

Abstract
Tangle-predominant dementia (TPD) patients exhibit cognitive decline that is clinically similar to early to moderate-stage Alzheimer disease (AD), yet autopsy reveals neurofibrillary tangles in the medial temporal lobe composed of the microtubule-associated protein tau without significant amyloid-beta (Aβ)-positive plaques. We performed a series of neuropathological, biochemical and genetic studies using autopsy brain tissue drawn from a cohort of 34 TPD, 50 AD and 56 control subjects to identify molecular and genetic signatures of this entity. Biochemical analysis demonstrates a similar tau protein isoform composition in TPD and AD, which is compatible with previous histological and ultrastructural studies. Further, biochemical analysis fails to uncover elevation of soluble Aβ in TPD frontal cortex and hippocampus compared to control subjects, demonstrating that non-plaque-associated Aβ is not a contributing factor. Unexpectedly, we also observed high levels of secretory amyloid precursor protein α (sAPPα) in the frontal cortex of some TPD patients compared to AD and control subjects, suggesting differences in APP processing. Finally, we tested whether TPD is associated with changes in the tau gene (MAPT). Haplotype analysis demonstrates a strong association between TPD and the MAPT H1 haplotype, a genomic inversion associated with some tauopathies and Parkinson disease (PD), when compared to age-matched control subjects with mild degenerative changes, i.e., successful cerebral aging. Next-generation resequencing of MAPT followed by association analysis shows an association between TPD and two polymorphisms in the MAPT 3′ untranslated region (UTR). These results support the hypothesis that haplotype-specific variation in the MAPT 3′ UTR underlies an Aβ-independent mechanism for neurodegeneration in TPD.
Keywords: Dementia, Neurofibrillary tangle, Tau, Amyloid, MAPT, 3′ Untranslated region, Aging, Alzheimer’s disease, sAPPα
Introduction
Classically, Alzheimer disease (AD) is associated with amyloid plaques and neurofibrillary tangles (NFT). Intriguingly, ~5 % of dementia patients develop a limbic tauopathy, termed tangle-predominant dementia (TPD) among numerous other names [7, 47, 61]. TPD patients exhibit NFT in the medial temporal lobe, progressing to a regional distribution corresponding to moderate-stage AD (Braak stages III–IV) [10]. However, the severity of NFT more closely resembles end-stage disease [48]. Neither clinical features nor diagnostic tests can differentiate TPD from early to moderate AD. Thus, the only way to diagnose TPD is post-mortem [33].
The etiology of TPD is not known. Authors have grouped TPD with frontotemporal lobar degeneration (FTLD), but this classification is imperfect given the differences in symptomatology and neuropathological features [12, 29]. Tauopathies are classified neuropathologically using the distribution, morphology and ultrastructure of NFTs, yet no features can differentiate the NFTs in TPD from those in moderate-stage AD. The prevalence of NFT in normal elderly individuals has prompted suggestions that TPD is a form of pathological or “accelerated” aging [9, 34, 50, 54]. Finally, TPD may be an AD variant [34]. The amyloid hypothesis posits that increased Aβ is the disease trigger in AD, leading to NFT formation and neurodegeneration [24]. The toxic species in AD may be soluble Aβ, rather than Aβ deposited in plaques [63]. While the possibility that APP or its catabolites contribute to TPD has not previously been tested experimentally, the consensus criteria require the presence of insoluble Aβ deposited in plaques together with NFT for the neuropathological diagnosis of AD [27, 42].
The discovery of highly penetrant MAPT mutations in rare families with FTLD demonstrates that tau dysfunction is sufficient to independently cause neurodegeneration [20]. Some MAPT mutations, clustered around exon 10, influence splicing, leading to accumulation of tau having four microtubule binding repeat domains (4R) over those with three repeat domains (3R) [62]. Over 40 mutations result in FTLD-tau, but previous studies on TPD have failed to detect a MAPT mutation [66]. MAPT is within a ~900 kb ancestral genomic inversion that defines two haplotypes, H1 and H2 [56]. These haplotypes are in complete linkage disequilibrium and do not recombine. Sporadic tauopathies such as progressive supranuclear palsy and corticobasal degeneration as well as Parkinson disease are associated with the H1 haplotype [6, 8, 18]. There are conflicting reports concerning an association of MAPT with AD [1, 43, 45]. How H1 confers risk for tauopathy is unclear, but increased expression of 4R tau mRNA isoforms has been implicated [46], albeit controversially [25]. Other factors may play a role. For example, elements in the tau 3′ UTR regulate mRNA stability and localization leading to speculation that polymorphisms in this region underlie disease risk [4, 5, 62].
We demonstrate here that TPD patients develop Alzheimer-type NFT that are biochemically identical to those in early to moderate-stage AD, yet soluble Aβ is not detectable. Furthermore, we observed evidence of preferential non-amyloidogenic APP processing in TPD brain. Our genetic analysis demonstrates that TPD is associated with the MAPT H1 haplotype in the absence of a coding region mutation. We also found a significant association between TPD and variation in the MAPT 3′ UTR, suggesting a novel mechanism whereby post-transcriptional regulation of MAPT contributes to tauopathy.
Materials and methods
Patient samples
Autopsy brain samples were obtained from seven centers (Table 1). The primary source of material was the brain bank at Columbia University Medical Center (New York, NY, USA; Supplementary Table 1). Secondary sources were the University of California San Diego (San Diego, CA, USA), the University of Kentucky (Lexington, KY, USA), the Banner Sun Health Research Institute (Sun City, AZ, USA), Northwestern University (Chicago, IL, USA), the University of Washington (Seattle, WA, USA) and Washington University (St. Louis, MO, USA). Patient data for each component of this study are summarized in Supplementary Table 2. Neuropathological examination was per the protocols of the respective institutions. Inclusion criteria for TPD were (1) frequent NFT corresponding to Braak NFT stage III–IV [11] and no or very rare NFT in the frontal, parietal or occipital cortex, (2) no more than sparse amyloid plaques (CERAD [41] score 0 or A) and (3) no other neuropathological substrate for dementia. All TPD cases had been clinically classified pre-mortem as either possible or probable AD (n = 31) or mild cognitive impairment (n = 3) by their respective source institutions. For APOE genotype comparisons, neuropathologically confirmed AD patients aged 75 years or higher from the CUMC cohort categorized as CERAD plaque score of C and Braak NFT stage of V–VI were used. Successful cerebral aging was defined as (1) age greater than or equal to 80 years, (2) CERAD plaque score of 0 and (3) Braak NFT stage of 0–II. All subjects were of Caucasian ancestry.
Table 1.
Summary of patient data
| Classification | n | Male | Female | Average age, years (range) | Braak NFT | CERAD plaque score | Clinical diagnosis |
|---|---|---|---|---|---|---|---|
| Control | 56 | 23 | 33 | 86.9 (67–108) | 0–II | 0–A | Normal |
| TPD | 34 | 11 | 23 | 90.2 (65–103) | III–IV | 0–A | AD or MCI |
| AD | 50 | 19 | 31 | 86.7 (73–104) | V–VI | B–C | AD |
TPD tangle-predominant dementia, AD Alzheimer disease, MCI mild cognitive impairment
Neuropathological analysis
Patient material from Columbia University was subjected to a detailed neuropathological analysis (Supplementary Table 1). Immunohistochemistry was performed on 6 μm paraffin-embedded sections as previously described [15] using various antisera (Supplementary Table 3). For transmission electron microscopy, a portion of CA1, previously fixed in 10 % neutral-buffered formalin, was post-fixed with 2.5 % glutaraldehyde in 0.1 M Sorenson’s buffer (pH 7.2) followed by 1 % OsO4 in Sorenson’s buffer for 1 h and embedded in Lx-112 (Ladd Research Industries, Inc.). 60 nm sections were stained with uranyl acetate and lead citrate and examined under a JEOL JEM-1200 EXII electron microscope and imaged using an ORCA-HR digital camera (Hamamatsu Photonics, Japan).
Biochemical analysis
For biochemical analysis of tau, protein extracts were prepared from fresh-frozen human brain as described by others with modifications [58]. Briefly, fresh-frozen brain tissue was homogenized using 10 volumes (wt/vol) of extraction buffer containing 20 mM (4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid]) (HEPES), pH 7.4, 100 mM NaCl, 20 mM NaF, 1 % Triton X-100, 1 mM sodium orthovanadate, 5 mM EDTA containing a Complete Mini protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA) supplemented with 2 mM PMSF using 15 strokes with a Teflon-coated pestle. Homogenates were centrifuged at 3000×g at 4 °C for 5 min. The supernatant (crude total tau fraction) was aliquoted and stored at −80 °C. Preparation of sarkosyl-soluble and insoluble fractions was carried out as described by others [37]. Briefly, homogenates were centrifuged at 27,000×g for 20 min. The pellet was resuspended in buffer containing 0.8 M NaCl and 10 % sucrose at 10 ml/g of initial tissue and recentrifuged at 27,000×g for 20 min. The supernatant was incubated in the presence of 1 % sarkosyl for 1 h at 25 °C. Finally, the sample was centrifuged at 100,000×g for 2 h. The resulting pellet was considered the sarkosyl-insoluble tau fraction. Samples were resolved by 10 % SDS-PAGE, and analyzed by immunoblot with various antisera (Supplementary Table 3). Some sarkosyl-insoluble fractions were applied to grids, allowed to dry for 20 min and negatively stained with 1 % lithium phosphotungstate and examined by electron microscopy as above.
For biochemical analysis of Aβ, APP and sAPPα/β, preparation of protein extracts was performed as described by others with modifications [55]. Fresh-frozen autopsy brain tissue was homogenized with 10 volumes (wt/vol) of buffer [250 mM sucrose, 20 mM Tris–HCl (pH 7.4), 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM ethylene glycol tetraacetic acid (EGTA)] containing a Complete Mini protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) using 20 strokes with a Teflon-coated pestle, aliquoted and frozen at −80 °C. Protein concentration was measured by bicinchoninic acid protein assay (Pierce, Rockford, IL, USA). For extraction of soluble Aβ, homogenate was combined with an equal volume of 0.4 % diethylamine (DEA) in 100 mM NaCl and centrifuged at 100,000×g for 1 h at 4 °C. Then, the supernatant was combined with an equal volume of 0.5 M Tris base, pH 6.8. ELISAs were performed using Aβ x-40 and x-42 BetaMark chemiluminescent kits (Covance Inc., Princeton, NJ, USA). For immunoblotting, the homogenates were separated into membrane (pellet) and cytosolic (supernatant) fractions by centrifugation at 100,000×g for 1 h at 4 °C. The pellet was resuspended in homogenization buffer, subjected to SDS-PAGE, transferred to nitrocellulose and probed with various antisera (Supplementary Table 3) and visualized by chemiluminescence. Densitometric analysis was performed using NIH Image J.
Resequencing and association analysis
For genomic DNA isolation, fresh-frozen brain was lysed overnight at 55 °C under continuous rotation in 500 μl of buffer [4 M urea, 10 mM of EDTA, 0.5 % sarkosyl, 0.1 M Tris HCl pH (8.0), 0.1 M NaCl and 20 mg/ml proteinase K]. DNA was purified by phenol–chloroform extraction. MAPT target enrichment was performed with the RDT1000 system (RainDance Technologies, Lexington, MA, USA) using 464 primer pairs spanning >99.9 % of MAPT totaling 140,252 bp (hg18; chr17: 41,324,942–41,465,194) and 243,995 bp of amplicons designed using Primer3 software (Supplementary Table 4). Amplicons were sequenced on the 454 platform (Roche 454 Life Sciences, Branford, CT, USA). Performance was analyzed using CLC Genomics Workbench (CLC bio, Cambridge, MA, USA; Supplementary Table 5). Variants were identified with gsMapper (Supplementary Table 6; Roche). Variants were called if identified on three or more reads, with a total read coverage of six and counted only if they were observed on forward and reverse reads. Variants on 10–90 % of reads were called as heterozygous and those on >90 % homozygous. Allele counts and frequencies were calculated and used to generate p values (Fisher’s exact test). Variants meeting one the following criteria (after filtering for H2 haplotype-tagging variants) were included for validation: (1) in coding regions or untranslated regions, (2) TPD specific and control specific, (3) p < 0.025 (Fisher’s exact test, allelic, unadjusted). Validation and genotyping of variants was performed on a Sequenom MassArray iPlex platform (Sequenom, San Diego, CA, USA) or Sanger sequencing. Subjects were also genotyped for APOE status using the rs7412 and rs429358 polymorphisms [13, 22].
Quantitative real-time polymerase chain reaction (QPCR)
Fresh-frozen brain tissue was pulverized under liquid nitrogen, lysed in QIAzol and homogenized using a QIA-shredder spin column (Qiagen, Valencia, CA, USA). RNA was extracted using an RNeasy Mini kit (Qiagen). cDNA synthesis was performed using a First Strand cDNA Synthesis Kit (Origene, Rockville, MD, USA) and used as template (1:4 dilution) in 20 μl reactions. Primers and probes specific for 3R tau, 4R tau, total tau and GAPDH and TaqMan Gene Expression Master Mix (Applied Biosystems, Foster, CA, USA) were used for reactions on a Mastercycler ep realplex (Eppendorf, Hauppauge, NY, USA), using the following settings: 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. For QPCR of APP mRNA, FastStart Universal SYBR Green Master (Roche Applied Science) was used with primers described by others [23]. The mRNA level was normalized to GAPDH.
Statistical analysis
For ELISA, quantitative immunoblotting, and QPCR experiments, the statistical significance was determined by one-way ANOVA and Tukey test or Student’s t test in GraphPad Prism (GraphPad Software, La Jolla, CA, USA). Statistical outliers and specimens with measurement errors were excluded. For APOE comparisons, a Fisher’s exact test performed in Microsoft Excel was used. For MAPT haplotype comparisons and single locus associations, a Chi-squared test was performed using plink [51].
Results
Neuropathological findings
We retrospectively reviewed a consecutive brain autopsy series of 992 patients performed at Columbia University Medical Center. Of the 336 patients clinically classified as possible or probable AD [40], 13 meet the neuropathological criteria for TPD [65]. This represents 3.8 % of all dementia patients, but increases to 7.3 % of patients 90 years or above. There is a female preponderance and an average age of death of 86.9 years (range 65–97 years), which is older than the average for AD patients in this series (74 years). While accurate estimates are unavailable, in part because of variability in the case definition, these data support the argument that TPD is more prevalent than recognized, which is consistent with previous reports [7, 30, 61]. Should estimates of 3–5 % of dementia patients prove accurate, TPD may be among the more common neurodegenerative disorders, affecting between 1.1 and 1.8 million individuals globally.
Post-mortem examination of these 13 TPD patients reveals a pattern reminiscent of early to moderate-stage AD. There is gross medial temporal lobe atrophy compared to controls. Unlike most late-stage AD patients, frontal, parietal and occipital cortices are preserved in TPD. Microscopically, TPD brains exhibit severe medial temporal lobe tauopathy with frequent NFT. All these cases exhibit numerous extracellular (“ghost”) tangles (Fig. 1a–g). Consistent with previous studies, NFT in TPD are immunopositive with specific antisera to 3R and 4R tau (Fig. 1h, i), as well as for various phospho-tau specific epitopes (Supplementary Table 7), which is the same profile seen in AD and certain rare tauopathies [28, 31, 48]. We confirmed that extracellular NFT have disproportionate immunolabeling for 3R tau in TPD (data not shown), as previously reported [31]. To determine whether there are unique ultrastructural features in TPD, electron microscopy was performed in the cornu ammonis 1 (CA1) sector of the hippocampal formation. Examination of epoxy resin ultrathin sections shows filaments that are suggestive of paired-helical filaments (PHFs) in TPD (n = 4), as is observed in AD (Fig. 1j–l) [36]. Ultrathin sections were also evaluated for insoluble amyloid deposits and inclusions, but none are observed. Aside from NFT, there are no ubiquitin-positive inclusions and no more than incidental α-synuclein-positive inclusions in the locus coeruleus, pars compacta of the substantia nigra, hypothalamus and substantia innominata in two patients. Vascular disease is frequent, as is common in aging, but there is no ischemic injury sufficient to cause dementia.
Fig 1.

Tangle-predominant dementia (TPD) patients exhibit neurofibrillary degeneration in a pattern similar to moderate-stage Alzheimer disease (AD). Immunohistochemical staining with phospho-tau (p-tau) specific antisera (AT8) demonstrates neurofibrillary tangles (NFT) and atrophy of the medial temporal lobe in TPD (a) and AD (b) compared to controls (c). Scale bar 7.5 mm. P-tau immunohistochemical (d) and Bielschowsky silver (e) stains demonstrate extensive neurofibrillary degeneration in TPD (CA1). Scale bar 200 μM. f Bielschowsky silver stain highlights the characteristic “flame” shape of the neuronal NFT. g Many NFT are silver-negative “ghost” tangles that are often associated with abnormal degenerating argyrophilic neurites. h, i NFT in the hippocampal formation are immunohistochemically positive for 3R (RD3, 8E6/C11) and 4R tau (RD4, 1E1/A6). Scale bar 50 μm. j Ultrastructural analysis of epoxy resin ultrathin sections from CA1 demonstrates an intracellular NFT (arrowheads). Scale bar 5 μm. k, l High-power imaging reveals filamentous structures suggestive of paired-helical filaments (PHF) in TPD and AD. Scale bar 200 nm
Biochemical and ultrastructural characterization of NFT in TPD
To determine whether differences in tau isoform expression occur in TPD, a biochemical analysis was performed in the Brodmann area 9 (BA9) region of the frontal lobe and CA1 using an expanded cohort that included specimens from multiple AD research centers in the United States. In both TPD (n = 13) and control (n = 12) subjects, immunoblots using total protein from BA9 and probed for tau reveal the three major bands at 55, 64 and 69 kD (Fig. 2a). In CA1, an additional high-molecular weight band is present at ~105 kD, likely representing tau aggregates. Immunoblots using antisera specifically recognizing 3R and 4R tau show no difference in the levels or ratio of tau isoforms between TPD (n = 13) and control (n = 11; Fig. 2b, c). Fractions enriched for insoluble tau isolated by sarkosyl extraction from CA1 and examined by immunoblot reveal no difference in the tau isoform expression in TPD (n = 5) and in AD (n = 6; Fig. 2d). Ultrastructural studies of sarkosyl fractions reveal predominantly PHFs (Fig. 2e). Together, the NFT in TPD are regionally, histologically, biochemically and ultrastructurally similar to those in early to moderate-stage AD.
Fig 2.

Tangle-predominant dementia (TPD) patients exhibit neurofibrillary tangles (NFT) that are biochemically similar to those occurring in Alzheimer disease (AD). a Representative immunoblot using antisera targeting total tau (HT7) and total protein samples from the frontal cortex (BA9) and hippocampus (CA1) demonstrate 69, 64 and 55 kD bands in TPD and control. A high-molecular weight ~105 kD band is observed in TPD in CA1. b Representative immunoblot using antisera to 3R and 4R tau show similar banding in TPD and controls. c Quantification of 3R and 4R tau shows no difference in the levels or ratio in TPD compared to control in BA9. Comparisons are by Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001. d Representative immunoblot with antisera recognizing total tau (HT7) on sarkosyl-insoluble tau fractions reveals similar banding patterns. e Ultrastructural examination of the sarkosyl-insoluble tau fractions confirms the presence of PHFs in TPD. Scale bar 200 nm
Biochemical characterization of Aβ and APP in TPD
Next, we assessed Aβ in TPD. Aβ is derived from the amyloid precursor protein (APP) [59]. Proteolysis of APP by α-secretase or β-secretase produces secreted N-terminal fragments termed sAPPα and sAPPβ, respectively, as well as C-terminal fragments (CTFs). Cleavage by γ-secretase of the β-CTF yields Aβ peptides of predominantly 40 or 42 amino acids. While, immunohistochemistry shows no significant Aβ deposition in TPD (Fig. 3a–c), soluble Aβ is histologically invisible. ELISAs show significantly lower soluble Aβ42 levels in TPD (n = 11, p < 0.001) and controls (n = 16, p < 0.05) as compared to AD (n = 8) in BA9 (Fig. 3d). ELISAs also show significantly lower levels of soluble Aβ42 in TPD (n = 6, p < 0.001) and controls (n = 5, p < 0.001) compared to AD (n = 5) in CA1, a region undergoing neurodegeneration in TPD (Fig. 3d). Aβ42 levels are lower in TPD when compared to control subjects in BA9, but this does not reach statistical significance. Measurements of the less fibrillogenic Aβ40 species [32] reveal significantly lower soluble Aβ40 levels in TPD (p < 0.001) and control (p < 0.01) than AD in BA9 but not CA1 (Fig. 3e). There is a higher Aβ42/40 ratio in AD in CA1 compared to TPD (p < 0.001) and control (p < 0.001), but there is no difference between TPD and control (Fig. 3f). These findings show that TPD brain parenchyma has low levels of soluble Aβ when compared to AD. The control brains have variable levels of soluble Aβ that overlap with those observed in AD and TPD.
Fig 3.

Tangle-predominant dementia (TPD) is associated with low levels of Aβ. a–c Immunohistochemistry with antisera targeting Aβ in the frontal cortex (BA9) reveals Aβ deposition in control and Alzheimer disease (AD), but not TPD. Scale bar 1 mm. d ELISA performed in BA9 and the hippocampus (CA1) shows that AD patients have significantly higher Aβ42 levels than TPD and controls. (e) In BA9, AD patients have significantly higher Aβ40 than TPD patients and control, but there is no difference in CA1. f The ratio of Aβ42/Aβ40 is not different in BA9 among TPD, AD and control, but is significantly increased in AD compared to TPD and controls in CA1. Comparisons are by one-way ANOVA and Tukey’s test, *p < 0.05, **p < 0.01, ***p < 0.001
Low Aβ may arise from decreased production, decreased fibrillization or increased clearance. Quantitative immunoblots reveal a significantly reduced level of the APP holoprotein in TPD (n = 14) compared to controls (n = 11, p < 0.001) in BA9 (Fig. 4a, b). We observed decreased levels of full-length APP in AD as well (data not shown), consistent with previous reports [16, 64]. TPD is unlike AD in that BA9 is preserved, leading us to conclude that low APP levels in TPD reflect differences in underlying APP metabolism rather than neuronal loss and gliosis. Using specific antisera recognizing neoepitopes formed by secretase cleavage, we also found significantly lower sAPPβ levels (p < 0.001) in TPD (n = 14) and significantly higher sAPPα (p < 0.05) compared to controls (n = 11) in BA9. Finally, there is no difference in the levels of APP mRNA among TPD (n = 8), AD (n = 6) and control (n = 9) in BA9 (Fig. 4c), suggesting that non-amyloidogenic processing contributes to decreased production of Aβ in TPD.
Fig 4.
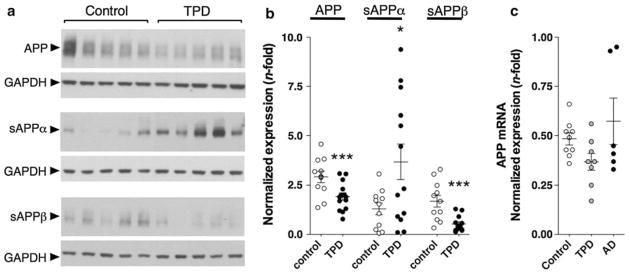
Tangle-predominant dementia (TPD) is associated with alterations in APP metabolism. a, b Representative quantitative immunoblot on extracts from BA9 normalized to GAPDH shows significantly lower levels of APP in TPD than controls. TPD patients have significantly higher levels of sAPPα and significantly lower levels of sAPPβ than controls in the frontal cortex. c There is no difference in the levels of the APP mRNA among TPD, AD and control. Comparisons are by Student’s t test (b) or one-way ANOVA/ Tukey’s test (c) or *p < 0.05, **p < 0.01, ***p < 0.001
Apolipoprotein E (ApoE) alleles correlate with AD risk and amyloid plaque load [14, 53]. We determined the ApoE allele frequency and found both a significant decrease in ε4 (p = 5.78 × 10−7) in TPD (n = 32) compared to age-matched AD (n = 50) and an increase in ε2 (p = 5.58 × 10−5) and ε3 (p = 0.037; Supplementary Table 8), which is consistent with previous studies [28, 34]. Compared to controls (n = 56), there are decreased ε4 and increased ε2 frequencies in TPD, but these differences are not significant. These data support the well-established finding that ApoE ε4 is associated with Aβ deposition and ε2 is protective [14].
Genetic analysis of MAPT in TPD
Next, we determined whether TPD is associated with the tau gene. Sequencing of the coding regions of all MAPT exons and directly flanking introns in 27 TPD patients fails to uncover a potential pathogenic mutation, which is consistent with previous reports [66]. Therefore, we hypothesized that TPD is associated with variation outside of the coding regions. Using markers previously employed to tag MAPT haplotype diversity [49], we do not observe a difference in the common H1 subhaplotypes, including H1c, between TPD (n = 34) and controls (n = 48). However, there is a difference in the H2 frequency in TPD compared to controls (p = 0.015, Table 2), but this finding is not significant following Bonferroni correction (p = 0.075). Post hoc analysis reveals that the trend toward significance is derived from differences between TPD patients and a subset of controls. Neurodegeneration is common in elderly individuals classified as cognitively normal [9, 50]. Employing a model proposed by Rowe and Kahn [52], we identified a subset of our oldest-old controls (average age = 89.3 years) that are exceptional based on their mild NFT burden and absence of amyloid plaques. This control group, that we termed successful cerebral aging, represents the ultimate in healthy brain aging. When successful cerebral aging controls (n = 28) are compared directly to TPD (Table 2), the difference in H1 allele frequency is highly significant (p = 0.004), even after adjusting for multiple comparisons (p = 0.022). These results are consistent with the hypothesis that the H1 haplotype is a risk factor for limbic NFT formation in TPD and that H2 is protective.
Table 2.
Association of common MAPT haplotypes and subhaplotypes with TPD
| TPD (n = 34)
|
v. Control (n = 48)
|
v. Successful cerebral aging (n = 28)
|
||||
|---|---|---|---|---|---|---|
| Haplotypea | Alleles | Frequency | Frequency | p (corrected) | Frequency | p (corrected) |
| H2a | AGGCCG | 0.21 | 0.39 | 0.015 (0.075) | 0.45 | 0.004 (0.022) |
| H1b | GGGCTA | 0.12 | 0.10 | 0.685 (1.000) | 0.09 | 0.551 (1.000) |
| H1c | AAGTTG | 0.16 | 0.09 | 0.228 (1.000) | 0.11 | 0.443 (1.000) |
| H1d | AAGCTA | 0.04 | 0.05 | 0.659 (1.000) | 0.06 | 0.646 (1.000) |
| H1e | AGGCTA | 0.15 | 0.10 | 0.275 (1.000) | 0.07 | 0.172 (0.860) |
Significant values in bold (Chi-squared test), Bonferroni correction for five tests
TPD tangle-predominant dementia
Haplotypes and subhaplotypes based on Pittman et al. [49] (rs1467967, rs242557, rs3 rs2471738, rs9468 and rs7521)
To identify genetic variation that may be associated with TPD risk, we resequenced MAPT in a discovery cohort of ten TPD patients and five successful cerebral aging controls. We focused on comparing TPD to successful cerebral aging controls, which represent the extremes of limbic neurofibrillary degeneration, to minimize misclassification of patients. Resequencing was performed using multiplex PCR for target enrichment followed by large-scale parallel pyrosequencing of these amplicons (Supplementary Table 5). The target region is 140 kb in total length and completely contained within the ancestral inversion, encompassing all of MAPT, including the promoter (which overlaps with LOC100128977), introns, exons and untranslated regions, as well as the saitohin gene (STH), LOC100130148, and ~2 kb of KIAA1267. We achieved a mean base coverage of 109 reads and an average completeness of 99.5 % (Supplementary Table 5). We identified a total of 1,236 variants, 705 of which are found in dbSNP and the remaining 531 are novel (Supplementary Table 6). 15 variants are within MAPT coding regions, 13 of which are known (rs10445337, rs1052551, rs1052553, rs11568305, rs17651549, rs17652121, rs2258689, rs62063786, rs62063787, rs62063845, rs63750072, rs63750222, rs63750417). Of these variants, 11 reside in exons that are not expressed in the central nervous system (i.e., exons 4a, 6 and 8) [3]. The remaining four coding region variants in exons 7 and 9 are synonymous. Consistent with previous reports, much of the variation is derived from differences between H1 and H2, which are in complete linkage disequilibrium [56].
Next, an association analysis was performed testing 20 variants identified by resequencing (2 coding and 18 non-coding). Variants were selected for validation and analysis based on their frequency, genomic location and p value. The two most significant associations (rs5820605; p = 0.032 and rs35134656; p = 0.015), identified when the TPD patients (n = 34) are compared to all controls (n = 48), do not survive strict Bonferroni correction for multiple testing (Table 3, Supplementary table 9). However, when TPD is directly compared to successful cerebral aging controls (n = 28), the differences in these two variants are highly significant, with both maintaining significance following correction: rs5820605 (p = 0.002, OR 0.32, 95 % CI = 0.15–0.67 and rs35134656 (p = 0.002, OR 4.76, CI = 1.66–13.66). The frequency of rs5820605 is 0.33 in TPD, but increases to 0.61 in successful cerebral aging, suggesting that it may be protective. In contrast, rs35134656 has a frequency of 0.09 in the controls, but increases to 0.32 in TPD, suggesting that it is a risk allele. Finally, we asked whether rs5820605 and rs35134656 are in linkage disequilibrium (LD) with any of the MAPT subhaplotype-tagging SNPs. We found that the H2-tagging SNP rs9486 is in complete LD with rs5820605 and rs35134656 suggesting that both these variants are on the H1 background (Fig. 5). Furthermore, rs5820605 and rs35134656 are in LD with each other.
Table 3.
MAPT single marker associations in TPD
| TPD (n = 34)
|
Control (n = 48)
|
Successful cerebral aging (n = 28)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Marker | MAF | MAF | p (corrected) | OR | CI (95 %) | MAF | p (corrected) | OR | CI (95 %) |
| rs5820605 | 0.33 | 0.50 | 0.032 (0.630) | 0.49 | 0.25–0.94 | 0.61 | 0.002 (0.044) | 0.32 | 0.15–0.67 |
| rs35134656 | 0.32 | 0.16 | 0.015 (0.297) | 2.52 | 1.18–5.37 | 0.09 | 0.002 (0.042) | 4.76 | 1.66–13.66 |
Significant associations in bold (Chi-squared test), Bonferroni correction for 20 tests
TPD tangle-predominant dementia, MAF minor allele frequency, OR odds ratio, CI confidence interval
Fig 5.

Linkage disequilibrium (LD) in MAPT. The genotypes for the MAPT haplotype-tagging SNPs (rs1467967, rs242557, rs3785883, rs2471738, rs9468 and rs7521) together with rs5820605 and rs35134656 from cases and controls (n = 82) were analyzed in Haploview v4.2 to generate the LD plot. The white horizontal bar represents the chromosomal distance between the polymorphisms. Pairwise D′ values are indicated within the diamonds. Variants with strong LD are shown in red. Those without LD are white and variants with uninformative data light blue
Discussion
TPD is a poorly understood and under-recognized tauopathy in need of a definitive neuropathological designation. Given the overlapping features with moderate AD and aging, and the absence of reliable markers, recognizing TPD continues to pose a challenge. This study demonstrates, for the first time, an association between TPD and the MAPT H1 haplotype. Obtaining large numbers of well-characterized neuropathologically confirmed TPD cases for genetic analysis poses a significant hurdle, and the results of this relatively small study should be interpreted with this in mind. However, should our findings be confirmed in a larger cohort, they provide a novel genetic foothold into the pathogenesis of this elusive tauopathy. Furthermore, TPD has the potential to provide a unique angle into understanding the mechanism of the contribution of the MAPT haplotypes to neurodegeneration in general, which remains unclear at this time. As the H1 haplotype is associated with a number of sporadic tauopathies as well as Parkinson disease, understanding the contribution of MAPT to neurodegeneration represents an important question. We identified two polymorphisms within the region of MAPT encoding the 3′ UTR that are associated with TPD. 3′ UTRs are critical cis-acting regulatory elements that are capable of regulating gene expression on the post-transcriptional level by influencing mRNA stability and localization, among other functions [4, 5]. Importantly, 3′ UTRs, through complementary base pairing, are the main binding sites for microRNA (miRNA) binding [2, 38]. These small non-coding RNAs silence mRNA and are implicated in neurodegeneration [35]. Future in vitro experiments addressing the functional role of variation in the MAPT 3′ UTR are required. Alternatively, these polymorphisms may be markers of functional genetic lesions residing in nearby chromosomal regions. Whether rs5820605 and rs35134656 are useful as genetic markers for diagnosis or risk stratification remains to be determined.
Our biochemical analysis of APP processing provides further evidence that TPD is Aβ independent. Namely, the low levels of soluble Aβ and increased sAPPα in TPD brain parenchyma indicate preferential non-amyloidogenic processing in these patients. However, we are unable to exclude the possibility that transient elevations in Aβ or other potentially toxic APP-derived factors trigger a cascade leading to NFT formation in TPD. These biochemical results, which are complementary to previous histological reports, are important because they raise the possibility that some dementia patients with amyloid plaques may also have a concurrent amyloid-independent limbic tauopathy capable of causing dementia. For example, it remains unclear at this time whether patients with limbic predominant AD, which exhibits an increased frequency of H1 compared to hippocampal sparing AD, share pathogenic determinants with TPD [44]. Improved methods to address patients with mixed pathologies would be useful.
The ultimate cause of TPD is unclear at this time. The increased levels of sAPPα in TPD is a surprising finding and we are unable to exclude the possibility that sAPPα may be pathogenic under certain circumstances. Previous research suggests that sAPPα attenuates excitotoxicity and Aβ-induced tau phosphorylation, perhaps serving to delay the onset and severity of TPD relative to AD [39, 57]. In this context, our data are consistent with a proposed role for Aβ in accelerating and amplifying an age-related tauopathy [50]. Nevertheless, the increased levels of sAPPα may serve as a useful biomarker, which is a possibility that can be addressed in future studies. Exogenous factors have also been proposed to cause TPD. For example, previous work has suggested an early-life viral etiology [47]. Other potential causes such as mild traumatic injury must be addressed in future studies [21].
As clinical trials of Aβ-reducing agents have been unsuccessful in showing clinical efficacy in AD, new approaches are urgently needed [26]. There is no way to clinically differentiate TPD from AD, yet this distinction is essential for developing novel therapies. The distinction between TPD and AD is fundamental as the low Aβ levels in TPD suggest that amyloid-targeting agents will not be useful for treating dementia patients with this tauopathy and may subject them to unnecessary risk. Unfortunately, the cerebrospinal fluid biomarkers for AD (i.e., low Aβ and high phospho-tau) are predicted to be positive in TPD [60]. Positron emission tomography (PET)-based amyloid imaging have demonstrated that many cognitively impaired patients are carbon 11-labeled Pittsburgh Compound B (11C-PiB) negative, suggesting that TPD and other amyloid-independent dementias are more widespread than recognized [17, 19].
Supplementary Material
Acknowledgments
We express our deepest gratitude to the patients and staff of the contributing centers and institutes, including the Taub Institute for Research on Alzheimer’s Disease & the Aging Brain at Columbia University [(P50AG08702/RO1AG037212), the Washington/Hamilton Heights-Inwood Columbia Aging Project (P01AG07232) and the Essential Tremor Centralized Brain Repository (R01NS042859)], the Sanders-Brown Center on Aging at the University of Kentucky (P30AG028383), the Shiley-Marcos Alzheimer’s Disease Research Center at the University of California San Diego (P50AG05131), the Northwestern University Alzheimer’s Disease Center (AG13854 and the NADC Neuropathology Core Tissue Bank), the Washington University School of Medicine, St. Louis, MO [NIA P50-AG05681, P01-AG03991), the Hope Center for Neurological Disorders, and the Charles F. & Joanne Knight Alzheimer’s Disease Research Center], the University of Washington Neuropathology Core [Alzheimer’s Disease Research Center (AG05136), the Adult Changes in Thought Study (AG006781), and Morris K Udall Center of Excellence for Parkinson’s Disease Research (NS062684)], and the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona [National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research]. We thank Drs. Michael L. Shelanski and Richard Mayeux. We also thank Drs. Jean-Paul Vonsattel and Lawrence Honig for providing the patients’ clinical and neuropathological data. We thank Dr. Etty Cortes for helpful comments and neuropathology core support. We thank Arlene Lawton for coordination of brain donations at Columbia University. We further thank Drs. Karen Duff and James E. Goldman for helpful comments. We thank Dr. Peter Davies for the phospho-tau antisera. Finally, we thank Kristy Brown for electron microscopy support. This project was funded in part by American Recovery and Reinvestment Act (ARRA) funds through Grant number P30AG036453 (MLS). This project was also supported by the Alzheimer’s Association (NIRG-11-204450) and the Louis V. Gerstner, Jr., Foundation (JFC).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-012-1017-1) contains supplementary material, which is available to authorized users.
Contributor Information
Ismael Santa-Maria, Department of Pathology and Cell Biology, Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, Columbia University, New York 10032, USA.
Aya Haggiagi, Department of Pathology and Cell Biology, Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, Columbia University, New York 10032, USA.
Xinmin Liu, Department of Pathology and Cell Biology, Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, Columbia University, New York 10032, USA.
Jessica Wasserscheid, Department of Human Genetics, McGill University, Montreal, QC H3A 1A4, Canada.
Peter T. Nelson, Division of Neuropathology, Department of Pathology, Sanders-Brown Center on Aging, University of Kentucky, 800 S. Limestone, Lexington, KY 40536, USA
Ken Dewar, Department of Human Genetics, McGill University, Montreal, QC H3A 1A4, Canada.
Lorraine N. Clark, Department of Pathology and Cell Biology, Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, Columbia University, New York 10032, USA
John F. Crary, Email: jc2892@columbia.edu, Department of Pathology and Cell Biology, Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, Columbia University, New York 10032, USA, Department of Pathology and Cell Biology, Columbia University Medical Center, 630 168th Street PH15-124, New York, NY 10032, USA
References
- 1.Abraham R, Sims R, Carroll L, Hollingworth P, O’Donovan MC, Williams J, Owen MJ. An association study of common variation at the MAPT locus with late-onset Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:1152–1155. doi: 10.1002/ajmg.b.30951. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V, Horvitz HR. The lin-14 locus of Caenorhabditis elegans controls the time of expression of specific postembryonic developmental events. Gene Dev. 1987;1:398–414. doi: 10.1101/gad.1.4.398. [DOI] [PubMed] [Google Scholar]
- 3.Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Aronov S, Aranda G, Behar L, Ginzburg I. Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J Neurosci Off J Soc Neurosci. 2001;21:6577–6587. doi: 10.1523/JNEUROSCI.21-17-06577.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aronov S, Marx R, Ginzburg I. Identification of 3′UTR region implicated in tau mRNA stabilization in neuronal cells. J Mol Neurosci. 1999;12:131–145. doi: 10.1007/BF02736927. [DOI] [PubMed] [Google Scholar]
- 6.Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- 7.Bancher C, Jellinger KA. Neurofibrillary tangle predominant form of senile dementia of Alzheimer type: a rare subtype in very old subjects. Acta Neuropathol. 1994;88:565–570. doi: 10.1007/BF00296494. [DOI] [PubMed] [Google Scholar]
- 8.Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23:228–242. doi: 10.1177/0891988710383572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex. 1994;4:138–150. doi: 10.1093/cercor/4.2.138. [DOI] [PubMed] [Google Scholar]
- 10.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 11.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–408. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- 12.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark LN, Kartsaklis LA, Wolf Gilbert R, et al. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch Neurol. 2009;66:578–583. doi: 10.1001/archneurol.2009.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 15.Crary JF, Shao CY, Mirra SS, Hernandez AI, Sacktor TC. Atypical protein kinase C in neurodegenerative disease I: PKMzeta aggregates with limbic neurofibrillary tangles and AMPA receptors in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:319–326. doi: 10.1097/01.jnen.0000218442.07664.04. [DOI] [PubMed] [Google Scholar]
- 16.Davidsson P, Bogdanovic N, Lannfelt L, Blennow K. Reduced expression of amyloid precursor protein, presenilin-1 and rab3a in cortical brain regions in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12:243–250. doi: 10.1159/000051266. [DOI] [PubMed] [Google Scholar]
- 17.Devanand DP, Mikhno A, Pelton GH, Cuasay K, Pradhaban G, Dileep Kumar JS, Upton N, Lai R, Gunn RN, Libri V, Liu X, van Heertum R, Mann JJ, Parsey RV. Pittsburgh compound B (11C-PIB) and fluorodeoxyglucose (18 F-FDG) PET in patients with Alzheimer disease, mild cognitive impairment, and healthy controls. J Geriatr Psychiatry Neurol. 2010;23:185–198. doi: 10.1177/0891988710363715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Maria E, Tabaton M, Vigo T, Abbruzzese G, Bellone E, Donati C, Frasson E, Marchese R, Montagna P, Munoz DG, Pramstaller PP, Zanusso G, Ajmar F, Mandich P. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol. 2000;47:374–377. doi: 10.1002/1531-8249(200003)47:3<374::aid-ana15>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Foster N, King R, Wang A, Landau S, Jagust W, Chen K, Reiman E. Diagnostic classification with amyloid PET and FDGPET among clinically diagnosed Alzheimer’s disease patients in the Alzheimer’s disease Neuroimaging Initiative. Human Amyloid Imaging Abstract. 2012 Jan 1; [Google Scholar]
- 20.Gasparini L, Terni B, Spillantini MG. Frontotemporal dementia with tau pathology. Neurodegener Dis. 2007;4:236–253. doi: 10.1159/000101848. [DOI] [PubMed] [Google Scholar]
- 21.Gavett BE, Stern RA, Cantu RC, Nowinski CJ, McKee AC. Mild traumatic brain injury: a risk factor for neurodegeneration. Alzheimers Res Ther. 2010;2:18. doi: 10.1186/alzrt42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghebranious N, Ivacic L, Mallum J, Dokken C. Detection of ApoE E2, E3 and E4 alleles using MALDI-TOF mass spectrometry and the homogeneous mass-extend technology. Nucleic Acids Res. 2005;33:e149. doi: 10.1093/nar/gni155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giliberto L, Zhou D, Weldon R, Tamagno E, De Luca P, Tabaton M, D’Adamio L. Evidence that the amyloid beta precursor protein-intracellular domain lowers the stress threshold of neurons and has a “regulated” transcriptional role. Mol Neurodegener. 2008;3:12. doi: 10.1186/1750-1326-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 25.Hayesmoore JB, Bray NJ, Cross WC, Owen MJ, O’Donovan MC, Morris HR. The effect of age and the H1c MAPT haplotype on MAPT expression in human brain. Neurobiol Aging. 2009;30:1652–1656. doi: 10.1016/j.neurobiolaging.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 26.Herrmann N, Chau SA, Kircanski I, Lanctot KL. Current and emerging drug treatment options for Alzheimer’s disease: a systematic review. Drugs. 2011;71:2031–2065. doi: 10.2165/11595870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 27.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement J Alzheimer’s Assoc. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikeda K, Akiyama H, Sahara N, Mori H, Usami M, Sakata M, Mizutani T, Wakabayashi K, Takahasi H. Senile dementia with abundant neurofibrillary tangles without accompanying senile plaques: a subset of senile dementia with high incidence of the APOE e2 Allele. In: Iqbal K, Winblad B, Nishimura T, Takeda M, Wisniewski HM, editors. Alzheimer’s disease: biology, diagnosis and therapeutics. 1. Wiley; New York: 1997. [Google Scholar]
- 29.Ikeda K, Akiyama H, Arai T, Oda T, Kato M, Iseki E, Kosaka K, Wakabayashi K, Takahashi H. Clinical aspects of ‘senile dementia of the tangle type’—a subset of dementia in the senium separable from late-onset Alzheimer’s disease. Dement Geriatr Cogn Disord. 1999;10:6–11. doi: 10.1159/000017091. [DOI] [PubMed] [Google Scholar]
- 30.Ikeda K, Akiyama H, Arai T, Sahara N, Mori H, Usami M, Sakata M, Mizutani T, Wakabayashi K, Takahashi H. A subset of senile dementia with high incidence of the apolipoprotein E epsilon2 allele. Ann Neurol. 1997;41:693–695. doi: 10.1002/ana.410410522. [DOI] [PubMed] [Google Scholar]
- 31.Iseki E, Yamamoto R, Murayama N, Minegishi M, Togo T, Katsuse O, Kosaka K, Akiyama H, Tsuchiya K, de Silva R, Andrew L, Arai H. Immunohistochemical investigation of neurofibrillary tangles and their tau isoforms in brains of limbic neurofibrillary tangle dementia. Neurosci Lett. 2006;405:29–33. doi: 10.1016/j.neulet.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 32.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 33.Jellinger KA, Attems J. Neurofibrillary tangle-predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol. 2007;113:107–117. doi: 10.1007/s00401-006-0156-7. [DOI] [PubMed] [Google Scholar]
- 34.Jellinger KA, Bancher C. Senile dementia with tangles (tangle predominant form of senile dementia) Brain Pathol. 1998;8:367–376. doi: 10.1111/j.1750-3639.1998.tb00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Junn E, Mouradian MM. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther. 2012;133:142–150. doi: 10.1016/j.pharmthera.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 37.Ksiezak-Reding H, Wall JS. Mass and physical dimensions of two distinct populations of paired helical filaments. Neurobiol Aging. 1994;15:11–19. doi: 10.1016/0197-4580(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 38.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 39.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 40.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 41.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 42.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukherjee O, Kauwe JS, Mayo K, Morris JC, Goate AM. Haplotype-based association analysis of the MAPT locus in late onset Alzheimer’s disease. BMC Genet. 2007;8:3. doi: 10.1186/1471-2156-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785–796. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myers AJ, Kaleem M, Marlowe L, Pittman AM, Lees AJ, Fung HC, Duckworth J, Leung D, Gibson A, Morris CM, de Silva R, Hardy J. The H1c haplotype at the MAPT locus is associated with Alzheimer’s disease. Hum Mol Genet. 2005;14:2399–2404. doi: 10.1093/hmg/ddi241. [DOI] [PubMed] [Google Scholar]
- 46.Myers AJ, Pittman AM, Zhao AS, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. 2007;25:561–570. doi: 10.1016/j.nbd.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 47.Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Santacruz K, Smith CD, Patel E, Markesbery WR. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have patho-genetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:774–784. doi: 10.1097/NEN.0b013e3181aacbe9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noda K, Sasaki K, Fujimi K, Wakisaka Y, Tanizaki Y, Wakugawa Y, Kiyohara Y, Iida M, Aizawa H, Iwaki T. Quantitative analysis of neurofibrillary pathology in a general population to reappraise neuropathological criteria for senile dementia of the neurofibrillary tangle type (tangle-only dementia): the Hisayama Study. Neuropathology. 2006;26:508–518. doi: 10.1111/j.1440-1789.2006.00722.x. [DOI] [PubMed] [Google Scholar]
- 49.Pittman AM, Myers AJ, Abou-Sleiman P, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. 2005;42:837–846. doi: 10.1136/jmg.2005.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 51.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rowe JW, Kahn RL. Human aging: usual and successful. Science. 1987;237:143–149. doi: 10.1126/science.3299702. [DOI] [PubMed] [Google Scholar]
- 53.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 54.Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302–2309. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-beta quantitation. Methods Mol Biol. 2005;299:267–278. doi: 10.1385/1-59259-874-9:267. [DOI] [PubMed] [Google Scholar]
- 56.Stefansson H, Helgason A, Thorleifsson G, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–137. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 57.Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takahashi M, Weidenheim KM, Dickson DW, Ksiezak-Reding H. Morphological and biochemical correlations of abnormal tau filaments in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2002;61:33–45. doi: 10.1093/jnen/61.1.33. [DOI] [PubMed] [Google Scholar]
- 59.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trojanowski JQ, Vandeerstichele H, Korecka M, et al. Update on the biomarker core of the Alzheimer’s disease neuroimaging initiative subjects. Alzheimer’s Dement J Alzheimer’s Assoc. 2010;6:230–238. doi: 10.1016/j.jalz.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ulrich J, Spillantini M, Goedert M, Dukas L, Staehelin H. Abundant neurofibrillary tangles without senile plaques in a subset of patients with senile dementia. Neurodegeneration. 1992;1:257–284. [Google Scholar]
- 62.Vandrovcova J, Anaya F, Kay V, Lees A, Hardy J, de Silva R. Disentangling the role of the tau gene locus in sporadic tauopathies. Curr Alzheimer Res. 2010;7:726–734. doi: 10.2174/156720510793611619. [DOI] [PubMed] [Google Scholar]
- 63.Walsh DM, Selkoe DJ. A beta oligomers—a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 64.Wu G, Sankaranarayanan S, Hsieh SH, Simon AJ, Savage MJ. Decrease in brain soluble amyloid precursor protein beta (sAPPbeta) in Alzheimer’s disease cortex. J Neurosci Res. 2011;89:822–832. doi: 10.1002/jnr.22618. [DOI] [PubMed] [Google Scholar]
- 65.Yamada M. Senile dementia of the neurofibrillary tangle type (tangle-only dementia): neuropathological criteria and clinical guidelines for diagnosis. Neuropathology. 2003;23:311–317. doi: 10.1046/j.1440-1789.2003.00522.x. [DOI] [PubMed] [Google Scholar]
- 66.Yamada M, Itoh Y, Sodeyama N, Suematsu N, Otomo E, Matsushita M, Mizusawa H. Senile dementia of the neurofibrillary tangle type: a comparison with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12:117–126. doi: 10.1159/000051245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.