, Poindexter J, Pak CYC, Pearle MS. Effect of ascorbic acid consumption on urinary stone risk factors. J Urol. 2003;170:397–401. doi: 10.1097/01.ju.0000076001.21606.53. [DOI] [PubMed] [Google Scholar]
- 62.Robitaille L, Mamer OA, Miller WHJ, Levine M, Assouline S, Melnychuk D, Rousseau C, Hoffer LJ. Oxalic acid excretion after intravenous ascorbic acid administration. Metabolism. 2009;58:263–269. doi: 10.1016/j.metabol.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asplin JR. Hyperoxaluric calcium nephrolithiasis. Endocrinol Metab Clin North Am. 2002;31:927–949. doi: 10.1016/s0889-8529(02)00030-0. [DOI] [PubMed] [Google Scholar]
- 64.Englard S, Seifter S. The biochemical functions of ascorbic acid. Annu Rev Nutr. 1986;6:365–406. doi: 10.1146/annurev.nu.06.070186.002053. [DOI] [PubMed] [Google Scholar]
- 65.Ozer A, Bruick RK. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3:144–153. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- 66.Kivirikko KI, Prockop DJ. Enzymatic hydroxylation of proline and lysine in protocollagen. Proc Natl Acad Sci U S A. 1967;57:782–789. doi: 10.1073/pnas.57.3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 68.Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liotta LA. Tumor invasion and metastases—role of the extracellular matrix: Rhoads Memorial Award lecture. Cancer Res. 1986;46:1–7. [PubMed] [Google Scholar]
- 70.Geesin JC, Darr D, Kaufman R, Murad S, Pinnell SR. Ascorbic acid specifically increases type I and type III procollagen messenger RNA levels in human skin fibroblasts. J Investig Dermatol. 1988;90:420–424. doi: 10.1111/1523-1747.ep12460849. [DOI] [PubMed] [Google Scholar]
- 71.Cameron E, Pauling L, Leibovitz B. Ascorbic acid and cancer: a review. Cancer Res. 1979;39:663–681. [PubMed] [Google Scholar]
- 72.Maeshima Y, Colorado PC, Torre A, Holthaus KA, Grunkemeyer JA, Ericksen MB, Hopfer H, Xiao Y, Stillman IE, Kalluri R. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J Biol Chem. 2000;275:21340–21348. doi: 10.1074/jbc.M001956200. [DOI] [PubMed] [Google Scholar]
- 73.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 74.Kallio PJ, Wilson WJ, O’Brien S, Makino Y, Poellinger L. Regulation of the hypoxia-inducible transcription factor 1alpha by the ubiquitin–proteasome pathway. J Biol Chem. 1999;274:6519–6525. doi: 10.1074/jbc.274.10.6519. [DOI] [PubMed] [Google Scholar]
- 75.Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 76.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 77.Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J, Lee LA, Semenza GL, Dang CV. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu H, Dalgard CL, Mohyeldin A, McFate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem. 2005;280:41928–41939. doi: 10.1074/jbc.M508718200. [DOI] [PubMed] [Google Scholar]
- 81.Kuiper C, Molenaar IG, Dachs GU, Currie MJ, Sykes PH, Vissers MC. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 2010;70:5749–5758. doi: 10.1158/0008-5472.CAN-10-0263. [DOI] [PubMed] [Google Scholar]
- 82.Mikirova N, Ichim T, Riordan N. Anti-angiogenic effect of high doses of ascorbic acid. J Transl Med. 2008;6:50. doi: 10.1186/1479-5876-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pathi SS, Lei P, Sreevalsan S, Chadalapaka G, Jutooru I, Safe S. Pharmacologic doses of ascorbic acid repress specificity protein (Sp) transcription factors and Sp-regulated genes in colon cancer cells. Nutr Cancer. 2011;63:1133–1142. doi: 10.1080/01635581.2011.605984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pagé EL, Chan DA, Giaccia AJ, Levine M, Richard DE. Hypoxia-inducible factor-1α stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol Biol Cell. 2008;19:86–94. doi: 10.1091/mbc.E07-06-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–1140. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chung TL, Brena RM, Kolle G, Grimmond SM, Berman BP, Laird PW, Pera MF, Wolvetang EJ. Vitamin C promotes widespread yet specific DNA demethylation of the epigenome in human embryonic stem cells. Stem Cells. 2010;28:1848–1855. doi: 10.1002/stem.493. [DOI] [PubMed] [Google Scholar]
- 87.Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. 2011;15:551–589. doi: 10.1089/ars.2010.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Esteban MA, Pei D. Vitamin C improves the quality of somatic cell reprogramming. Nat Genet. 2012;44:366–367. doi: 10.1038/ng.2222. [DOI] [PubMed] [Google Scholar]
- 89.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 90.Verma M. Cancer control and prevention by nutrition and epigenetic approaches. Antioxid Redox Signal. 2012;17:355–364. doi: 10.1089/ars.2011.4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Block G. Vitamin C and cancer prevention: the epidemiologic evidence. Am J Clin Nutr. 1991;53:270S–282S. doi: 10.1093/ajcn/53.1.270S. [DOI] [PubMed] [Google Scholar]
- 92.Loria CM, Klag MJ, Caulfield LE, Whelton PK. Vitamin C status and mortality in US adults. Am J Clin Nutr. 2000;72:139–145. doi: 10.1093/ajcn/72.1.139. [DOI] [PubMed] [Google Scholar]
- 93.Patterson RE, White E, Kristal AR, Neuhouser ML, Potter JD. Vitamin supplements and cancer risk: the epidemiologic evidence. Cancer Causes Control. 1997;8:786–802. doi: 10.1023/a:1018443724293. [DOI] [PubMed] [Google Scholar]
- 94.Gey KF. Vitamins E plus C and interacting conutrients required for optimal health. A critical and constructive review of epidemiology and supplementation data regarding cardiovascular disease and cancer. Biofactors. 1998;7:113–174. doi: 10.1002/biof.5520070115. [DOI] [PubMed] [Google Scholar]
- 95.Gonzalez CA, Riboli E. Diet and cancer prevention: contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Euro J Cancer. 2010;46:2555–2562. doi: 10.1016/j.ejca.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 96.Musil F, Zadák Z, Solichová D, Hyšpler R, Kaška M, Sobotka L, Manák J. Dynamics of antioxidants in patients with acute pancreatitis and in patients operated for colorectal cancer: a clinical study. Nutrition. 2005;21:118–124. doi: 10.1016/j.nut.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 97.Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: a systematic review and meta-analysis. Lancet. 2004;364:1219–1228. doi: 10.1016/S0140-6736(04)17138-9. [DOI] [PubMed] [Google Scholar]
- 98.Gaziano JM, Glynn RJ, Christen WG, Kurth T, Belanger C, MacFadyen J, Bubes V, Manson JE, Sesso HD, Buring JE. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2009;301:52–62. doi: 10.1001/jama.2008.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lin J, Cook NR, Albert C, Zaharris E, Gaziano JM, Van Denburgh M, Buring JE, Manson JE. Vitamins C and E and beta carotene supplementation and cancer risk: a randomized controlled trial. J Natl Cancer Inst. 2009;101:14–23. doi: 10.1093/jnci/djn438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heaney RP. Nutrition, chronic disease, and the problem of proof. Am J Clin Nutr. 2006;84:471–472. doi: 10.1093/ajcn/84.3.471. [DOI] [PubMed] [Google Scholar]
- 101.Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR, Park JB, Lazarev A, Graumlich J, King J, Cantilena LR. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93:3704–3709. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Burri BJ, Jacob RA. Human metabolism and the requirement for vitamin C. In: Packer L, Fuchs J, editors. Vitamin C in Health and Disease. Marcel Dekker; New York: 1997. pp. 341–366. [Google Scholar]
- 103.Ames BN. Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage. Proc Natl Acad Sci U S A. 2006;103:17589–17594. doi: 10.1073/pnas.0608757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Levine M, Eck P. Vitamin C: working on the x-axis. Am J Clin Nutr. 2009;90:1121–1123. doi: 10.3945/ajcn.2009.28687. [DOI] [PubMed] [Google Scholar]
- 105.Ito A, Hayashi S-i, Yoshida T. Participation of a cytochrome b5-like hemoprotein of outer mitochondrial membrane (OM cytochrome b) in NADH-semidehydroascorbic acid reductase activity of rat liver. Biochem Biophys Res Commun. 1981;101:591–598. doi: 10.1016/0006-291x(81)91300-0. [DOI] [PubMed] [Google Scholar]
- 106.Borgese N, Pietrini G. Distribution of the integral membrane protein NADH-cytochrome b5 reductase in rat liver cells, studied with a quantitative radioimmunoblotting assay. Biochem J. 1986;239:393–403. doi: 10.1042/bj2390393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.May JM, Qu Z, Cobb CE. Recycling of the ascorbate free radical by human erythrocyte membranes. Free Radic Biol Med. 2001;31:117–124. doi: 10.1016/s0891-5849(01)00566-4. [DOI] [PubMed] [Google Scholar]
- 108.Bode AM, Cunningham L, Rose RC. Spontaneous decay of oxidized ascorbic acid (dehydro-L-ascorbic acid) evaluated by high-pressure liquid chromatography. Clin Chem. 1990;36:1807–1809. [PubMed] [Google Scholar]
- 109.Borsook H, Davenport HW, Jeffreys CEP, Warner RC. The oxidation of ascorbic acid and its reduction in vitro and in vivo. J Biol Chem. 1937;117:237–279. [Google Scholar]
- 110.Bogdanski K, Bogdanska H. Sur la stabilite de l’acide L-dehdroascorbique dans les solutions cbeaqueses. Bull Acad Pol Sci. 1955;3:41–44. [Google Scholar]
- 111.Winkler BS, Orselli SM, Rex TS. The redox couple between glutathione and ascorbic acid: a chemical and physiological perspective. Free Radic Biol Med. 1994;17:333–349. doi: 10.1016/0891-5849(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 112.Wells WW, Xu DP, Yang YF, Rocque PA. Mammalian thioltransferase (glutaredoxin) and protein disulfide isomerase have dehydroascorbate reductase activity. J Biol Chem. 1990;265:15361–15364. [PubMed] [Google Scholar]
- 113.May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J Biol Chem. 1997;272:22607–22610. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- 114.Del Bello B, Maellaro E, Sugherini L, Santucci A, Comporti M, Casini AF. Purification of NADPH-dependent dehydroascorbate reductase from rat liver and its identification with 3 alpha-hydroxysteroid dehydrogenase. Biochem J. 1994;304:385–390. doi: 10.1042/bj3040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Frei B, England L, Ames BN. Ascorbate is an outstanding antioxidant in human blood plasma. Proc Natl Acad Sci U S A. 1989;86:6377–6381. doi: 10.1073/pnas.86.16.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Linster CL, Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 117.Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch Biochem Biophys. 2010;500:107–115. doi: 10.1016/j.abb.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 118.Bielski BHJ, Allen AO, Schwart HA. Mechanism of disproportionation of ascorbate radicals. J Am Chem Soc. 1981;103:3516–3518. [Google Scholar]
- 119.Niki E, Noguchi N. Protection of human low-density lipoprotein from oxidative modification by vitamin C. In: Packer L, Fuchs J, editors. Vitamin C in Health and Disease. Marcel Dekker; New York: 1997. pp. 183–192. [Google Scholar]
- 120.Lynch SM, Morrow JD, Roberts LJ, II, Frei B. Formation of non-cyclooxygenase-derived prostanoids (F2-isoprostanes) in plasma and low density lipoprotein exposed to oxidative stress in vitro. J Clin Invest. 1994;93:998–1004. doi: 10.1172/JCI117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Golumbic C, Mattill HA. Antioxidants and the autoxidation of fats. XIII. The antioxygenic action of ascorbic acid in association with tocopherols, hydroquinones and related compounds. J Am Chem Soc. 1941;63:1279–1280. [Google Scholar]
- 122.Burton GW, Ingold KU. Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function. Acc Chem Res. 1986;19:194–201. [Google Scholar]
- 123.Csallany AS, Draper HH, Shah SN. Conversion of D-alpha-tocopherol-C14 to tocopheryl-p-quinone in vivo. Arch Biochem Biophys. 1962;98:142–145. doi: 10.1016/0003-9861(62)90159-5. [DOI] [PubMed] [Google Scholar]
- 124.Bruno RS, Leonard SW, Atkinson J, Montine TJ, Ramakrishnan R, Bray TM, Traber MG. Faster plasma vitamin E disappearance in smokers is normalized by vitamin C supplementation. Free Radic Biol Med. 2006;40:689–697. doi: 10.1016/j.freeradbiomed.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 125.Berger TM, Polidori MC, Dabbagh A, Evans PJ, Halliwell B, Morrow JD, Roberts LJ, II, Frei B. Antioxidant activity of vitamin C in iron-overloaded human plasma. J Biol Chem. 1997;272:15656–15660. doi: 10.1074/jbc.272.25.15656. [DOI] [PubMed] [Google Scholar]
- 126.Suh J, Zhu BZ, Frei B. Ascorbate does not act as a pro-oxidant towards lipids and proteins in human plasma exposed to redox-active transition metal ions and hydrogen peroxide. Free Radic Biol Med. 2003;34:1306–1314. doi: 10.1016/s0891-5849(03)00147-3. [DOI] [PubMed] [Google Scholar]
- 127.Chen K, Suh J, Carr AC, Morrow JD, Zeind J, Frei B. Vitamin C suppresses oxidative lipid damage in vivo, even in the presence of iron overload. Am J Physiol Endocrinol Metab. 2000;279:E1406–E1412. doi: 10.1152/ajpendo.2000.279.6.E1406. [DOI] [PubMed] [Google Scholar]
- 128.Halliwell B. Vitamin C: poison, prophylactic or panacea? Trends Biochem Sci. 1999;24:255–259. doi: 10.1016/s0968-0004(99)01418-8. [DOI] [PubMed] [Google Scholar]
- 129.Frei B, Lawson S. Vitamin C and cancer revisited. Proc Natl Acad Sci U S A. 1998;105:11037–11038. doi: 10.1073/pnas.0806433105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rees S, Slater TF. Ascorbic acid and lipid peroxidation: the cross-over effect. Acta Biochim Biophys Hung. 1987;22:241–249. [Google Scholar]
- 131.Buettner GR, Jurkiewicz BA. Chemistry and biochemistry of ascorbic acid. In: Cadenas E, Packer L, editors. Handbook of Antioxidants. Marcel Dekker; New York: 1996. pp. 91–115. [Google Scholar]
- 132.Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV. Serum ferritin: past, present and future. Biochim Biophys Acta — Gen Subj. 2010;1800:760–769. doi: 10.1016/j.bbagen.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Richardson DR, Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta Rev Biomembr. 1997;1331:1–40. doi: 10.1016/s0304-4157(96)00014-7. [DOI] [PubMed] [Google Scholar]
- 134.Buettner GR. The reaction of superoxide, formate radical, and hydrated electron with transferrin and its model compound, Fe(III)-ethylenediamine-N,N′-bis [2-(2-hydroxyphenyl) acetic acid] as studied by pulse radiolysis. J Biol Chem. 1987;262:11995–11998. [PubMed] [Google Scholar]
- 135.Sirivech S, Frieden E, Osaki S. The release of iron from horse spleen ferritin by reduced flavins. Biochem J. 1974;143:311–315. doi: 10.1042/bj1430311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boyer RF, MacCleary CJ. Superoxide ion as a primary reductant in ascorbate-mediated ferritin iron release. Free Radic Biol Med. 1987;3:389–395. doi: 10.1016/0891-5849(87)90017-7. [DOI] [PubMed] [Google Scholar]
- 137.Keyer K, Imlay JA. Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci U S A. 1996;93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Biemond P, van Eijk HG, Swaak AJ, Koster JF. Iron mobilization from ferritin by superoxide derived from stimulated polymorphonuclear leukocytes. Possible mechanism in inflammation diseases. J Clin Invest. 1984;73:1576–1579. doi: 10.1172/JCI111364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Coudray C, Pucheu S, Boucher F, Arnaud J, de Leiris J, Favier A. Effect of ischemia/reperfusion sequence on cytosolic iron status and its release in the coronary effluent in isolated rat hearts. Biol Trace Elem Res. 1994;41:69–75. doi: 10.1007/BF02917218. [DOI] [PubMed] [Google Scholar]
- 140.Bailey DM, Raman S, McEneny J, Young IS, Parham KL, Hullin DA, Davies B, McKeeman G, McCord JM, Lewis MH. Vitamin C prophylaxis promotes oxidative lipid damage during surgical ischemia–reperfusion. Free Radic Biol Med. 2006;40:591–600. doi: 10.1016/j.freeradbiomed.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 141.Buettner GR, Chamulitrat W. The catalytic activity of iron in synovial fluid as monitored by the ascorbate free radical. Free Radic Biol Med. 1990;8:55–56. doi: 10.1016/0891-5849(90)90144-8. [DOI] [PubMed] [Google Scholar]
- 142.Dabbagh AJ, Trenam CW, Morris CJ, Blake DR. Iron in joint inflammation. Ann Rheum Dis. 1993;52:67–73. doi: 10.1136/ard.52.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Galley HF, Davies MJ, Webster NR. Ascorbyl radical formation in patients with sepsis: effect of ascorbate loading. Free Radic Biol Med. 1996;20:139–143. doi: 10.1016/0891-5849(95)02022-5. [DOI] [PubMed] [Google Scholar]
- 144.Güner G, Kirkali G, Yenisey Ç, Töre İR. Cytosol and serum ferritin in breast carcinoma. Cancer Lett. 1992;67:103–112. doi: 10.1016/0304-3835(92)90132-f. [DOI] [PubMed] [Google Scholar]
- 145.Miyata Y, Koga S, Nishikido M, Hayashi T, Kanetake H. Relationship between serum ferritin levels and tumour status in patients with renal cell carcinoma. BJU Int. 2001;88:974–977. doi: 10.1046/j.1464-4096.2001.01323.x. [DOI] [PubMed] [Google Scholar]
- 146.Basso D, Fabris C, Del Pavero G, Meggiato T, Panozzo MP, Vianello D, Plebani M, Maccarato R. Hepatic changes and serum ferritin in pancreatic cancer and other gastrointestinal diseases: the role of cholestasis. Ann Clin Biochem. 1991;28:34–38. doi: 10.1177/000456329102800105. [DOI] [PubMed] [Google Scholar]
- 147.Hann HW, Lange B, Stahlhut MW, McGlynn KA. Prognostic importance of serum transferrin and ferritin in childhood Hodgkin’s disease. Cancer. 1990;66:313–316. doi: 10.1002/1097-0142(19900715)66:2<313::aid-cncr2820660219>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 148.Jacobs A. Serum ferritin and malignant tumours. Med Oncol Tumor Pharmacother. 1984;1:149–156. doi: 10.1007/BF02934136. [DOI] [PubMed] [Google Scholar]
- 149.Feng D, Nagy JA, Dvorak AM, Dvorak HF. Different pathways of macromolecule extravasation from hyperpermeable tumor vessels. Microvasc Res. 2000;59:24–37. doi: 10.1006/mvre.1999.2207. [DOI] [PubMed] [Google Scholar]
- 150.Rossiello R, Carriero MV, Giordano GG. Distribution of ferritin, transferrin and lactoferrin in breast carcinoma tissue. J Clin Pathol. 1984;37:51–55. doi: 10.1136/jcp.37.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Deubzer B, Mayer F, Kuci Z, Niewisch M, Merkel G, Handgretinger R, Bruchelt G. H2O2-mediated cytotoxicity of pharmacologic ascorbate concentrations to neuroblastoma cells: potential role of lactate and ferritin. Cell Physiol Biochem. 2010;25:767–774. doi: 10.1159/000315098. [DOI] [PubMed] [Google Scholar]
- 152.Rousseau I, Puntarulo S. Ferritin-dependent radical generation in rat liver homogenates. Toxicology. 2009;264:155–161. doi: 10.1016/j.tox.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 153.Baader SL, Bill E, Trautwein AX, Bruchelt G, Matzanke BF. Mobilization of iron from cellular ferritin by ascorbic acid in neuroblastoma SK-N-SH cells: an EPR study. FEBS Lett. 1996;381:131–134. doi: 10.1016/0014-5793(96)00098-1. [DOI] [PubMed] [Google Scholar]
- 154.Olakanmi O, Stokes JB, Britigan BE. Acquisition of iron bound to low molecular weight chelates by human monocyte-derived macrophages. J Immunol. 1994;153:2691–2703. [PubMed] [Google Scholar]
- 155.Cameron E, Campbell A. The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem Biol Interact. 1974;9:285–315. doi: 10.1016/0009-2797(74)90019-2. [DOI] [PubMed] [Google Scholar]
- 156.Cameron E, Campbell A, Jack T. The orthomolecular treatment of cancer. III. Reticulum cell sarcoma: double complete regression induced by high-dose ascorbic acid therapy. Chem Biol Interact. 1975;11:387–393. doi: 10.1016/0009-2797(75)90007-1. [DOI] [PubMed] [Google Scholar]
- 157.Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A. 1976;73:3685–3689. doi: 10.1073/pnas.73.10.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A. 1978;75:4538–4542. doi: 10.1073/pnas.75.9.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Creagan ET, Moertel CG, O’Fallon JR, Schutt AJ, O’Connell MJ, Rubin J, Frytak S. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N Engl J Med. 1979;301:687–690. doi: 10.1056/NEJM197909273011303. [DOI] [PubMed] [Google Scholar]
- 160.Moertel CG, Fleming TR, Creagan ET, Rubin J, O’Connell MJ, Ames MM. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N Engl J Med. 1985;312:137–141. doi: 10.1056/NEJM198501173120301. [DOI] [PubMed] [Google Scholar]
- 161.Drisko JA, Chapman J, Hunter VJ. The use of antioxidants with first-line chemotherapy in two cases of ovarian cancer. J Am Coll Nutr. 2003;22:118–123. doi: 10.1080/07315724.2003.10719284. [DOI] [PubMed] [Google Scholar]
- 162.Riordan HD, Casciari JJ, Gonzalez MJ, Riordan NH, Miranda-Massari JR, Taylor P, Jackson JA. A pilot clinical study of continuous intravenous ascorbate in terminal cancer patients. P R Health Sci J. 2005;24:269–276. [PubMed] [Google Scholar]
- 163.Sestili P, Brandi G, Brambilla L, Cattabeni F, Cantoni O. Hydrogen peroxide mediates the killing of U937 tumor cells elicited by pharmacologically attainable concentrations of ascorbic acid: cell death prevention by extracellular catalase or catalase from cocultured erythrocytes or fibroblasts. J Pharmacol Exp Ther. 1996;277:1719–1725. [PubMed] [Google Scholar]
- 164.Verrax J, Calderon PB. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic Biol Med. 2009;47:32–40. doi: 10.1016/j.freeradbiomed.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 165.Clement MV, Ramalingam J, Long LH, Halliwell B. The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxid Redox Signal. 2001;3:157–163. doi: 10.1089/152308601750100687. [DOI] [PubMed] [Google Scholar]
- 166.Wagner BA, Teesch LM, Buettner GR, Britigan BE, Burns CP, Reszka KJ. Inactivation of anthracyclines by serum heme proteins. Chem Res Toxicol. 2007;20:920–926. doi: 10.1021/tx700002f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Giandomenico AR, Cerniglia GE, Biaglow JE, Stevens CW, Koch CJ. The importance of sodium pyruvate in assessing damage produced by hydrogen peroxide. Free Radic Biol Med. 1997;23:426–434. doi: 10.1016/s0891-5849(97)00113-5. [DOI] [PubMed] [Google Scholar]
- 168.Long LH, Halliwell B. Artefacts in cell culture: pyruvate as a scavenger of hydrogen peroxide generated by ascorbate or epigallocatechin gallate in cell culture media. Biochem Biophys Res Commun. 2009;388:700–704. doi: 10.1016/j.bbrc.2009.08.069. [DOI] [PubMed] [Google Scholar]
- 169.Casciari JJ, Riordan NH, Schmidt TL, Meng XL, Jackson JA, Riordan HD. Cytotoxicity of ascorbate, lipoic acid, and other antioxidants in hollow fibre in vitro tumours. Br J Cancer. 2001;84:1544–1550. doi: 10.1054/bjoc.2001.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem. 1991;266:22028–22034. [PubMed] [Google Scholar]
- 171.Benade L, Howard T, Burk D. Synergistic killing of Ehrlich ascites carcinoma cells by ascorbate and 3-amino-1,2,4,-triazole. Oncology. 1969;23:33–43. doi: 10.1159/000224465. [DOI] [PubMed] [Google Scholar]
- 172.Gaetani GF, Galiano S, Canepa L, Ferraris AM, Kirkman HN. Catalase and glutathione peroxidase are equally active in detoxification of hydrogen peroxide in human erythrocytes. Blood. 1989;73:334–339. [PubMed] [Google Scholar]
- 173.Gaetani GF, Kirkman HN, Mangerini R, Ferraris AM. Importance of catalase in the disposal of hydrogen peroxide within human erythrocytes. Blood. 1994;84:325–330. [PubMed] [Google Scholar]
- 174.Ng CF, Schafer FQ, Buettner GR, Rodgers VGJ. The rate of cellular hydrogen peroxide removal shows dependency on GSH: mathematical insight into in vivo H2O2 and GPx concentrations. Free Radic Res. 2007;41:1201–1211. doi: 10.1080/10715760701625075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sasaki K, Bannai S, Makino N. Kinetics of hydrogen peroxide elimination by human umbilical vein endothelial cells in culture. Biochim Biophys Acta Gen Subj. 1998;1380:275–288. doi: 10.1016/s0304-4165(97)00152-9. [DOI] [PubMed] [Google Scholar]
- 176.Low FM, Hampton MB, Winterbourn CC. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxid Redox Signal. 2008;10:1621–1630. doi: 10.1089/ars.2008.2081. [DOI] [PubMed] [Google Scholar]
- 177.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 178.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 179.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 180.Liu J, Hinkhouse MM, Sun W, Weydert CJ, Ritchie JM, Oberley LW, Cullen JJ. Redox regulation of pancreatic cancer cell growth: role of glutathione peroxidase in the suppression of the malignant phenotype. Hum Gene Ther. 2004;15:239–250. doi: 10.1089/104303404322886093. [DOI] [PubMed] [Google Scholar]
- 181.Oberley LW. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed Pharmacother. 2005;59:143–148. doi: 10.1016/j.biopha.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 182.Glickstein H, Ben El R, Shvartsman M, Cabantchik ZI. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood. 2005;106:3242–3250. doi: 10.1182/blood-2005-02-0460. [DOI] [PubMed] [Google Scholar]
- 183.Browne P, Shalev O, Hebbel RP. The molecular pathobiology of cell membrane iron: the sickle red cell as a model. Free Radic Biol Med. 1998;24:1040–1048. doi: 10.1016/s0891-5849(97)00391-2. [DOI] [PubMed] [Google Scholar]
- 184.Hempel SL, Buettner GR, Wessels DA, Galvan GM, O’Malley YQ. Extracellular Iron(II) can protect cells from hydrogen peroxide. Arch Biochem Biophys. 1996;330:401–408. doi: 10.1006/abbi.1996.0268. [DOI] [PubMed] [Google Scholar]
- 185.Schafer FQ, Qian SY, Buettner GR. Iron and free radical oxidations in cell membranes. Cell Mol Biol (Noisy-le-grand) 2000;46:657–662. [PMC free article] [PubMed] [Google Scholar]
- 186.Jurkiewicz BA, Buettner GR. EPR detection of free radicals in UV-irradiated skin: mouse versus human. Photochem Photobiol. 1996;64 doi: 10.1111/j.1751-1097.1996.tb01856.x. [DOI] [PubMed] [Google Scholar]
- 187.Bloomer SA, Brown KE, Buettner GR, Kregel KC. Dysregulation of hepatic iron with aging: implications for heat stress-induced oxidative liver injury. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1165–R1174. doi: 10.1152/ajpregu.00719.2007. [DOI] [PubMed] [Google Scholar]
- 188.Spencer KT, Lindower PD, Buettner GR, Kerber RE. Transition metal chelators reduce directly measured myocardial free radical production during reperfusion. J Cardiovas Pharmacol. 1998;32:343–348. doi: 10.1097/00005344-199809000-00002. [DOI] [PubMed] [Google Scholar]
- 189.Reif DW. Ferritin as a source of iron for oxidative damage. Free Radic Biol Med. 1992;12:417–427. doi: 10.1016/0891-5849(92)90091-t. [DOI] [PubMed] [Google Scholar]
- 190.Carmine TC, Evans P, Bruchelt G, Evans R, Handgretinger R, Niethammer D, Halliwell B. Presence of iron catalytic for free radical reactions in patients undergoing chemotherapy: implications for therapeutic management. Cancer Lett. 1995;94:219–226. doi: 10.1016/0304-3835(95)03852-n. [DOI] [PubMed] [Google Scholar]
- 191.Persson HL. Radiation-induced lysosomal iron reactivity: implications for radioprotective therapy. IUBMB Life. 2006;58:395–401. doi: 10.1080/15216540600755998. [DOI] [PubMed] [Google Scholar]
- 192.Buettner GR. Ascorbate autoxidation in the presence of iron and copper chelates. Free Radic Res Commun. 1986;1:349–353. doi: 10.3109/10715768609051638. [DOI] [PubMed] [Google Scholar]
- 193.Miller DM, Buettner GR, Aust SD. Transition metals as catalysts of “autoxidation” reactions. Free Radic Biol Med. 1990;8:95–108. doi: 10.1016/0891-5849(90)90148-c. [DOI] [PubMed] [Google Scholar]
- 194.Kimoto E, Tanaka H, Gyotoku J, Morishige F, Pauling L. Enhancement of antitumor activity of ascorbate against Ehrlich ascites tumor cells by the copper: glycylglycylhistidine complex. Cancer Res. 1983;43:824–828. [PubMed] [Google Scholar]
- 195.Batinić-Haberle I, Rebouças JS, Spasojević I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antioxid Redox Signal. 2010;13:877–918. doi: 10.1089/ars.2009.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gardner PR, Nguyen DDH, White CW. Superoxide scavenging by Mn(II/III) tetrakis (1-Methyl-4-pyridyl) porphyrin in mammalian cells. Arch Biochem Biophys. 1996;325:20–28. doi: 10.1006/abbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 197.Zhong W, Yan T, Webber MM, Oberley TD. Alteration of cellular phenotype and responses to oxidative stress by manganese superoxide dismutase and a superoxide dismutase mimic in RWPE-2 human prostate adenocarcinoma cells. Antioxid Redox Signal. 2004;6:513–522. doi: 10.1089/152308604773934279. [DOI] [PubMed] [Google Scholar]
- 198.Tian J, Peehl DM, Knox SJ. Metalloporphyrin synergizes with ascorbic acid to inhibit cancer cell growth through Fenton chemistry. Cancer Biother Radiopharm. 2010;25:439–448. doi: 10.1089/cbr.2009.0756. [DOI] [PubMed] [Google Scholar]
- 199.Batinic-Haberle I, Rajic Z, Tovmasyan A, Reboucas JS, Ye X, Leong KW, Dewhirst MW, Vujaskovic Z, Benov L, Spasojevic I. Diverse functions of cationic Mn(III) N-substituted pyridylporphyrins, recognized as SOD mimics. Free Radic Biol Med. 2011;51:1035–1053. doi: 10.1016/j.freeradbiomed.2011.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ye X, Fels D, Tovmasyan A, Aird KM, Dedeugd C, Allensworth JL, Kos I, Park W, Spasojevic I, Devi GR, Dewhirst MW, Leong KW, Batinic-Haberle I. Cytotoxic effects of Mn(III) N-alkylpyridylporphyrins in the presence of cellular reductant, ascorbate. Free Radic Res. 2011;45:1289–1306. doi: 10.3109/10715762.2011.616199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Klockars M, Weber T, Tanner P, Hellstrom PE, Pettersson T. Pleural fluid ferritin concentrations in human disease. J Clin Pathol. 1985;38:818–824. doi: 10.1136/jcp.38.7.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Weinberger A, Simkin P. Plasma proteins in synovial fluids of normal human joints. Semin Arthritis Rheum. 1989;19:66–76. doi: 10.1016/0049-0172(89)90087-5. [DOI] [PubMed] [Google Scholar]
- 203.Halliwell B. Oxygen radicals, nitric oxide and human inflammatory joint disease. Ann Rheum Dis. 1995;54:505–510. doi: 10.1136/ard.54.6.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Baronzio G, Schwartz L, Kiselevsky M, Guais A, Sanders E, Milanesi G, Baronzio M, Freitas I. Tumor interstitial fluid as modulator of cancer inflammation, thrombosis, immunity and angiogenesis. Anticancer Res. 2012;32:405–414. [PubMed] [Google Scholar]
- 205.Nagy JA, Masse EM, Herzberg KT, Meyers MS, Yeo KT, Yeo TK, Sioussat TM, Dvorak HF. Pathogenesis of ascites tumor growth: vascular permeability factor, vascular hyperpermeability, and ascites fluid accumulation. Cancer Res. 1995;55:360–368. [PubMed] [Google Scholar]
- 206.Nagy JA, Herzberg KT, Dvorak JM, Dvorak HF. Pathogenesis of malignant ascites formation: initiating events that lead to fluid accumulation. Cancer Res. 1993;53:2631–2643. [PubMed] [Google Scholar]
- 207.Roche M, Rondeau P, Singh NR, Tarnus E, Bourdon E. The antioxidant properties of serum albumin. FEBS Lett. 2008;582:1783–1787. doi: 10.1016/j.febslet.2008.04.057. [DOI] [PubMed] [Google Scholar]
- 208.Halliwell B, Gutteridge JMC. The antioxidants of human extracellular fluids. Arch Biochem Biophys. 1990;280:1–8. doi: 10.1016/0003-9861(90)90510-6. [DOI] [PubMed] [Google Scholar]
- 209.Espey MG, Chen P, Chalmers B, Drisko J, Sun AY, Levine M, Chen Q. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic Biol Med. 2011;50:1610–1619. doi: 10.1016/j.freeradbiomed.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Riordan NH, Riordan HD, Meng X, Jackson JA. Intravenous ascorbate as a tumor cytotoxic chemotherapeutic agent. Med Hypotheses. 1995;44:207–213. doi: 10.1016/0306-9877(95)90137-x. [DOI] [PubMed] [Google Scholar]
- 211.Riordan HD, Riordan NH, Jackson JA, Casciari JJ, Hunninghake R, Gonzalez MJ, Mora EM, Miranda-Massari JR, Roscario N. Intravenous vitamin C as a chemotherapy agent: a report on clinical cases. P R Health Sci J. 2004;23:115–118. [PubMed] [Google Scholar]
- 212.Padayatty SJ, Riordan HD, Hewitt SM, Katz A, Hoffer LJ, Levine M. Intravenously administered vitamin C as cancer therapy: three cases. CMAJ. 2006;174:937–942. doi: 10.1503/cmaj.050346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Padayatty SJ, Sun AY, Chen Q, Espey MG, Drisko J, Levine M. Vitamin C: intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS One. 2010;5:e11414. doi: 10.1371/journal.pone.0011414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tian WN, Braunstein LD, Apse K, Pang J, Rose M, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am J Physiol. 1999;276:C1121–C1131. doi: 10.1152/ajpcell.1999.276.5.C1121. [DOI] [PubMed] [Google Scholar]
- 215.Campbell GD, Steinberg MH, Bower JD. Letter: Ascorbic acid-induced hemolysis in G-6-PD deficiency. Ann Intern Med. 1975;82:810–815. doi: 10.7326/0003-4819-82-6-810_1. [DOI] [PubMed] [Google Scholar]
- 216.Mehta JB, Singhal SB, Mehta BC. Ascorbic-acid-induced haemolysis in G-6-PD deficiency. Lancet. 1990;336:944. doi: 10.1016/0140-6736(90)92317-b. [DOI] [PubMed] [Google Scholar]
- 217.Rees DC, Kelsey H, Richards JD. Acute haemolysis induced by high dose ascorbic acid in glucose-6-phosphate dehydrogenase deficiency. BMJ. 1993;306:841–842. doi: 10.1136/bmj.306.6881.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wong K, Thomson C, Bailey RR, McDiarmid S, Gardner J. Acute oxalate nephropathy after a massive intravenous dose of vitamin C. Aust N Z J Med. 1994;24:410–411. doi: 10.1111/j.1445-5994.1994.tb01477.x. [DOI] [PubMed] [Google Scholar]
- 219.Campbell A, Jack T. Acute reactions to mega ascorbic acid therapy in malignant disease. Scott Med J. 1979;24:151–153. doi: 10.1177/003693307902400210. [DOI] [PubMed] [Google Scholar]
- 220.Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiule K, Rousseau C, Robitaille L, Miller WHJ. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol. 2008;19:1969–1974. doi: 10.1093/annonc/mdn377. [DOI] [PubMed] [Google Scholar]
- 221.Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, Pillai MV, Newberg AB, Deshmukh S, Levine M. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One. 2012;7:e29794. doi: 10.1371/journal.pone.0029794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Prasad KN, Sinha PK, Ramanujam M, Sakamoto A. Sodium ascorbate potentiates the growth inhibitory effect of certain agents on neuroblastoma cells in culture. Proc Natl Acad Sci U S A. 1979;76:829–832. doi: 10.1073/pnas.76.2.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kurbacher CM, Wagner U, Kolster B, Andretti PE, Krebs D, Bruckner HW. Ascorbic acid (vitamin C) improves the antineoplastic activity of doxorubicin, cisplatin, and paclitaxel in human breast carcinoma cells in vitro. Cancer Lett. 1996;103:183–189. doi: 10.1016/0304-3835(96)04212-7. [DOI] [PubMed] [Google Scholar]
- 224.Herst PM, Broadley KWR, Harper JL, McConnell MJ. Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radic Biol Med. 2012;52:1486–1493. doi: 10.1016/j.freeradbiomed.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 225.Vollbracht C, Schneider B, Leendert V, Weiss G, Auerbach L, Beuth J. Intravenous vitamin C administration improves quality of life in breast cancer patients during chemo-/radiotherapy and aftercare: results of a retrospective, multicentre, epidemiological cohort study in Germany. In Vivo. 2011;25:983–990. [PubMed] [Google Scholar]
- 226.Terpstra M, Ugurbil K, Tkac I. Noninvasive quantification of human brain ascorbate concentration using 1H NMR spectroscopy at 7 T. NMR Biomed. 2010;23:227–232. doi: 10.1002/nbm.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Terpstra M, Torkelson C, Emir U, Hodges JS, Raatz S. Noninvasive quantification of human brain antioxidant concentrations after an intravenous bolus of vitamin C. NMR Biomed. 2011;24:521–528. doi: 10.1002/nbm.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Omaye ST, Skala JH, Jacob RA. Rebound effect with ascorbic acid in adult males. Am J Clin Nutr. 1988;48:379–380. doi: 10.1093/ajcn/48.2.379. [DOI] [PubMed] [Google Scholar]
- 229.Tsao CS, Leung PY. Urinary ascorbic acid levels following the withdrawal of large doses of ascorbic acid in guinea pigs. J Nutr. 1988;118:895–900. doi: 10.1093/jn/118.7.895. [DOI] [PubMed] [Google Scholar]
- 230.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- 232.Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, Nyska A, Wachsman JT, Ames BN, Basu S, Brot N, FitzGerald GA, Floyd RA, George M, Heinecke JW, Hatch GE, Hensley K, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, II, Rokach J, Shigenaga MK, Sohal RS, Sun J, Tice RR, Van Thiel DH, Wellner D, Walter PB, Tomer KB, Mason RP, Barrett JC. Biomarkers of oxidative stress study II: are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radic Biol Med. 2005;38:698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 233.Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2:78–88. doi: 10.3945/an.110.000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Emir UE, Raatz S, McPherson S, Hodges JS, Torkelson C, Tawfik P, White T, Terpstra M. Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed. 2011;24:888–894. doi: 10.1002/nbm.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lönnrot K, MetsÄ-KetelÄ T, Molnár G, Ahonen JP, Latvala M, Peltola J, PietilÄ T, Alho H. The effect of ascorbate and ubiquinone supplementation on plasma and CSF total antioxidant capacity. Free Radic Biol Med. 1996;21:211–217. doi: 10.1016/0891-5849(95)02207-4. [DOI] [PubMed] [Google Scholar]
- 236.Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51:571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- 237.Voigt K, Kontush A, Stuerenburg HJ, Muench-Harrach D, Hansen HC, Kunze K. Decreased plasma and cerebrospinal fluid ascorbate levels in patients with septic encephalopathy. Free Radic Res. 2002;36:735–739. doi: 10.1080/10715760290032557. [DOI] [PubMed] [Google Scholar]
- 238.Harrison FE, May JM. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic Biol Med. 2009;46:719–730. doi: 10.1016/j.freeradbiomed.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Vissers MCM, Gunningham SP, Morrison MJ, Dachs GU, Currie MJ. Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radic Biol Med. 2007;42:765–772. doi: 10.1016/j.freeradbiomed.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 240.Koh WS, Choi WH, Lee SJ, Park CS, Park CH. Enhancement of plasmacytoma cell growth by ascorbic acid is mediated via glucose 6-phosphate dehydrogenase. Cancer Res Treat. 2007;39:22–29. doi: 10.4143/crt.2007.39.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Castranova V, Wright JR, Colby HD, Miles PR. Ascorbate uptake by isolated rat alveolar macrophages and type II cells. J Appl Physiol. 1983;54:208–214. doi: 10.1152/jappl.1983.54.1.208. [DOI] [PubMed] [Google Scholar]
- 242.Korcok J, Dixon SJ, Lo TCY, Wilson JX. Differential effects of glucose on dehydroascorbic acid transport and intracellular ascorbate accumulation in astrocytes and skeletal myocytes. Brain Res. 2003;993:201–207. doi: 10.1016/j.brainres.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 243.Bergsten P, Yu R, Kehrl J, Levine M. Ascorbic acid transport and distribution in human B lymphocytes. Arch Biochem Biophys. 1995;317:208–214. doi: 10.1006/abbi.1995.1155. [DOI] [PubMed] [Google Scholar]
- 244.May JM, Qu ZC. Chelation of intracellular iron enhances endothelial barrier function: a role for vitamin C? Arch Biochem Biophys. 2010;500:162–168. doi: 10.1016/j.abb.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Reidling JC, Subramanian VS, Dahhan T, Sadat M, Said HM. Mechanisms and regulation of vitamin C uptake: studies of the hSVCT systems in human liver epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1217–G1227. doi: 10.1152/ajpgi.90399.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kramarenko GG, Wilke WW, Dayal D, Buettner GR, Schafer FQ. Ascorbate enhances the toxicity of the photodynamic action of Verteporfin in HL-60 cells. Free Radic Biol Med. 2006;40:1615–1627. doi: 10.1016/j.freeradbiomed.2005.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Steffner RJ, Wu L, Powers AC, May JM. Ascorbic acid recycling by cultured β cells: effects of increased glucose metabolism. Free Radic Biol Med. 2004;37:1612–1621. doi: 10.1016/j.freeradbiomed.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 248.May JM, Qu ZC. Ascorbate-dependent electron transfer across the human erythrocyte membrane. Biochim Biophys Acta Biomembr. 1999;1421:19–31. doi: 10.1016/s0005-2736(99)00107-8. [DOI] [PubMed] [Google Scholar]
- 249.Han M, Pendem S, Teh SL, Sukumaran DK, Wu F, Wilson JX. Ascorbate protects endothelial barrier function during septic insult: role of protein phosphatase type 2A. Free Radic Biol Med. 2010;48:128–135. doi: 10.1016/j.freeradbiomed.2009.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.May J, Li L, Hayslett K, Qu Z-c. Ascorbate transport and recycling by SH-SY5Y neuroblastoma cells: response to glutamate toxicity. Neurochem Res. 2006;31:785–794. doi: 10.1007/s11064-006-9077-z. [DOI] [PubMed] [Google Scholar]
- 251.Duarte TL, Almeida GM, Jones GD. Investigation of the role of extracellular H2O2 and transition metal ions in the genotoxic action of ascorbic acid in cell culture models. Toxicol Lett. 2007;170:57–65. doi: 10.1016/j.toxlet.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 252.Peterkofsky B, Prather W. Cytotoxicity of ascorbate and other reducing agents towards cultured fibroblasts as a result of hydrogen peroxide formation. J Cell Physiol. 1977;90:61–70. doi: 10.1002/jcp.1040900109. [DOI] [PubMed] [Google Scholar]
- 253.Johnson NA, Chen BH, Sung SY, Liao CH, Hsiao WC, Chung LWK, Hsieh CL. A novel targeting modality for renal cell carcinoma: human osteocalcin promoter-mediated gene therapy synergistically induced by vitamin C and vitamin D3. J Gene Med. 2010;12:892–903. doi: 10.1002/jgm.1516. [DOI] [PubMed] [Google Scholar]
- 254.Park CH, Amare M, Savin MA. Growth suppression of human leukemic cells in vitro by L-ascorbic acid. Cancer Res. 1980;40:1062–1065. [PubMed] [Google Scholar]
- 255.Jamison JM, Gilloteaux J, Nassiri MR, Venugopal M, Neal DR, Summers JL. Cell cycle arrest and autoschizis in a human bladder carcinoma cell line following vitamin C and vitamin K3 treatment. Biochem Pharmacol. 2004;67:337–351. doi: 10.1016/j.bcp.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 256.Rozanova N, Zhang JZ, Heck DE. Catalytic therapy of cancer with porphyrins and ascorbate. Cancer Lett. 2007;252:216–224. doi: 10.1016/j.canlet.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 257.Berndt C, Kurz T, Bannenberg S, Jacob R, Holmgren A, Brunk UT. Ascorbate and endocytosed Motexafin gadolinium induce lysosomal rupture. Cancer Lett. 2011;307:119–123. doi: 10.1016/j.canlet.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 258.Solovieva ME, Soloviev VV, Akatov VS. Vitamin B12b increases the cytotoxicity of short-time exposure to ascorbic acid, inducing oxidative burst and iron-dependent DNA damage. Euro J Phmacol. 2007;566:206–214. doi: 10.1016/j.ejphar.2007.03.035. [DOI] [PubMed] [Google Scholar]
- 259.Biswas S, Zhao X, Mone AP, Mo X, Vargo M, Jarjoura D, Byrd JC, Muthusamy N. Arsenic trioxide and ascorbic acid demonstrate promising activity against primary human CLL cells in vitro. Leuk Res. 2010;34:925–931. doi: 10.1016/j.leukres.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hahm E, Jin DH, Kang JS, Kim YI, Hong SW, Lee SK, Kim HN, Jung DJ, Kim JE, Shin DH, Hwang YI, Kim YS, Hur DY, Yang Y, Cho D, Lee MS, Lee WJ. The molecular mechanisms of vitamin C on cell cycle regulation in B16F10 murine melanoma. J Cell Biochem. 2007;102:1002–1010. doi: 10.1002/jcb.21336. [DOI] [PubMed] [Google Scholar]
- 261.Josephy PD, Palcic B, Skarsgard LD. Ascorbate-enhanced cytotoxicity of misonidazole. Nature. 1978;271:370–372. doi: 10.1038/271370a0. [DOI] [PubMed] [Google Scholar]
- 262.Belin S, Kaya F, Duisit G, Giacometti S, Ciccolini J, Fontes M. Antiproliferative effect of ascorbic acid is associated with the inhibition of genes necessary to cell cycle progression. PLoS One. 2009;4:e4409. doi: 10.1371/journal.pone.0004409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Verrax J, Stockis J, Tison A, Taper HS, Calderon PB. Oxidative stress by ascorbate/menadione association kills K562 human chronic myelogenous leukaemia cells and inhibits its tumour growth in nude mice. Biochem Pharmacol. 2006;72:671–680. doi: 10.1016/j.bcp.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 264.Ohtani S, Iwamaru A, Deng W, Ueda K, Wu G, Jayachandran G, Kondo S, Atkinson EN, Minna JD, Roth JA, Ji L. Tumor suppressor 101F6 and ascorbate synergistically and selectively inhibit non-small cell lung cancer growth by caspase-independent apoptosis and autophagy. Cancer Res. 2007;67:6293–6303. doi: 10.1158/0008-5472.CAN-06-3884. [DOI] [PubMed] [Google Scholar]
- 265.Takemura Y, Satoh M, Satoh K, Hamada H, Sekido Y, Kubota S. High dose of ascorbic acid induces cell death in mesothelioma cells. Biochem Biophys Res Commun. 2010;394:249–253. doi: 10.1016/j.bbrc.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 266.Yeom CH, Lee G, Park JH, Yu J, Park SI, Yi SY, Lee HR, Hong YS, Yang J, Lee S. High dose concentration administration of ascorbic acid inhibits tumor growth in BALB/C mice implanted with sarcoma 180 cancer cells via the restriction of angiogenesis. J Transl Med. 2009;7:70–79. doi: 10.1186/1479-5876-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Park S, Ahn ES, Lee S, Jung M, Park JH, Yi SY, Yeom CH. Proteomic analysis reveals upregulation of RKIP in S-180 implanted BALB/C mouse after treatment with ascorbic acid. J Cell Biochem. 2009;106:1136–1145. doi: 10.1002/jcb.22097. [DOI] [PubMed] [Google Scholar]