

Ubiquitin-protein ligases (E3s) are often in the precarious position of ubiquitinating themselves, mediating their own destruction. The intrinsically disordered E3 San1 prevents its own autoubiquitination and degradation by minimizing Lys residues and hydrophobic stretches in its disordered regions.
Abstract
Ubiquitin-protein ligases (E3s) that ubiquitinate substrates for proteasomal degradation are often in the position of ubiquitinating themselves due to interactions with a charged ubiquitin-conjugating enzyme (E2). This can mediate the E3’s proteasomal degradation. Many E3s have evolved means to avoid autoubiquitination, including protection by partner or substrate binding, preventative modifications, and deubiquitinating enzyme reversal of ubiquitination. Here we describe another adaptation for E3 self-protection discovered while exploring San1, which ubiquitinates misfolded nuclear proteins in yeast for proteasomal degradation. San1 is highly disordered in its substrate-binding regions N- and C-terminal to its RING domain. In cis autoubiquitination could occur if these flexible regions come in proximity to the E2. San1 prevents this by containing no lysines in its disordered regions; thus the canonical residue used for ubiquitin attachment has been selectively eliminated. San1’s target substrates have lost their native structures and expose hydrophobicity. To avoid in trans autoubiquitination, San1 possesses little concentrated hydrophobicity in its disordered regions, and thus the that feature San1 recognizes in misfolded substrates has also been selectively eliminated. Overall the presence of key residues in San1 have been evolutionarily minimized to avoid self-destruction either in cis or in trans. Our work expands the ways in which E3s protect themselves from autoubiquitination.
INTRODUCTION
The ability to destroy proteins with a high degree of specificity is essential to cellular function. One means by which eukaryotes specifically degrade proteins is through the ubiquitin-proteasome pathway, which is used typically for one of two cellular functions: 1) spatial or temporal regulation of a functional protein or 2) removal of a misfolded protein among a pool of normally folded proteins. Ubiquitination of substrates occurs via an enzymatic cascade of a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin-protein ligase (E3), which typically confers substrate specificity within the cascade (Ciechanover, 2006). Because E3s interact with E2s charged with ubiquitin, there is the potential for an E3 to cause its own autoubiquitination, which can lead to the E3’s proteasomal degradation in vivo. Unnecessary autoubiquitination and degradation would reduce the functional levels of active E3s, and thus means of protection must have evolved to prevent or regulate this possibility.
Four ways have been identified that minimize E3 autoubiquitination and maintain E3 stability in vivo. The first is through the interaction of the E3 with its complex partners. For example, interaction of the yeast E3 Hrd1 with Hrd3 prevents autoubiquitination of Hrd1 and its degradation by the proteasome (Plemper et al., 1999; Gardner et al., 2000). The second is protection of the E3 by the binding of a substrate. This is most notable in cullin E3s, where the binding of a substrate to the adapter F-box protein protects the F-box protein from ubiquitination (Galan and Peter, 1999; Li et al., 2004). The third is through posttranslational modification of the E3. For instance, phosphorylation or acetylation of the mammalian E3 Mdm2 decreases its autoubiquitination and degradation (Feng et al., 2004; Wang et al., 2004). The fourth and final way is removal of ubiquitin from the E3 by the activity of a deubiquitinating enzyme. For example, the stability of the mammalian E3 Nrd1 is dependent on the catalytic activity of the deubiquitinating enzyme USP8 (Wu et al., 2004). In each case of E3 autoubiquitination, it is not clear whether autoubiquitination is a functional means to regulate the E3, similar to the specific ubiquitination of a substrate, or is simply due to the inherent activity of the E3 against itself.
In this article, we describe an additional mode of E3 self-protection that we discovered while studying the yeast E3 San1, which mediates the ubiquitination of misfolded nuclear proteins for proteasome-mediated degradation (Gardner et al., 2005). Our previous studies revealed that San1 possesses highly disordered N- and C-terminal regions that contain interspersed substrate-binding sites used for misfolded substrate recognition (Rosenbaum et al., 2011). In particular, San1 recognizes exposed hydrophobicity within its misfolded substrates (Fredrickson et al., 2011). These observations led to two questions concerning how San1 prevents its own ubiquitination and subsequent proteasome degradation. First, how does San1 prohibit its highly disordered and conformationally flexible regions from in cis autoubiquitination if/when they become positioned near the charged E2 bound by San1’s RING domain? Second, if San1 lacks structure, why is one San1 molecule not recognized for in trans autoubiquitination by another San1 molecule if San1 is capable of targeting proteins that have lost their native structures? Here we reveal the molecular means by which San1 has evolved to prevent its own in cis and in trans autoubiquitination and degradation.
RESULTS
San1 lacks Lys residues in its N- and C-terminal regions
The first clue for how San1 might be protected from in cis autoubiquitination came from examining the amino acid distribution of San1’s primary sequence. On initial inspection, we noticed that the overall Lys content for San1 is atypical. San1 possesses only 13 Lys residues, whereas a typical protein the size of San1 is predicted to have ∼40 Lys residues based on average codon usage in yeast (Saccharomyces Genome Database; www.yeastgenome.org). On closer inspection, we found that this unusual feature could be ascribed to the fact that San1’s highly disordered N- and C-terminal regions were devoid of Lys residues (Figure 1A). The 13 Lys residues present in San1 are clustered in or near the RING domain (Figure 1A), with nearly half comprising San1’s nuclear localization sequence (NLS; Figure 1A). Because E3s typically mediate the covalent attachment of ubiquitin to the free amino group in the side chains of Lys residues in their substrates, we hypothesized that the lack of Lys residues in San1’s N- and C- terminal regions protects San1 from in cis autoubiquitination (Figure 1B).
FIGURE 1:
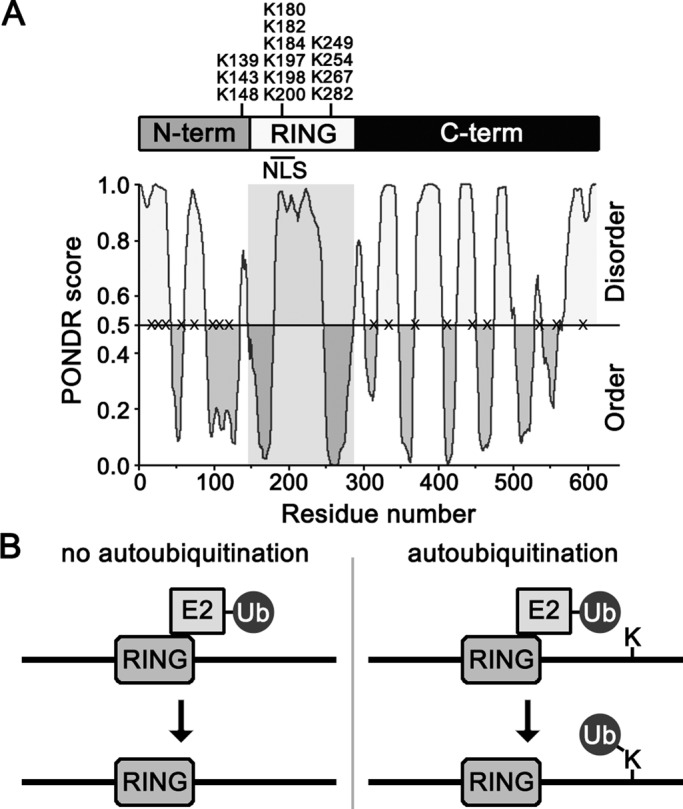
Sequence features of San1. (A) Representation of the overall domain topology of San1. Endogenous Lys residue positions are marked on top. A line denotes the presence of an NLS in the RING domain (Gardner et al., 2005). PONDR (www.pondr.com/) evaluation of San1’s intrinsic disorder is on the bottom. Gray box highlights the RING domain. The X's on the 0.5 line mark the location of the “plus Lys” mutants. (B) Model of in cis autoubiquitination upon addition of a Lys residue in the intrinsically disordered C-terminal region of San1.
Introduction of a single Lys residue in San1’s N- and C-terminal regions results in rapid degradation
If a lack of Lys residues prevents San1 in cis autoubiquitination, we predicted that the addition of a single Lys residue in San1’s N- and C-terminal regions would disrupt San1’s stability. To test our prediction, we randomly made 17 single Arg- or Asn-to-Lys point mutations along the length of San1’s N- and C-terminal regions (Figure 1A, X's). Because the N- and C- terminal regions are not completely disordered, the mutations were made in predicted regions of order and disorder. We then examined the stability of the “plus Lys” mutant San1s in cells with the endogenous SAN1 gene intact (SAN1) or deleted (san1∆). To monitor stability and distinguish the “plus Lys” mutant San1s from endogenous San1, we tagged each San1 mutant at its C-terminus with a 3× herpes simplex virus (HSV) epitope, which does not alter the function of San1 (Gardner et al., 2005). In every case, we found that the “plus Lys” San1 mutants were rapidly degraded in both the presence and absence of endogenous San1, whereas wild-type San1 was stable (Figure 2A). We believed it possible that degradation of these mutants could be independent of the Lys residue and that an analogous conservative mutation would be similarly sufficient to cause degradation. However, mutation of the same 17 residues to either Asn or Arg, the most conservative changes in relation to Lys, did not appreciably alter the stability of San1 (Figure 2, B vs. A). Thus specific addition a single Lys residue in San1’s highly disordered N- and C-terminal regions consistently disrupted San1’s stability.
FIGURE 2:

Introduction of Lys residues in San1’s highly disordered N- and C- terminal regions leads to degradation via three distinct mechanisms. (A) Cycloheximide-chase assays were performed to assess the stability of 17 “plus Lys” mutants in the presence or absence of SAN1. Time after cycloheximide addition is indicated. Western blots were probed with anti-HSV antibodies. (B) Identical experiment as in A, except that each mutant was now either an Arg or Asn residue. (C) Identical experiment as in A, except that each San1 contains a RING-inactivating mutant, R280A.
The underlying assumption of the hypothesis is that addition of a single Lys residue in the N- and C-terminal regions should result in degradation through in cis autoubiquitination. However, it is possible that addition of a Lys residue could also result in the recognition of a “plus Lys” San1 mutant as a substrate by another San1 molecule, leading to degradation through in trans autoubiquitination. Therefore, it was important to distinguish between an in cis and an in trans mode for the degradation of the “plus Lys” San1 mutants. If degradation occurs as a result of in cis autoubiquitination, we predicted that introducing an additional RING-inactivating mutation would result in the stability of the “plus Lys” San1 mutant even in the presence of functional endogenous San1. Conversely, if degradation occurred as a result of in trans autoubiquitination, we predicted that introducing a RING-inactivating mutation would result in degradation of the “plus Lys” San1 mutant only when endogenous San1 was intact. To inactivate the RING domain, we chose to mutate the Arg residue at position 280 to an Ala residue. R280 is the next position after the final Cys residue (C279) in San1’s RING domain (Rosenbaum et al., 2011), and this position in other RING E3s is important for interactions with the charged E2 but is not required for the intact RING structure (Pruneda et al., 2012). Accordingly, mutation of R280 will abrogate E2 recruitment but will not result in the misfolding of the San1 RING domain.
After introduction of the R280A mutation, we observed three distinct behaviors for the “plus Lys” San1 mutants. In the first case, we found “plus Lys” San1 mutants whose degradation pointed to a predominantly in cis autoubiquitination mechanism, where the “plus Lys” mutants were now highly stable in the presence or absence of endogenous San1. The majority of the “plus Lys” San1 mutants (11 of 17) fell into this class, which included the N13K, N23K, R311K, N332K, R371K, R407N N444K, R458K, R535K, R556K, and N596K mutants (Figure 2C). The second class consisted of “plus Lys” San1 mutants that were rapidly degraded in the presence of endogenous San1 but were stable in its absence, and this is consistent with a predominantly in trans autoubiquitination mechanism. These mutants included N71K, R105K, and R115K (Figure 2C). The third class comprised “plus Lys” San1 mutants whose degradation was not stabilized by the creation of the R280A mutation even in san1∆ cells, which indicated that a San1-independent ubiquitination mechanism was primarily operating. This class included N33K, N61K, and N91K (Figure 2C). Because all members of this third class lie within the same portion of the N-terminal region, perhaps this region has attributes different from those of the rest of San1’s N- and C- terminal portions (see Discussion). Finally, it is important to note that all “plus Lys” San1 mutants had very low steady-state levels in both SAN1 and san1∆ cells when the RING domain was intact due to their rapid degradation and these levels increased with the introduction of the R280A mutation. An increase in steady-state levels upon RING mutation suggests that all the “plus Lys” mutants are degraded via an in cis mechanism at least in part. Thus an in cis autoubiquitination mechanism occurred to some extent with every “plus Lys” mutant.
“Plus Lys” mutants do not alter San1–substrate interactions
Because the subunits of some E3s are protected from autoubiquitination by interaction with their substrates, it is possible that the “plus Lys” mutations impair San1–substrate interactions and this is the reason for their degradation. To test the substrate-interaction capabilities of the “plus Lys” mutants, we used a two-hybrid interaction assay that we previously developed for San1 and its substrates (Rosenbaum et al., 2011). This two-hybrid assay is very sensitive and can detect alterations in San1–substrate interactions that have only modest effects on substrate degradation rates (Rosenbaum et al., 2011). To perform the two-hybrid assay, we previously fused San1 carrying a RING-inactivating mutation (to prevent ubiquitination of interacting substrates) to the Gal4 DNA-binding domain (GBD), and we fused San1 substrates to the Gal4 activation domain (GAD; Rosenbaum et al., 2011). For the studies here, we again chose to use a RING-inactive variant of San1 fused to the GBD, both to prevent the ubiquitination of San1’s substrates and, more important, to prevent the autoubiquitination of San1 itself. The latter reason is crucial because alterations in the steady-state levels of the GBD-San1 constructs can affect the two-hybrid interaction. Inactivating San1’s RING domain prevents the autoubiquitination San1 mutants that self-destruct via an in cis mechanism (Figure 2C) and thus allows these San1 mutants to have the same steady-state levels as wild-type San1. For the two-hybrid interaction assay, we chose to use the San1 substrates Cdc13-1 and Sir4-9, which we previously showed interacted with San1 by the two-hybrid assay (Rosenbaum et al., 2011). San1’s interactions with these particular substrates are the most sensitive of all the substrates to small deletions within San1’s N- and C-terminal regions (Rosenbaum et al., 2011) and thus should be the most capable in revealing whether the “plus Lys” mutations have any effect on San1–substrate interactions.
We introduced into the GBD-San1R280A construct each of the “plus Lys” mutants that were degraded via a primarily in cis mechanism (N13K, N23K, R311K, N332K, R371K, N444K, R458K, R535K, R556K, and N596K). We chose these “plus Lys” mutations in particular because we wanted to avoid the complication of low steady-state levels that occur when San1–in trans and San1-independent mechanisms also operated. After spotting cells on selective media that reveals the two-hybrid interaction, we saw that each “plus Lys” mutant interacted with Cdc13-1 and Sir4-9 equivalently to the parent San1 construct (Figure 3). Conversely, a San1 mutant with residues 402–422 deleted (∆402–422) did not interact with Cdc13-1 or Sir4-9 (Figure 3), as we previously demonstrated (Rosenbaum et al., 2011). These results indicate that San1–substrate interactions are unaltered by introduction of the “plus Lys” mutations.
FIGURE 3:

San1 “plus Lys” mutants interact with substrates normally. Cells expressing GAD-Cdc13-1 (left two) or GAD-Sir4-9 (right two) and the indicated San1R280A “plus Lys” mutants fused to the GBD (left) were spotted onto media with or without histidine to measure spotting efficiency and two-hybrid interaction.
A sole Lys residue in San1 is sufficient to cause in cis instability
We initially hypothesized that the addition of a single Lys residue in San1’s N- or C-terminal regions would be sufficient to cause in cis San1 autoubiquitination and subsequent degradation (Figure 1B), which we observed in Figure 2. However, San1 contains 13 endogenous Lys residues in and near the RING domain (Figure 1A), and it could be that the creation of a Lys residue in the N- or C-terminal regions now results in the ubiquitination of the endogenous Lys residues rather than the new additional residue. To test whether a sole Lys residue in the N- or C-terminal regions is sufficient, we mutated all 13 endogenous Lys residues in San1 to Arg residues, creating a “no Lys” San1 mutant.
To verify that the Lys to Arg mutations did not disrupt intrinsic San1 function, we tested whether the “no Lys” San1 mutant was capable of complementing the san1∆ allele for cdc68-1 temperature sensitivity (Figure 4A). cdc68-1 cells have a restrictive temperature of 34°C that can be suppressed by deletion of SAN1 (Xu et al., 1993; Gardner et al., 2005). Replacement of the san1∆ allele with a functional copy of SAN1 rescues the temperature suppression (Gardner et al., 2005). In the case of the “no Lys” San1 mutant, we observed near-complete rescue of the san1∆ allele for cdc68-1 temperature sensitivity (Figure 4A). We anticipated that a lack of full complementation is likely the result of mutating the six Lys residues that constitute San1’s NLS (Figure 1A), thus weakening San1’s transport to the nucleus, where it normally functions. To alleviate this potential problem, we added the SV40 NLS to the C-terminus of the “no Lys” San1 mutant and found that this version restored San1 function completely in terms of rescuing cdc68-1 temperature sensitivity (Figure 4A). We conclude that mutating all 13 endogenous Lys residues in San1 to Arg residues does not appreciably affect San1’s E3 activity, only its nuclear localization.
FIGURE 4:

A single introduced Lys residue is sufficient and necessary for degradation. (A) Growth assays of cdc68-1 cells were performed to determine the ability of the San1no Lys mutant to complement a san1∆ allele. Cells were spotted onto synthetic nutrient plates in 10-fold serial dilutions, and the plates were incubated for 3 d at the permissive (25°C) or restrictive (34°C) temperatures. (B) Cycloheximide-chase assays were performed to assess the stability of the San1N13K and San1N444K mutants with either San1’s endogenous Lys residues intact (top three, “endogenous Lys”) or mutated (bottom three, “no Lys”) in the presence or absence of SAN1. Time after cycloheximide addition is indicated. Western blots were probed with anti-HSV antibodies. (C) Decay curves for degradation assays in B were determined using ImageJ. (D) Identical experiment as in B, except that each San1 construct also contained a RING-inactivating R280A mutation.
Having verified the “no Lys” San1 mutant functions normally if properly localized, we examined whether the degradation caused by introducing a single Lys residue in the N- and C-terminal regions was due solely to that new Lys residue. For these studies, we chose to focus on one representative “plus Lys” mutant example for each the N- and C- terminal regions (N13K and N444K). We found that introduction of a Lys residue (N13K or N444K) in the “no Lys” San1’s N- or C-terminal regions resulted in degradation of San1 similar to that seen when San1’s endogenous Lys residues were intact (Figure 4, B and C). Furthermore, the degradation of the “no Lys” San1N13K or San1N444K mutants required an intact RING domain (Figure 4D), indicating that an in cis mechanism was operative. Taken together, the results show that the specific presence of a single Lys residue in San1’s highly disordered N- or C- terminal regions is sufficient to cause San1 degradation.
Ubiquitination states of the “plus Lys” mutants correlates with their degradation
The majority of the “plus Lys” mutants were degraded in a manner that is consistent with an in cis autoubiquitination mechanism (Figure 2). To determine the actual mode of autoubiquitination, we examined the in vivo ubiquitination states of representative “plus Lys” San1 mutants to see whether they correlated with the degradation results. To query in vivo ubiquitination, we expressed versions of wild-type San1, San1N13K, or San1N444K (each with the RING intact or mutated) in cells that also coexpressed 6His-1Myc-ubiquitin, which allowed us to purify ubiquitin conjugates from cells using metal affinity chromatography (Spence et al., 2000). Subsequent probing of the purified ubiquitin conjugates with an HSV antibody allowed us to assess San1’s ubiquitination levels. As expected from their degradation behavior, the RING-intact San1N13K and San1N444K mutants showed significant ubiquitination in SAN1 cells that was not observed with wild-type San1 (Figure 5A). Introduction of the RING-inactivating R280A mutation abrogated the ubiquitination of the San1N13K and San1N444K mutants (Figure 5A). To verify that the ubiquitination requires solely N13K or N444K, we examined the ubiquitination status of the “no Lys” versions of San1, San1N13K, or San1N444K in SAN1 cells (Figure 5B). The ubiquitination of the “no Lys” mutants mirrored that of those with endogenous Lys intact. Overall the in vivo ubiquitination results support the hypothesis that San1 prevents its own in cis autoubiquitination by a lack of Lys residues in its N- and C-terminal substrate-interaction regions.
FIGURE 5:
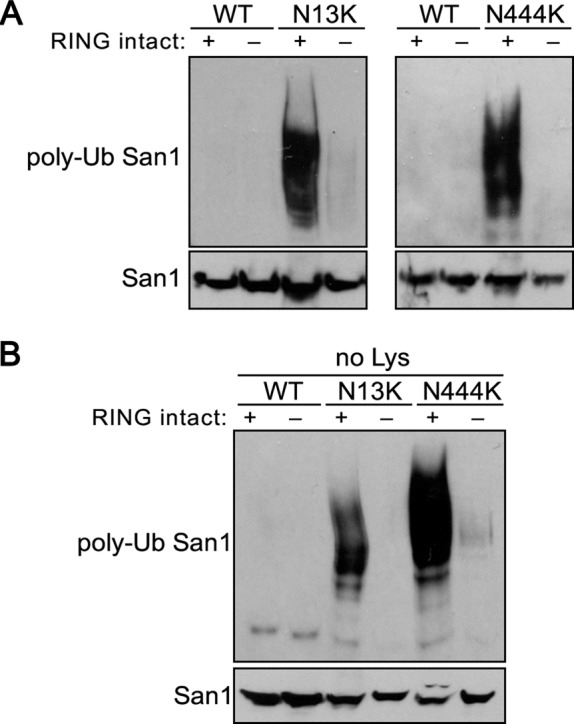
Ubiquitination of the “plus Lys” mutants demonstrates an in cis mechanism. (A) Ubiquitination assays were performed to assess the extent of San1, San1N13K, and San1N444K ubiquitination in cells with endogenous SAN1 intact. Each San1 variant was expressed in cells that coexpressed 6His-1Myc-ubiquitin. 6His-1Myc-ubiquitin conjugates were purified from cells using metal affinity chromatography. Total San1 in lysates (bottom) and in the purified 6His-1Myc-ubiquitin conjugates (top) were determined by Western analysis using anti-HSV antibodies. (B) Same experiment as in A, except “no Lys” mutants were examined.
San1 possesses low hydrophobic content in its N- and C-terminal domains
As mentioned in the Introduction, San1’s primary function is to bind misfolded proteins that have lost their native structures and expose hydrophobicity that would have been otherwise buried within a protein's structure (Fredrickson et al., 2011). However, San1 itself is highly disordered and lacks significant structure in the majority of its sequence (Rosenbaum et al., 2011). Because San1 is stable (Gardner et al., 2005; Figure 2A), it must have evolved a means to avoid recognizing another San1 as a misfolded protein.
The initial hint as to how San1 manages to avert in trans autoubiquitination and degradation once again came from an examination of the residue content of San1’s N- and C- terminal regions. We noticed that San1 had a low hydrophobic residue content in these regions, which was revealed when we plotted Kyte–Doolittle hydrophobicity values (Kyte and Doolittle, 1982) along San1’s sequence length (Figure 6A). Low hydrophobic residue content is a common feature for disordered proteins (Tompa, 2002), which do not form extensive globular structures in large part due to the lack of sufficient hydrophobic residues to form a core. Because San1 targets exposed hydrophobicity in its misfolded protein substrates (Fredrickson et al., 2011), the lack of sufficient exposed hydrophobicity in the highly disordered San1 could explain why one San1 molecule does not target another.
FIGURE 6:
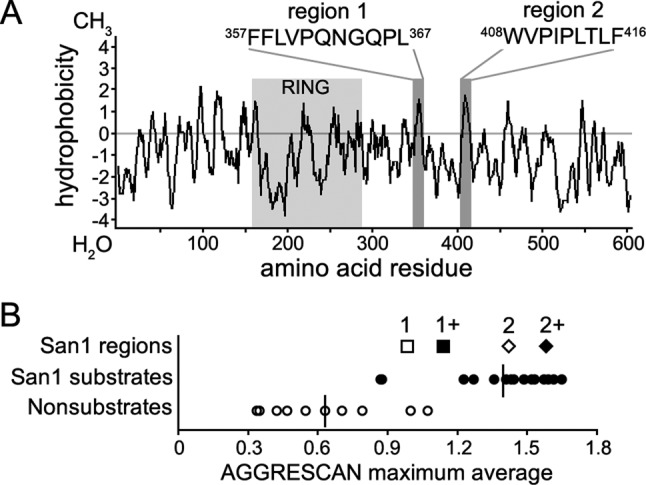
San1 possesses low hydrophobic residue content. (A) The hydrophobic content of San1 as determined by Kyte–Doolittle hydrophobicity analysis. RING domain is highlighted in light gray. Two of the most hydrophobic regions are highlighted in dark gray. (B) AGGRESCAN sliding-window analyses were performed to identify windows of hydrophobicity in San1. Previously described ranges of hydrophobicity observed in San1 substrates and nonsubstrates are shown for reference (Fredrickson et al., 2011). San1 regions 1 and 2 are marked with an open square and diamond, respectively. Regions 1 and 2 with increased stretches of hydrophobicity are indicated by a closed square and diamond, respectively.
Previously we used AGGRESCAN—an algorithm that predicts aggregation propensity of protein regions from their hydrophobic content (Conchillo-Sole et al., 2007)—to measure the local window of exposed hydrophobicity that San1 recognizes in its substrates (Fredrickson et al., 2011). This method provided a significant statistical separation between San1 substrates and nonsubstrates in terms of the maximal hydrophobicity score in a five-residue moving window (Fredrickson et al., 2011; Figure 6B). When we applied a similar analysis to San1’s sequence we found, surprisingly, that the maximum hydrophobicity scores within San1’s highly disordered N- and C- terminal domains (Figure 6A, regions 1 and 2) were within the range observed with San1 substrates (Figure 6B, 1 and 2). Some hydrophobic content in San1’s N- and C- terminal domains is expected, as small regions of increased hydrophobicity within disordered domains are predicted to be protein-binding sites (Dosztanyi et al., 2009), and the disordered N- and C-terminal regions contain San1 substrate-binding sites (Rosenbaum et al., 2011).
In the same previous hydrophobic analysis, we also found that San1 prefers a minimum stretch of five contiguous hydrophobic residues within its substrates (Fredrickson et al., 2011). Examination of the sequences of regions 1 and 2 in San1 revealed that the maximum contiguous stretch of hydrophobic residues was four (Figure 6A), below San1’s preference of five or more. Thus there are two hypotheses as to why one San1 molecule does not target another San1 molecule in trans as a misfolded substrate. First, it could be that San1 lacks the necessary extent of exposed hydrophobicity in its disordered regions to register as a misfolded protein. Second, the hydrophobicity in San1 might form a local structure and thus not be exposed in the context of the disordered regions, which would mask the potential recognition feature for in trans recognition.
Introduction of an appropriate hydrophobicity window in San1’s disordered N- and C-terminal domains causes degradation via an in trans mechanism
If an insufficient degree of hydrophobicity in San1’s highly disordered N- and C- terminal regions prevents one San1 from recognizing another San1, we predicted that increasing the hydrophobicity of these regions above the threshold San1 targets in its misfolded substrates would lead to San1 instability via an in trans mechanism. To test our prediction, we increased the number of contiguous hydrophobic residues in regions 1 and 2 of San1’s disordered C-terminal region (Figure 6A). Mutation of hydrophilic residues to hydrophobic residues in these regions (P361L, Q362I, N363V or P410L, P412I) resulted in the new contiguous hydrophobic sequences of 357FFLVLIV363 and 408WVLLIL416, both now above the threshold of five contiguous hydrophobic residues required for San1 recognition of its substrates (Fredrickson et al., 2011). We chose to make the stretches slightly above the threshold (six and seven contiguous hydrophobic residues rather than five) because we did not know how the hydrophilic flanking disordered segments would influence the hydrophobicity of the new hydrophobic stretches. In recent studies, we found that flanking hydrophilicity can alter the threshold of San1 recognition in its substrates (Fredrickson et al., 2013).
Because RING-inactivating mutant variants of the “plus Lys” San1 mutants allowed us to separate in cis versus in trans (Figure 2C), we used RING-inactivating mutant variants of the “plus hydro” mutants to determine whether increased hydrophobicity causes San1 to become unstable via an in trans mechanism. In both cases, we found that the RING-mutant versions of San1P361L, Q362I, N363V and San1P410L, P412I were now degraded in SAN1 cells, whereas RING-mutant normal San1 was stable (Figure 7A). Both RING-mutant San1P361L, Q362I, N363V and San1P410L, P412I were stable in san1∆ cells (Figure 7A), indicating that endogenous San1 recognizes each “plus hydro” San1 mutant in trans. Ubiquitination assays confirmed that the ubiquitination of RING-mutant San1P361L, Q362I, N363V and San1P410L, P412I occurred only when endogenous San1 was intact (Figure 7B), supporting an in trans recognition model.
FIGURE 7:

Increasing hydrophobic residue content in San1’s highly disordered N- and C-terminal regions leads to degradation via an in trans mechanism. (A) Cycloheximide-chase assays were performed to assess the stability of San1P361L, Q362I, N363V, San1P410L, P412I, and San1P410S, P412S possessing the RING-inactivating R280A mutation in the presence or absence of SAN1. Time after cycloheximide addition is indicated. Western blots were probed with anti-HSV antibodies. (B) Ubiquitination assays were performed to assess the extent of wild-type and mutant San1 ubiquitination. Each San1 variant was expressed in cells that coexpressed 6His-1Myc-ubiquitin. 6His-1Myc-ubiquitin conjugates were purified from cells using metal affinity chromatography. Total San1 in cell lysates (bottom) and in the purified 6His-1Myc-ubiquitin conjugates (top) were determined by Western analysis using anti-HSV antibodies. (C) Cycloheximide-chase assays were performed to assess the stability of “no Lys” San1P361L, Q362I, N363V, “no Lys” San1P410L, P412I, San1N332K, P361L, Q362I, N363V, San1P361L, Q362I, N363V, R371K, San1R371K, P410L, P412I, and San1P410L, P412I, N444K possessing the RING-inactivating R280A mutation in the presence or absence of SAN1. Time after cycloheximide addition is indicated. Western blots were probed with anti-HSV antibodies. (D) Decay curves for degradation assays in A and C were determined using ImageJ.
In each “plus hydro” San1 mutant, we replaced at least one Pro residue, which would normally be predicted to disrupt the structure of an ordered protein. Thus it is possible that we altered potential San1 structure such that San1 now exposes sufficient hydrophobicity rather than creating sufficient hydrophobicity with the substitutions. However, when we made the P410S and P412S substitutions, this did not alter the stability of the mutant San1 (Figure 7A), indicating that it was the introduction of an appropriate exposed hydrophobicity window that caused in trans recognition. Overall the results support the idea that San1 averts in trans ubiquitination and degradation by possessing low localized hydrophobicity in its disordered regions.
We do note that the degradation of the “plus hydro” San1 mutants was sluggish compared with degradation of the “plus Lys” San1 mutants, which showed a predominantly in trans mechanism (Figure 2C, N71K, R105K, and R115K). These “plus Lys” San1 mutants are proximal to San1’s endogenous Lys residues, whereas the “plus hydro” San1 mutants are distal to the endogenous Lys residues. Thus one model for the sluggish degradation rates of the “plus hydro” San1 mutants is that San1 requires Lys residues proximal to exposed hydrophobicity in its substrates to achieve optimal ubiquitination and rapid degradation. Before testing this hypothesis, we verified that the endogenous Lys residues were in fact necessary for the “plus hydro” mutants’ degradation (Figure 7C), indicating that the endogenous Lys residues are the sites of ubiquitin conjugation for the “plus hydro” mutants. We then examined the degradation of the “plus hydro” mutants with a single Arg- or Asn-to-Lys mutation created either upstream or downstream of the introduced hydrophobic region. We chose “plus Lys” mutants from Figure 2 that were the most proximal to hydro regions 1 or 2. In each case, the addition of a proximal Lys residue led to in trans degradation of each “plus hydro” San1 mutant that was more rapid than the degradation of each “plus hydro” San1 mutant without a proximal Lys residue (Figure 7, C and D). It is important to note that, in the context of the RING-inactivating mutation, the “plus Lys” mutants individually were stable (Figure 2C), and so their enhancing effect when combined with the “plus hydro” mutations was to allow more efficient in trans San1 degradation.
Stabilizing features are evolutionarily conserved in San1 orthologues
We have now described two key features of San1’s disordered N- and C-terminal regions that are required for maintaining San1’s stability in relation to San1’s function: 1) elimination of Lys residues to prevent in cis autoubiquitination and 2) minimization of local hydrophobicity to avert in trans autoubiquitination. Although it is clear that Saccharomyces cerevisiae San1 possesses these features, we wanted to know whether they are solely a function of S. cerevisiae San1 or are evolutionarily conserved in San1’s orthologues. Previously we demonstrated that SAN1’s orthologues from the closely related Saccharomyces bayanus and Saccharomyces mikatae species were capable of complementing the san1∆ allele for misfolded protein degradation in S. cerevisiae (Rosenbaum et al., 2011), suggesting the possibility of conservation.
To explore further the conservation of sequence characteristics, we examined San1 orthologue sequences from the more distantly related species Saccharomyces castellii, Ashbya gossypii, Candida albicans, and Schizosaccharomyces pombe (Figure 8A). These orthologues ranged from 23 to 73% identity and 39 to 80% similarity (Figure 8A). Of greatest interest, the most distant San1 orthologue from S. pombe has been shown to function in the nuclear protein quality control degradation of misfolded proteins in its native species (Matsuo et al., 2011). To assess each orthologue's characteristics as they compare to S. cerevisiae San1, we first determined the degree of intrinsic disorder in each San1 orthologue using PONDR (Romero et al., 1997). In every case, we found that the San1 orthologue was predicted to be highly disordered in the regions N- and C-terminal to the RING domain (Figure 8, B–F). Consistent with their predicted disorder, each San1 orthologue also contained low localized hydrophobic residue content when we plotted their Kyte–Doolittle hydrophobicity scores (Figure 8, G–K). Moreover, when we looked at the distribution of Lys residues along the length of each San1 orthologue, we found that the disordered N- and C-terminal regions in S. castellii, A. gossypii, and C. albicans were devoid of Lys residues (Figure 8, C–E). The San1 orthologue in S. pombe was not entirely devoid of Lys residues, as it possessed three in the distal portion of its C-terminal region (Figure 8F). Of interest, this region of S. pombe San1 possesses what appears to be a noncanonical second RING domain (residues 687–735), which is not seen in the other San1 orthologues. Overall the sequence features of S. cerevisiae San1 important for maintaining its stability appear to be conserved in its distantly related fungal orthologues.
FIGURE 8:

Lack of Lys residues, high disorder, and low hydrophobic residue content are conserved among San1 orthologues. (A) Species tree of related Ascomycota fungi. Figure was adapted from Wapinski et al. (2007). Listed below each species is the predicted evolutionary distance, percentage identity, and percentage similarity to S. cerevisiae. (B–F) Representation of the overall disorder of S. cerevisiae San1 (B) and orthologues from S. castellii (C), A. gossypii (D), C. albicans (E), and S. pombe (F). Endogenous Lys residue positions are marked on top. PONDR (www.pondr.com/) evaluation of disorder is on the bottom. (G–K) Representation of the overall Kyte–Doolittle hydropathy of S. cerevisiae San1 (G) and orthologues from S. castellii (H), A. gossypii (I), C. albicans (J), and S. pombe (K). Gray box highlights the RING domain in each orthologue.
To verify that the lack of Lys residues is also important for maintaining the stability of San1’s orthologues, we conducted studies to examine S. pombe San1 stability in S. cerevisiae. First, we tested whether S. pombe SAN1 can complement the san1∆ allele in S. cerevisiae for cdc68-1 temperature sensitivity. By itself S. pombe SAN1 did not complement (Figure 9A), but addition of the SV40 NLS to its C-terminus did result in complementation (Figure 9A), indicating that function was conserved if properly localized to the S. cerevisiae nucleus. S. pombe San1 is nuclear localized in S. pombe cells (Matsuo et al., 2011), so whatever internal sequence and import method it uses for optimal nuclear localization is not conserved between S. pombe and S. cerevisiae.
FIGURE 9:
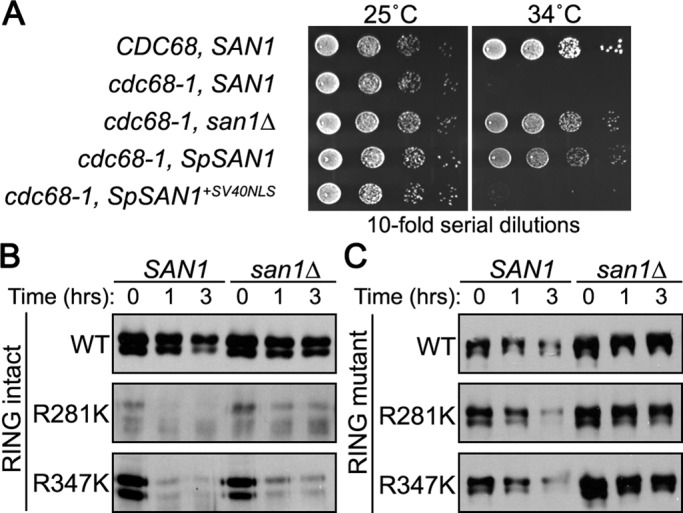
The San1 orthologue from S. pombe is degraded when Lys residues are introduced into the highly disordered C-terminal region. (A) Growth assays of cdc68-1 cells were performed to determine the ability of the S. pombe San1 orthologue to complement a san1∆ allele. Cells were spotted onto synthetic nutrient plates in 10-fold serial dilutions, and the plates were incubated for 3 d at the permissive (25°C) or restrictive (34°C) temperature. (B) Cycloheximide-chase assays were performed to assess the stability of S. pombe San1R281K and San1R347K in the presence or absence of SAN1. Time after cycloheximide addition is indicated. Western blots were probed with anti-HSV antibodies. (C) Identical experiment as in B, except that each S. pombe San1 construct also contained a RING-inactivating R149A mutation.
Next we examined S. pombe San1 stability in S. cerevisiae cells. We did observe some degradation of S. pombe San1 in SAN1 cells that was absent in san1∆ cells (Figure 9B), indicating that S. pombe San1 is recognized to some extent as a misfolded protein by S. cerevisiae San1. We next tested whether placement of additional Lys residues caused enhanced degradation by an in cis mechanism. We introduced single Arg-to-Lys point mutations in S. pombe San1’s disordered C-terminal region (R281K and R347K) and examined their degradation in cells with the endogenous S. cerevisiae SAN1 gene intact or deleted. Similar to the S. cerevisiae “plus Lys” San1 mutants, the S. pombe “plus Lys” San1 mutants were rapidly degraded in an identical manner in the presence or absence of endogenous S. cerevisiae San1 (Figure 9B). Introduction of a RING-inactivating mutation (R149A) to the S. pombe “plus Lys” San1 mutants abrogated the rapid degradation acquired upon addition of a Lys residue (Figure 9C). We conclude that S. pombe San1 has also minimized Lys residues in its disordered regions as a means of in cis self-protection identical to that of S. cerevisiae San1.
DISCUSSION
San1’s physiological function is to recognize misfolded proteins in the nucleus, targeting them for ubiquitination and proteasome degradation (Gardner et al., 2005). San1 accomplishes substrate recognition using highly disordered N- and C-terminal arms that interact with substrates (Rosenbaum et al., 2011). Within its substrates, San1 recognizes exposed hydrophobicity (Fredrickson et al., 2011). Thus the means of San1 self-protection that we discovered here are consistent with San1’s function. Selective loss of Lys residues in its disordered N- and C-terminal regions provides a simple means for San1 to avoid in cis autoubiquitination should these flexible regions come within close proximity to a bound, activated E2. Minimization of hydrophobicity in localized stretches within San1’s sequence keeps San1 from possessing the feature it recognizes in its substrates, which in turn prevents in trans autoubiquitination of one San1 molecule by another San1 molecule.
Although the majority of the San1 “plus Lys” mutants we examined were predominantly degraded via a San1-dependent in cis mechanism, there was one peculiar class of “plus Lys” mutants whose degradation was primarily independent of San1 (N33K, N61K, and N91K). Every member of this class of mutants is within the same portion of the N-terminal region (residues 33–91), suggesting that perturbation of this region has consequences distinct from other regions in San1. However, identical to all other regions of San1, general mutation of these positions to a residue other than Lys did not lead to rapid degradation, indicating that the specific addition of a Lys residue at these positions is essential for degradation. This brings up the perplexing question as to why these particular “plus Lys” mutations undergo San1-independent degradation. One possibility is that the ubiquitination of the “plus Lys” residue specifically in this region causes structural perturbations such that another quality control E3 now targets San1. This is hard to reconcile because, if we assume the initial ubiquitination event is caused by either in cis or in trans San1-dependent ubiquitination, deletion of endogenous SAN1 and introduction of a RING-inactivating mutation should have caused complete stability of these “plus Lys” mutants. Alternatively, perhaps San1 engages another E3 (or E4) as a partner in this region, and placement of a Lys residue within this region now allows for uncontrollable ubiquitination by this partner. Future experiments are required to address how and why these specific “plus Lys” mutants are rapidly degraded via a San1-independent degradation mechanism.
The “plus Lys” San1 mutants that were subject to a primarily San1-dependent in trans degradation mechanism (N71K, R105K, R115K) also clustered in the region immediately N-terminal to the RING domain. This region is predicted to have the highest order/structure outside of the RING domain (Figure 1A). It could be that these Lys mutations cause exposure of hydrophobicity that would normally be buried within a local structure, thus allowing them to be recognized as a misfolded protein by another San1 molecule. Again, this is hard to reconcile with the fact that mutation of these positions to either an Asn or Arg residue had very little effect on San1 stability (Figure 2A). It is therefore unlikely that the structure is perturbed by a Lys mutation. Perhaps this region is one that normally allows a functional interaction between two San1 molecules, such that placement of a Lys residue does not alter the structure but does improperly allow in trans ubiquitination to occur. We previously explored whether one San1 molecule interacts with another, but we were unable to gather any evidence to support this possibility using either coimmunoprecipitation or two-hybrid assays (Rosenbaum et al., 2011). It is possible that the interaction of one San1 molecule with another only occurs transiently in the context of substrate engagement to facilitate substrate polyubiquitination. If so, the highly transient San1–San1 substrate-mediated interaction would not be capable of being queried by traditional methods used to identify protein–protein interactions. Further studies will be required to reveal how and why these specific “plus Lys” San1 mutants are degraded via a San1-dependent in trans mechanism.
As we noted, the in trans degradation of the “plus hydro” San1 mutants was comparatively sluggish (Figure 7A). However, we could engineer the rapid degradation of the “plus hydro” San1 mutants by simply inserting a Lys residue proximal to their expanded, exposed hydrophobicity (Figure 7C). Previously it was found that the E3 Hrd1, which functions in the degradation of misfolded proteins in the ER (Smith et al., 2011), uses Ser and Thr residues in addition to Lys residues for substrate ubiquitination (Ishikura et al., 2010; Shimizu et al., 2010). Clearly this is not the case for the “plus hydro” San1 mutants, as the endogenous Lys residues alone were essential for their degradation. We believe that our new observations have important implications for how San1 normally operates in targeting its misfolded substrates. Although we do not think that the exposed hydrophobicity in San1’s misfolded substrates must be presented in the context of disorder as appears to be the case for misfolded proteins in the endoplasmic reticulum (Xie et al., 2009), we do now believe that the exposed hydrophobicity likely has to be proximal to Lys residues for efficient ubiquitination and rapid degradation, and no residue other than Lys can be used for San1-mediated ubiquitination. In support of this, in each San1 substrate for which we identified the regions of exposed hydrophobicity recognized by San1 (Fredrickson et al., 2011), the exposed hydrophobic region has proximally located Lys residues (our unpublished observations), and each substrate is rapidly degraded (Fredrickson et al., 2011).
Yeast possess ∼50 RING E3s by homology (Li et al., 2008). We examined this group for those that display high intrinsic disorder and contain fewer Lys residues than predicted for an average protein. Only the E3 Slx5 shared the similar characteristics seen with San1 (our unpublished observations). This is intriguing because Slx5 has also been implicated as a nuclear protein quality control E3 (Wang and Prelich, 2009). Other protein quality control E3s in the yeast endoplasmic reticulum (Hrd1 and Doa10) or cytoplasm (Ubr1) are not highly disordered, nor do they have a below-average complement of Lys residues (our unpublished observations). Thus these sequence features might be exclusive to the nucleus, where recognition of misfolded proteins could require a different mechanism than in the endoplasmic reticulum or cytoplasm.
There are also ∼300 RING E3s predicted by homology to exist in humans (Li et al., 2008). No human E3 has been identified that functions analogously to San1 in general nuclear protein quality control degradation. One issue with identifying a human orthologue of San1 is that highly disordered proteins often have low conservation of linear sequence identity (Brown et al., 2002), which is apparent in the fungal orthologues of San1 (Figure 8A). A lack of linear sequence identity conservation would confound orthologue identification searches, and so additional approaches are necessary to discover a bona fide human orthologue of San1. Because high intrinsic disorder and lack of Lys residues are conserved among San1’s fungal orthologues, we predict San1 human orthologues should also have these features. Of the ∼300 human RING E3s, we identified 6 RING E3s with large regions of disorder and a reduced number of Lys residues (our unpublished observations), suggesting that these disordered E3s likely employ a similar self-protective mechanism to that of San1. It is not known whether any of these candidates functions in protein quality control degradation in the human nucleus.
Finally, we believe that our findings might also be broadly applicable to E3s that are structured. It is conceivable that some structured E3s have evolved to protect themselves by having reduced Lys residue content on their surfaces in or near the E2-binding interface or substrate-interaction regions. This idea is difficult to assess, as the large majority of solved E3/E2 structures lack the full-length E3, which means we cannot precisely know where Lys residues are located in the complete E3 tertiary structure. Although it is not clear whether Lys-residue minimization is a broader means for structured E3 self-protection, it remains a possibility to be explored as more complete structures are determined.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strains used in this study are BY4741 (Brachmann et al., 1998), RGY506 (BY4741 san1∆) (Gardner et al., 2005), RGY3408 (RGY506 trp1∆::HIS3), and PJ69-3D (James et al., 1996). Standard yeast growth media and yeast genetic methods were used (Guthrie and Fink, 1991). Standard cloning protocols were used to construct each San1 mutant plasmid. The relevant portion of each plasmid was sequenced to verify the mutations. Exact oligonucleotide sequences, plasmids, and cloning details will be provided upon request.
Two-hybrid assays
All two-hybrid interaction tests were performed in duplicate using two independent isolates. Synthetic media plates lacking tryptophan and leucine (to verify spotting efficiency) or lacking tryptophan, leucine, and histidine (to test the two-hybrid interaction) were incubated at 30°C for 3–6 d.
Degradation assays
Cycloheximide-chase assays were performed similar to those previously described (Gardner et al., 2005). Briefly, cells were grown in synthetic liquid media with 2% glucose to a culture density of ∼1 × 107 cells/ml. Cycloheximide was added to 50 μg/ml, and the cultures were further incubated for 0–4 h. Cells were lysed at the appropriate time point in 200 μl of SUMEB (8 M urea, 1% SDS, 10 mM 3-(N-morpholino)propanesulfonic acid, pH 6.8, 10 mM EDTA, 0.01% bromophenol blue) by vortexing for 5 min with 100 μl of 0.5-mm acid-washed glass beads. Lysates were incubated at 65°C for 10 min and clarified for 5 min by centrifugation at 12,800 × g. Proteins were resolved on 8% SDS–PAGE gels, transferred to nitrocellulose, and immunoblotted with anti-HSV (Novagen, Gibbstown, NJ) antibodies in 2% milk in 1× Tris-buffered saline (TBS).
Ubiquitination assays
San1 variants were coexpressed with 6His-1Myc-ubiquitin (Spence et al., 2000) in cells with either endogenous SAN1 intact or deleted (san1∆). Cells were grown in 40 ml of synthetic liquid media at 30°C to a culture density of ∼1 × 107 cells/ml. Cells were harvested by centrifugation and resuspended in 1 ml of lysis buffer (50 mM Tris, pH 8.5, 8 M urea, 0.1% SDS) with phenylmethylsulfonyl fluoride. Resuspended cells were lysed by vortexing at 4°C for 20 min with 100 μl of 0.5-mm acid-washed glass beads. Cell lysates were clarified by centrifugation at 4000 × g for 10 min at 4°C and then added to 100 μl of TALON resin (Clontech, Mountain View, CA) that was equilibrated in lysis buffer. After overnight rotation at 4°C, the resin was collected by centrifugation for 2 min at 500 × g. Resin was washed 3× in lysis buffer plus 7.5 mM imidazole. Proteins were eluted by addition of 75 μl of SUMEB, followed by incubation at 65°C for 10 min. Lysate and eluate fractions were resolved on 4–12% gradient gels (Lonza, Basel, Switzerland), transferred to nitrocellulose, and immunoblotted with anti-HSV (Novagen) antibodies in 2% milk in 1× TBS.
Image processing
Western blots were scanned using an Epson Perfection V350 Photo scanner at 300 dpi (Epson, Long Beach, CA). All images were processed with a Mac iMac or Pro computer (Apple, Cupertino, CA) using Photoshop CS (Adobe, San Jose, CA). Decay curves for degradation assays were determined using ImageJ (National Institutes of Health, Bethesda, MD).
Acknowledgments
We thank Makoto Kawamukai for providing plasmids and Ning Zheng for key discussions and experimental advice on San1’s sequence characteristics. R.G.G. thanks Chris Hague for many illuminating conversations on averting in cis and in trans degradation. This work was supported by National Institutes of Health/National Institute of General Medical Sciences Training Grant 5T32GM007750 (E.K.F.), National Institutes of Health/National Institute on Aging Grant R01AG031136 (R.G.G.), an Ellison Medical Foundation New Scholar Award in Aging (R.G.G.), and a Marian E. Smith Junior Faculty Award (R.G.G.).
Abbreviations used:
- HSV
herpes simplex virus
- NLS
nuclear localization sequence
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-11-0811) on January 30, 2013.
REFERENCES
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, Williams CJ, Dunker AK. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55:104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology. 2006;66:S7–19. doi: 10.1212/01.wnl.0000192261.02023.b8. [DOI] [PubMed] [Google Scholar]
- Conchillo-Sole O, de Groot NS, Aviles FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics. 2007;8:65. doi: 10.1186/1471-2105-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z, Meszaros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–2746. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D, Hemmings BA. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem. 2004;279:35510–35517. doi: 10.1074/jbc.M404936200. [DOI] [PubMed] [Google Scholar]
- Fredrickson EK, Gallagher PS, Clowes Candadai SV, Gardner RG. Substrate recognition in nuclear protein quality control degradation is governed by exposed hydrophobicity that correlates with aggregation and insolubility. J Biol Chem. 2013;288:6130–6139. doi: 10.1074/jbc.M112.406710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson EK, Rosenbaum JC, Locke MN, Milac TI, Gardner RG. Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol Biol Cell. 2011;22:2384–2395. doi: 10.1091/mbc.E11-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JM, Peter M. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc Natl Acad Sci USA. 1999;96:9124–9129. doi: 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Nelson ZW, Gottschling DE. Degradation-mediated protein quality control in the nucleus. Cell. 2005;120:803–815. doi: 10.1016/j.cell.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. Methods Enzymol. 1991;194:1–863. [PubMed] [Google Scholar]
- Ishikura S, Weissman AM, Bonifacino JS. Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J Biol Chem. 2010;285:23916–23924. doi: 10.1074/jbc.M110.127936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CA. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS One. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gazdoiu S, Pan ZQ, Fuchs SY. Stability of homologue of Slimb F-box protein is regulated by availability of its substrate. J Biol Chem. 2004;279:11074–11080. doi: 10.1074/jbc.M312301200. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Kishimoto H, Tanae K, Kitamura K, Katayama S, Kawamukai M. Nuclear protein quality is regulated by the ubiquitin-proteasome system through the activity of Ubc4 and San1 in fission yeast. J Biol Chem. 2011;286:13775–13790. doi: 10.1074/jbc.M110.169953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper RK, Bordallo J, Deak PM, Taxis C, Hitt R, Wolf DH. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J Cell Sci. 1999;112(Pt 22):4123–4134. doi: 10.1242/jcs.112.22.4123. [DOI] [PubMed] [Google Scholar]
- Pruneda JN, Littlefield PJ, Soss SE, Nordquist KA, Chazin WJ, Brzovic PS, Klevit RE. Structure of an E3:E2∼Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol Cell. 2012;47:933–942. doi: 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P, Obradovic Z, Dunker AK. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Inform Ser Workshop Genome Inform. 1997;8:110–124. [PubMed] [Google Scholar]
- Rosenbaum JC, et al. Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol Cell. 2011;41:93–106. doi: 10.1016/j.molcel.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Okuda-Shimizu Y, Hendershot LM. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol Cell. 2010;40:917–926. doi: 10.1016/j.molcel.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J, Gali RR, Dittmar G, Sherman F, Karin M, Finley D. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell. 2000;102:67–76. doi: 10.1016/s0092-8674(00)00011-8. [DOI] [PubMed] [Google Scholar]
- Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- Wang X, Taplick J, Geva N, Oren M. Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett. 2004;561:195–201. doi: 10.1016/S0014-5793(04)00168-1. [DOI] [PubMed] [Google Scholar]
- Wang Z, Prelich G. Quality control of a transcriptional regulator by SUMO-targeted degradation. Mol Cell Biol. 2009;29:1694–1706. doi: 10.1128/MCB.01470-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A. Natural history and evolutionary principles of gene duplication in fungi. Nature. 2007;449:54–61. doi: 10.1038/nature06107. [DOI] [PubMed] [Google Scholar]
- Wu X, Yen L, Irwin L, Sweeney C, Carraway KL., 3rd Stabilization of the E3 ubiquitin ligase Nrdp1 by the deubiquitinating enzyme USP8. Mol Cell Biol. 2004;24:7748–7757. doi: 10.1128/MCB.24.17.7748-7757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Kanehara K, Sayeed A, Ng DT. Intrinsic conformational determinants signal protein misfolding to the Hrd1/Htm1 endoplasmic reticulum–associated degradation system. Mol Biol Cell. 2009;20:3317–3329. doi: 10.1091/mbc.E09-03-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Johnston GC, Singer RA. The Saccharomyces cerevisiae Cdc68 transcription activator is antagonized by San1, a protein implicated in transcriptional silencing. Mol Cell Biol. 1993;13:7553–7565. doi: 10.1128/mcb.13.12.7553. [DOI] [PMC free article] [PubMed] [Google Scholar]