

Tumor necrosis factor-α activates the enzyme TACE/ADAM17 through the guanine nucleotide exchange factor GEF-H1, Rac, and p38, leading to activation of the epidermal growth factor. GEF-H1 mediates hierarchical activation of Rac and RhoA through differential phosphorylation.
Abstract
Transactivation of the epidermal growth factor receptor (EGFR) by tumor necrosis factor-α (TNF-α) is a key step in mediating RhoA activation and cytoskeleton and junction remodeling in the tubular epithelium. In this study we explore the mechanisms underlying TNF-α–induced EGFR activation. We show that TNF-α stimulates the TNF-α convertase enzyme (TACE/a disintegrin and metalloproteinase-17), leading to activation of the EGFR/ERK pathway. TACE activation requires the mitogen-activated protein kinase p38, which is activated through the small GTPase Rac. TNF-α stimulates both Rac and RhoA through the guanine nucleotide exchange factor (GEF)-H1 but by different mechanisms. EGFR- and ERK-dependent phosphorylation at the T678 site of GEF-H1 is a prerequisite for RhoA activation only, whereas both Rac and RhoA activation require GEF-H1 phosphorylation on S885. Of interest, GEF-H1-mediated Rac activation is upstream from the TACE/EGFR/ERK pathway and regulates T678 phosphorylation. We also show that TNF-α enhances epithelial wound healing through TACE, ERK, and GEF-H1. Taken together, our findings can explain the mechanisms leading to hierarchical activation of Rac and RhoA by TNF-α through a single GEF. This mechanism could coordinate GEF functions and fine-tune Rac and RhoA activation in epithelial cells, thereby promoting complex functions such as sheet migration.
INTRODUCTION
The Rho-family small GTPases RhoA and Rac are key regulators of the cytoskeleton and affect a variety of vital cellular functions, including growth, adhesion, polarity, and migration (Jaffe and Hall, 2005). In epithelial cells RhoA and Rac are also major regulators of the intercellular junctions and transepithelial transport (Kapus and Szaszi, 2006; Samarin and Nusrat, 2009; Citi et al., 2011). Despite their important roles, the mechanisms underlying activation/inactivation of Rho-family GTPases are not fully understood. Rho proteins cycle between GDP-bound (inactive) and GTP-bound (active) states, and this cycle is controlled by the coordinated action of guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and GDP dissociation inhibitors. Specific members of the large RhoGEF family mediate activation of Rho proteins in response to different extracellular stimuli (Rossman et al., 2005). Understanding of the pathway- and context-specific regulation of individual GEFs, however, is incomplete.
The pleiotropic inflammatory cytokine tumor necrosis factor-α (TNF-α) has emerged as an important pathogenic factor in a number of chronic diseases, including rheumatoid arthritis and inflammatory bowel disease (Clark, 2007), as well as acute renal injury and chronic kidney disease (Pascher and Klupp, 2005; Vielhauer and Mayadas, 2007). TNF-α disrupts epithelial intercellular junctions and elevates transepithelial permeability, and these effects are believed to contribute to disease pathogenesis. In previous work we showed that in kidney tubular cells TNF-α activates RhoA and induces Rho kinase–dependent myosin phosphorylation, leading to cytoskeleton remodeling and enhanced paracellular permeability (Kakiashvili et al., 2009). We also identified GEF-H1 (ArhGEF2) as the exchange factor mediating TNF-α–induced RhoA activation and showed that it was activated toward RhoA by ERK-mediated phosphorylation on its threonine 678 site (Kakiashvili et al., 2009).
GEF-H1 (and its murine homologue lfc) is a guanine nucleotide exchange factor that can activate RhoA and Rac. It binds to and is regulated by microtubules and tight junctions (Ren et al., 1998; Benais-Pont et al., 2003; reviewed in Birkenfeld et al., 2008). GEF-H1 has been implicated in a remarkable array of functions in different cell types, including the regulation of cytokinesis, cell growth, migration, epithelial and endothelial barrier, dendritic spine morphology, and mechanical signal transduction (Birkenfeld et al., 2007; Kang et al., 2009; Nalbant et al., 2009; Nie et al., 2009; Guilluy et al., 2011). In previous studies we found that in tubular cells physical and chemical stimuli, such as depolarization of the plasma membrane potential (Waheed et al., 2010), exposure to the immunosuppressant drugs cyclosporine and sirolimus (Martin-Martin et al., 2012), and hyperosmotic shock (Ly et al., 2013), also require GEF-H1 for RhoA activation. Of interest, most of the described effects of GEF-H1 were attributed to RhoA activation. However, in vitro exchange activity assays demonstrated that GEF-H1 can also act as a GEF for Rac (Ren et al., 1998; Gao et al., 2001). Moreover, PAK4-dependent phosphorylation was suggested to act as a switch that blocks GEF-H1–dependent stress fiber formation and promotes lamellipodia generation in fibroblasts. However, the functional significance and stimulus-dependent regulation of the Rac GEF activity of GEF-H1 is largely unexplored.
In search for the signaling pathway that mediates TNF-α–induced GEF-H1 activation toward RhoA, we found that TNF-α transactivates the epidermal growth factor receptor (EGFR), leading to stimulation of ERK, GEF-H1, and RhoA (Kakiashvili et al., 2011). Recently EGFR transactivation has emerged as a common mechanism through which many stimuli, including G protein–coupled receptor activation and changes in the physical environment, indirectly activate ERK and enhance proliferation (Higashiyama et al., 2008; Liebmann, 2011). EGFR activation is caused by the release of epidermal growth factor (EGF)–family ligands by proteases belonging to the a disintegrin and metalloproteinase (ADAM) enzyme family (Edwards et al., 2008). We found that EGFR transactivation by TNF-α in tubular cells was inhibited by TAPI-1, suggesting the involvement of an ADAM family enzyme (Kakiashvili et al., 2011). However, the identity of the enzyme and the mechanisms through which it is activated by TNF-α remained unknown. The TAPI-1–sensitive TNF-α convertase enzyme (TACE), also known as ADAM17, is a good candidate. This enzyme was named after its role in the release of TNF-α (Black et al., 1997) but was later found to cleave a multitude of other substrates, including proforms of EGFR ligands (Doedens et al., 2003; Gooz, 2010). TACE is activated by many stimuli, but the underlying mechanisms are not entirely understood. Phosphorylation is likely involved in regulation, as the cytosolic tail of TACE can be phosphorylated by ERK (Soond et al., 2005; Gooz et al., 2006), PDK1 (Zhang et al., 2006), p38 (Killock and Ivetic, 2010; Xu and Derynck, 2010), and Src kinase (Maretzky et al., 2011).
The aim of this study is to explore the mechanism through which TNF-α induces EGFR/ERK/RhoA activation in tubular cells. We present evidence that GEF-H1, Rac, and p38 have a central role in TNF-α–induced TACE activation. Our data also reveal a hierarchical activation of Rac and RhoA induced by differential regulation of GEF-H1 toward these two small GTPases through phosphorylation on different sites.
RESULTS
TNF-α activates TACE in tubular cells, which in turn mediates ERK activation
Our previous work suggested that TNF-α exerts some of its effects in tubular cells through the activation of a TAPI-1–sensitive metalloprotease (MMP; Kakiashvili et al., 2009). To identify this enzyme and explore the potential role of TACE, we assessed the effect of TNF-α on MMP activity. The assay we used follows the cleavage of a fluorigenic peptide that is an excellent substrate for TACE. In the uncleaved peptide the fluorescence of the 7-methoxycoumarin group is efficiently quenched by resonance energy transfer to the 2,4-dinitrophenyl group. Cleaving of the amide bond between the fluorescent group and the quencher group by ADAM family MMPs, including TACE, causes an increase in fluorescence. LLC-PK1 or normal rat kidney (NRK) cells were treated with TNF-α and lysed, and MMP activity was measured. As shown in Figure 1, A–D, TNF-α induced a significant increase in the cleavage of the fluorigenic substrate in both cell types. Of importance, this enhanced cleavage was prevented by the addition of TAPI-1, an inhibitor of TACE (Figure 1, A and C). Because neither the fluorigenic substrate nor the inhibitor is exclusively specific for TACE, we used a small interfering RNA (siRNA) approach as well. As shown in Figure 1, E and F, transfection with the TACE-specific siRNA achieved >90% decrease in levels of the protein in both LLC-PK1 and NRK cells. The overall basal MMP activity was unaffected by TACE silencing. Of importance, however, and consistent with the results obtained using TAPI, TACE down-regulation prevented TNF-α–induced enhanced substrate cleavage in both cell lines (Figure 1, B and D). These data therefore suggest that TACE is indeed activated in TNF-α–stimulated tubular cells.
FIGURE 1:

(A–D) TNF-α activates TACE. Confluent LLC-PK1 (A, B) or NRK (C, D) cells were treated with 50 ng/ml TNF-α for 30 min, and MMP activity was measured in the cell lysates using a fluorigenic peptide substrate, as described in Materials and Methods. In A and C cells were pretreated for 30 min with 10 or 20 μM TAPI-1 as indicated. In B and D cells were transfected with NR or TACE-specific siRNA 48 h before TNF-α addition. After subtraction of the background fluorescence, the control fluorescence values in each experiment were taken as unity, and the fluorescence in the treated samples was expressed as fold increase. The graphs represent mean ± SEM from three independent experiments performed in triplicate. (E, F) TACE mediates TNF-α–induced ERK activation. LLC-PK1 (E) or NRK (F) cells were transfected with nonrelated siRNA or siRNA directed against pig (E) or rat (F) TACE. Forty-eight hours later the cells were treated with 10 ng/ml TNF-α for 10 min, and the levels of phospho-ERK, total ERK, and TACE were detected by Western blotting. The graphs show quantification of the blots using densitometry. The amount of phospho-ERK was normalized to total ERK in the corresponding cell lysates. The results in each experiment were expressed as percentage compared with the TNF–α–treated sample, taken as 100%. The graphs show mean ± SE from n = 3 independent experiments. Statistical analysis is described in Materials and Methods.
Using the specific siRNA, we next sought to ascertain the role of TACE in the TNF-α–induced activation of ERK, which according to our earlier finding was inhibited by TAPI-1 (Kakiashvili et al., 2011). TNF-α induced an elevation in pERK levels in both LLC-PK1 and NRK cells (Figure 1, E and F), and this effect was abolished by the TACE-specific siRNA. Taken together, these experiments show that TNF-α activates TACE, which is necessary for the ensuing ERK activation.
TNF-α–induced TACE activation is mediated by p38
We next explored the mechanisms through which TNF-α activates TACE. Because TNF-α is a potent activator of the stress kinase p38 (Leonard et al., 1999), which regulates TACE (Killock and Ivetic, 2010; Xu and Derynck, 2010), we first studied the potential role of this kinase. Western blotting using a phospho-p38–specific antibody verified activation of p38 by TNF-α in LLC-PK1 cells (Figure 3B). Of importance, the p38 inhibitor SB203580 prevented TNF-α–induced TACE activation (Figure 2, A and B), as well as TNF-α-induced ERK activation (Figure 2C). Next we asked whether p38 activation is sufficient to stimulate the TACE/EGFR/ERK pathway. To address this, we transfected cells with a p38 construct along with hemagglutinin (HA)–tagged ERK2, which was used as an indicator of TACE and EGFR activation downstream from p38. Transfected ERK2 was precipitated through the tag, and its phosphorylation was detected using a phospho-ERK–specific antibody. Because under the applied conditions cotransfection efficiency is high, this method allowed us to detect ERK phosphorylation exclusively in cells that also expressed the active p38 construct. As shown in Figure 2D, coexpression of an active p38 with ERK induced significant ERK phosphorylation. Of importance, when cells were treated with the TACE inhibitor TAPI-1 before precipitation of HA-ERK, the p38-induced ERK phosphorylation was no longer detectable, suggesting that p38 indeed enhances ERK phosphorylation through TACE.
FIGURE 3:

Rac is activated by TNF-α and mediates p38 and TACE activation. (A) TNF-α activates Rac. LLC-PK1 cells were treated with 10 ng/ml TNF-α for the indicated times. Cells were lysed, and active Rac was precipitated using GST-PBD. Rac in the precipitates and total cell lysates (active and total, respectively) was detected by Western blotting and quantified by densitometry. The amount of active Rac in each sample was normalized to the corresponding total Rac. The data obtained in each experiment are expressed as percentage compared with the level of the 5-min TNF-α–treated sample, which is taken as 100%. (B, C) LLC-PK1 cells were transfected with NR or porcine Rac1/2-specific siRNA. Forty-eight hours later the cells were treated with 10 ng/ml TNF-α for 5 min (B) or 30 min (C). In B, total cell lysates were probed on Western blots with antibodies against phospho-p38, p38, Rac, and the loading control GAPDH. The blots were quantified and phospho-p38 normalized with p38 in the same samples, as described for pERK in Figure 1. In C, TACE activity was measured as described in Figure 1. The graphs show mean ± SE from n = 5 (A), 8 (B), or 3 (C) independent experiments.
FIGURE 2:

TNF-α–induced TACE and ERK activation is mediated by p38. (A–C) Confluent LLC-PK1 (A, C) or NRK (B) cells were treated with 10 μM SB203580 for 30 min, followed by 50 ng/ml TNF-α (30 min [A, B] or 5 min [C]). In A and B, MMP activity was measured and expressed as in Figure 1. In C, pERK and ERK levels were determined as in Figure 1. (D) LLC-PK1 cells grown in 10-cm dishes were transfected with HA-tagged ERK2 with or without cotransfection of FLAG-p38. Forty-eight hours later, where indicated, cells were treated with 10 μM TAPI for 30 min. Next the cells were lysed and HA-ERK was precipitated through the tag, and pERK and HA in the precipitates were detected by Western blotting. The graphs in C and D show quantification of the blots by densitometry. Density values of pERK were normalized using the corresponding total ERK (C) or HA (D) signal and were expressed as in Figure 1. All graphs show mean ± SE from n = 3 independent experiments. Note that in C the samples were run on the same gel, and unrelated lanes were cut from the scanned gel.
TNF-α–induced TACE activation is mediated by Rac
The small GTPase Rac can activate p38 through Pak1 (Zhang et al., 1995). The possible role of Rac in TACE regulation, however, has not been studied. Therefore we next asked whether Rac could contribute to the TNF-α–induced activation of TACE. First we tested whether TNF-α activates Rac, using an affinity precipitation assay with glutathione S-transferase (GST)–p21-binding domain (PBD)–coupled beads. The antibody that we used to visualize precipitated Rac is able to detect all three Rac isoforms. Our results revealed that TNF-α induced Rac activation as early as after 0.5 min of stimulation (Figure 3A), with some further increase at the 5-min time point. To test the role of Rac in mediating the effects of TNF-α, we used a specific siRNA against Rac 1 and 2. As shown in Figure 3B, the siRNA induced a marked decrease in Rac expression. Of importance, Rac silencing prevented both TNF-α–induced p38 (Figure 3B) and TACE activation (Figure 3C). Rac silencing, however, did not interfere with TNF-α–induced activation of NFκB (Supplemental Figure S1, A and B), verifying that it did not cause an overall inhibition of all TNF-α–induced signaling.
Rac is required for TNF-α–induced ERK activation
To ensure that Rac and p38 are indeed necessary for TNF-α–induced activation of the ERK pathway, we used two different approaches to interfere with Rac activation. First, we silenced Rac using an siRNA as before. Figure 4A demonstrates that Rac silencing prevented TNF-α–induced ERK activation. We verified this finding by using a dominant-negative Rac (RacT17A, DN-Rac), which was cotransfected with HA-tagged ERK2. After treatment with TNF-α, HA-ERK was immunoprecipitated, and its phosphorylation status was evaluated by Western blotting with a pERK-specific antibody. As described earlier, this method allowed us to study ERK activation exclusively in the transfected cells. As shown in Figure 4B, whereas TNF-α induced phosphorylation of HA-ERK in cells transfected with HA-ERK alone, this was prevented by the coexpression of DN-Rac.
FIGURE 4:

Rac mediates ERK and RhoA activation induced by TNF-α. (A) LLC-PK1 cells were transfected with NR siRNA or porcine Rac1/2 siRNA. Forty-eight hours later the cells were incubated in Na+ medium for 15 min, followed by the addition of 10 ng/ml TNF-α in Na+ medium or exchange of the medium for K+ medium (5 min). pERK was detected and quantified as in Figure 1. The blot was stripped and reprobed with anti-Rac. (B) Cells were transfected with HA-ERK2 with or without cotransfection of DN-Rac and 48 h later treated with TNF-α (5 min). HA-ERK was precipitated and its phosphorylation detected as in Figure 2D. (C) Cells were transfected with NR siRNA or porcine Rac1/2-specific siRNA for 48 h. Cells were treated with TNF-α (5 min), and active RhoA was precipitated with GST-RBD and quantified as described for Rac in Figure 3. The graphs show mean ± SE from n = 3 (A, B) or 5 (C) independent experiments.
Next we asked whether the requirement for Rac is specific for TNF-α–induced ERK activation. We compared the effect of Rac silencing on ERK activation induced by TNF-α and plasma membrane depolarization. Depolarization also activates RhoA through an ERK- and GEF-H1–dependent mechanism (Waheed et al., 2010). As expected, depolarization induced by 130 mM KCl potently stimulated ERK phosphorylation in LLC-PK1 cells (Figure 4A). In contrast to the TNF-α–induced ERK activation, however, the effect of depolarization was not affected by Rac silencing, suggesting that Rac does not mediate ERK activation by all stimuli but is specific for the TNF-α–induced pathway.
Rac is required for TNF-α–induced RhoA activation
ERK activation is necessary for TNF-α–induced RhoA activation, suggesting that Rac might also be required for RhoA activation induced by this cytokine. To test this assumption, we explored RhoA activation using the Rho-binding domain (RBD)–GST precipitation assay after Rac silencing. TNF-α induced a well-detectable RhoA activation in LLC-PK1 cells transfected with a nonrelated control siRNA. In contrast, RhoA was not activated in cells transfected with the Rac siRNA (Figure 4C). These findings suggest that in tubular epithelial cells TNF-α–induced RhoA activation depends on Rac and that activation of the two small GTPases occurs as a sequential event with a hierarchy between Rac and RhoA.
TNF-α activates Rac in a GEF-H1–dependent manner
We next wished to identify the exchange factor mediating TNF-α–induced Rac activation. GEF-H1, the exchange factor required for TNF-α-induced RhoA activation, was also found to have Rac exchange activity (Ren et al., 1998). This prompted us to explore its potential role in TNF-α–induced activation of Rac. Activated Rac GEFs were precipitated from control and TNF-α–treated cell lysates using the nucleotide-free Rac(G15A) mutant, and the presence of GEF-H1 was tested by Western blotting. Figure 5A shows that only a small amount of GEF-H1 was captured by GST-Rac(G15A) from untreated cells. Of importance, when cells were stimulated with TNF-α, the Rac(G15A)-associated GEF-H1 was significantly enhanced. Moreover, this was detectable as early as 0.5 min after the addition of TNF-α, similar to the rapid activation of Rac. These data suggest that GEF-H1 is activated toward Rac. To substantiate that GEF-H1 indeed mediates TNF-α–induced Rac activation, we silenced it using a specific siRNA. The siRNA transfection achieved ≥90% reduction in GEF-H1 protein expression (Figure 5B). No change in the small basal activity of Rac was evident in cells transfected with the GEF-H1–specific siRNA. Of importance, however, TNF-α–induced Rac activation was prevented by GEF-H1 silencing. In fact, Rac activity in TNF-α–treated and GEF-H1–down-regulated cells was consistently lower than the control level. We verified that GEF-H1 silencing did not prevent TNF-α–induced activation of NFκB, suggesting that it did not prevent activation of the TNF receptors (Supplemental Figure S1; Kakiashvili et al., 2009). Taken together, these data suggest that GEF-H1 mediates not only the TNF-α–induced activation of RhoA but also that of Rac.
FIGURE 5:

TNF-α activates Rac, TACE, and ERK through GEF-H1. (A) LLC-PK1 cells were treated with 10 ng/ml TNF-α for the indicated times. Active GEF-H1 was precipitated using GST-Rac(G15A). GEF-H1 in the precipitates and total cell lysates (active and total, respectively) was detected by Western blotting. The blots were quantified as described for Rac. (B–E, G) LLC-PK1 cells were transfected with NR siRNA or GEF-H1-specific siRNA. Forty-eight hours later the cells were treated with 10 ng/ml TNF-α for 5 min (B–D, G) or 30 min (E). In B, active Rac was detected and quantified as in Figure 3. In C and D, pERK, ERK, phospho-p38, p38, GEF-H1, and GAPDH were detected by Western blotting. In E, MMP activity was determined as in Figure 1. In G, pEGFR was detected using an antibody against phospho-Y845 EGFR. For all blots quantification was done using densitometry as described earlier. The data for phospho-p38, pERK, and pEGFR were normalized to the corresponding total levels of these proteins, and the data for EGFR were normalized using GAPDH. (F) LLC-PK1 cells were transfected with HA-ERK2 with cotransfection of NR siRNA, GEF-H1 siRNA, or FLAG-p38, as indicated. Where indicated, cells were treated with TNF-α for 5 min. HA-ERK was precipitated and its phosphorylation assessed using a pERK antibody. The top two blots show the immunoprecipitated pERK and HA signals (IP), and the bottom two blots demonstrate GEF-H1 and tubulin in the corresponding total cell lysates. The graphs show mean ± SE from n = 3 (E–G), 4 (A, B), or 8 (C, D) independent experiments.
TNF-α activates p38, TACE, and ERK through GEF-H1
We next sought to ascertain whether GEF-H1 is a mediator of TNF-α–induced activation of the p38/TACE/ERK pathway, as anticipated from its role in Rac activation. GEF-H1 silencing indeed reduced TNF-α-induced activation of ERK and p38 (Figure 5, C and D) and prevented TACE activation (Figure 5E). These effects were similar to those observed with Rac down-regulation (Figure 3, B and C). Of interest, the basal activity of TACE was not affected by GEF-H1 silencing, suggesting that the GEF-H1/Rac/p38 pathway has no role in regulating basal MMP activity but is key for TNF-α–induced stimulation of TACE.
To verify that p38 activation is indeed an effector of GEF-H1 in mediating ERK activation, we asked whether the inhibition of TNF-α–induced ERK activation observed when GEF-H1 was silenced can be overcome by overexpressing p38. First, we verified the effectiveness of GEF-H1 silencing in cells cotransfected with GEF-H1 siRNA and HA-ERK with or without active p38. As shown in Figure 5F (left), GEF-H1 was potently down-regulated, and this abolished TNF-α–induced HA-ERK phosphorylation. Figure 5F (right) demonstrates that coexpression of an active p38 construct together with the nonrelated (NR) siRNA enhanced HA-ERK phosphorylation (see also Figure 2D). FLAG-p38–induced ERK phosphorylation was not prevented by GEF-H1 silencing, suggesting that p38 is downstream from GEF-H1.
Because GEF-H1 also mediates TNF-α–induced RhoA activation, we next asked whether RhoA contributes to stimulation of TACE and ERK. Of interest, silencing of RhoA using a specific siRNA also reduced TNF-α–induced ERK activation, although to a lesser extent than Rac silencing (Supplemental Figure S2). Further, TACE activation was also prevented by RhoA silencing. Of importance, we found that in cells transfected with RhoA siRNA GEF-H1 levels were also reduced, which could partly explain this finding (see Discussion).
TACE activates the EGFR through GEF-H1
Having seen that TACE activation was regulated by GEF-H1, we also sought to verify that GEF-H1 indeed regulates EGFR activation. Therefore we explored how GEF-H1 silencing affects TNF-α–induced EGFR activation. TNF-α-induced phosphorylation of the EGFR was detected using an antibody against the phosphorylated Y845 site. TNF-α induced a well-detectable increase in phospho-EGFR in cells transfected with the control siRNA (Figure 5G). GEF-H1 down-regulation prevented this increase. Surprisingly, GEF-H1 silencing also induced a significant drop in the levels of the total EGFR protein. Normalizing EGFR phosphorylation to the total EGFR levels, however, revealed that GEF-H1 silencing also prevented TNF-α–induced phosphorylation of the remaining EGFR. Taken together, these data verify that GEF-H1 regulates EGFR activation induced by TNF-α through TACE. In addition, GEF-H1 silencing also reduces EGFR expression.
Rac activation and GEF-H1 stimulation toward Rac are independent of the EGFR and ERK
In previous work we showed that TNF-α–induced, GEF-H1-dependent RhoA activation was mediated by the EGFR and ERK. Our data presented so far, however, suggest that in contrast to RhoA activation, GEF-H1–dependent Rac activation is upstream of the EGFR and ERK. To substantiate this notion, we explored how inhibition of the EGFR and ERK affected Rac activation and the stimulation of GEF-H1 toward Rac. In contrast to RhoA, which was inhibited by the MEK1/2 inhibitor PD98059 or the EGFR inhibitor AG1478 (Kakiashvili et al., 2011), neither TNF-α–induced Rac activation nor stimulation of GEF-H1 toward Rac was affected by these inhibitors (Figure 6, A–D). These data suggest that the Rac- and RhoA-specific exchange activities of GEF-H1 are indeed differentially regulated.
FIGURE 6:

TNF–α-induced Rac activation and stimulation of GEF-H1 toward Rac do not require EGFR and ERK. LLC-PK1 cells were treated with 20 μM PD98059 (A, B) or 10 μM AG1478 (C, D) for 15 min, followed by addition of 10 ng/ml TNF-α for 5 min (A, C) or 2 min (B, D). In A and C, active Rac was precipitated using GST-PBD. In B and D, active GEFs were precipitated using GST-Rac(G15A), and GEF-H1 was detected by Western blotting. Densitometric analysis was done as described. The graphs show mean ± SE from n = 4 (A–C) or 3 (D) independent experiments.
TNF-α–induced Rac activation and GEF-H1 stimulation toward Rac do not require phosphorylation on T678
A possible mechanism for the differential regulation of GEF-H1 is through specific phosphorylation sites. RhoA activation requires phosphorylation on T678 (Fujishiro et al., 2008; Kakiashvili et al., 2009). Therefore we tested the role of this site in the differential regulation of GEF-H1 toward Rac and RhoA by comparing how a point mutant GEF-H1 that lacks the ERK-target T678 is activated toward Rac and RhoA. LLC-PK1 cells were transiently transfected with a GFP-tagged wild-type or GEF-H1T678A point-mutant protein. RhoA and Rac GEFs were precipitated using GST-tagged RhoA(G17A) or Rac(G15A), respectively, and the presence of the GFP-tagged WT or mutant GEF-H1 was detected using an antibody against GFP. TNF-α enhanced the association of WT-GEF-H1 with both nucleotide-free small GTPases (Figure 7, A and B). Consistent with our previously reported findings, elimination of the T678 site prevented TNF-α–induced activation of GEF-H1 toward RhoA (Figure 7A). In contrast, TNF-α stimulated the association of GEF-H1T678A with Rac(G15A) to a similar extent as the WT protein. Taken together, these data support the role of differential phosphorylation in GEF-H1 activation toward Rac and RhoA. Whereas GEF-H1 activation toward RhoA is mediated by EGFR- and ERK-dependent phosphorylation on T678, its activation toward Rac does not require this phosphorylation.
FIGURE 7:

Differential role of the GEF-H1 T678 and S885 phosphorylation sites in GEF-H1 activation toward Rac and RhoA. LLC-PK1 cells were transfected with GFP-tagged wild-type GEF-H1 (GEF-H1WT) or the nonphosphorylatable point mutant GEF-H1T678A or GEF-H1S885A as indicated. At 48 h posttransfection, cells were treated with 10 ng/ml TNF-α (5 min), and activated GEFs were precipitated using RhoA(G17A) (A, C) or Rac(G15A) (B, D). The GFP-tagged GEF-H1 protein was detected by Western blotting using anti-GFP. The blots were quantified as described earlier. The graphs show mean ± SE from n = 4 (A, D) or 3 (B and C) independent experiments.
TNF-α–induced GEF-H1 activation toward Rac requires phosphorylation on Ser-885
Next we wished to gain insight into the mechanisms that mediate TNF-α–induced activation of GEF-H1 toward Rac. In a mass spectrometry analysis of the phosphorylated amino acids in GEF-H1 precipitated from TNF-α–stimulated cells (Kakiashvili et al., 2009) we found the S885 site to be phosphorylated. This site is the target of numerous kinases and has been implicated in GEF-H1 regulation (Birkenfeld et al., 2008). Therefore we explored the role of this site in the TNF-α–induced effects. First, using an HA-tagged GEF-H1, we investigated the basal and TNF-α–induced phosphorylation of the S885 site. The HA-tagged GEF-H1 was precipitated from control and TNF-α–treated cells, and its phosphorylation was tested using an antibody specific for phospho-S885 GEF-H1. In most (but not all) experiments we found a trend for increased phosphorylation in the TNF-α–treated samples (Supplemental Figure S3).
To gain further insight into the role of S885, we generated a point mutant lacking this phosphorylation site (GFP-GEF-H1S885A). Using the GST-Rac(G15A) precipitation assay, we tested whether this mutant can be activated toward Rac. As shown in Figure 7D, in contrast to GEF-H1T678A, GFP-GEF-H1S885A showed significantly reduced TNF-α–induced activation toward Rac. To further substantiate the differential role of the T678 and S885 sites in Rac and RhoA activation, we tested the effect of the phosphorylation-incompetent point mutant GEF-H1 molecules on ERK activation.
To eliminate the confounding effect of endogenous GEF-H1 in these experiments, we silenced GEF-H1 using either a porcine-specific siRNA (Figure 8A) or a DNA vector–based short hairpin RNA (shRNA; Figure 8B). Both approaches efficiently prevented TNF-α–induced ERK activation (Figure 8, A and B, compare lanes 1–4). Expression of the human (siRNA resistant) GFP-GEF-H1T678A in cells in which endogenous GEF-H1 was silenced resulted in the restoration of TNF-α–induced ERK phosphorylation (Figure 8, A and B, lanes 5 and 6). In contrast, expression of GFP-GEF-H1S885A did not promote TNF-α–induced ERK activation (Figure 8, A and B, lanes 7 and 8). Taken together, the data verify that phosphorylation of S885 but not T678 plays a key role in TNF-α–induced GEF-H1 and Rac-dependent ERK activation.
FIGURE 8:
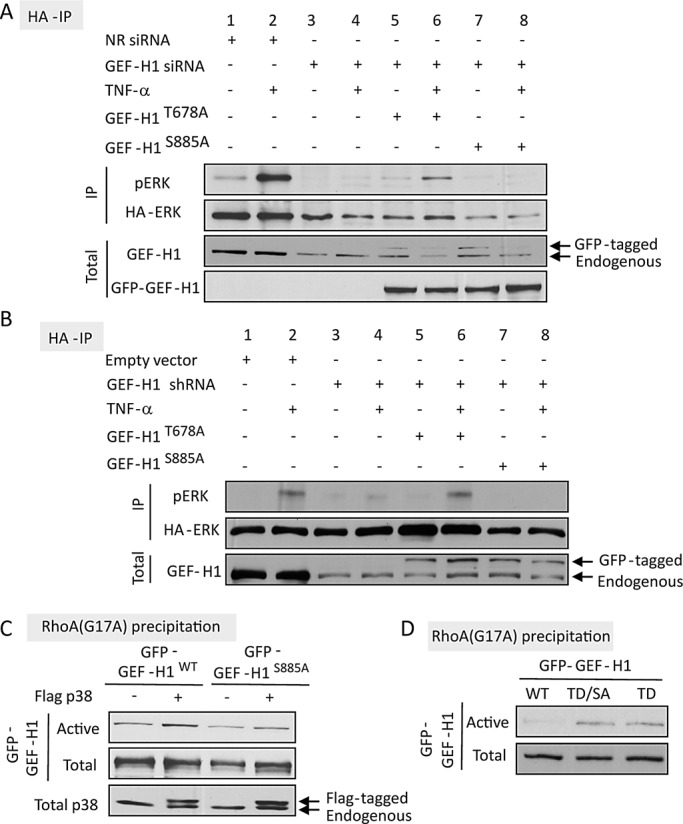
(A, B) Differential role of T678 and S885 in TNF-α–induced ERK activation. (A) LLC-PK1 cells grown in 6-cm dishes were transfected with 100 nM NR or GEF-H1–specific siRNA and 24 h later with GFP-GEF-H1T678A or GFP- GEF-H1TS885A along with HA-ERK2. (B) Cells were transfected with GEF-H1 shRNA along with HA-ERK with or without GFP- GEF-H1T678A or GFP- GEF-H1TS885A. Details of the transfection are described in Materials and Methods. Cells were treated with 10 ng/ml TNF-α as indicated, and HA-ERK was immunoprecipitated and its phosphorylation detected using Western blotting as in Figure 2D. GEF-H1 and GFP were also detected in the cell lysates to assess down-regulation of endogenous GEF-H1 and expression of the GFP-tagged mutants. (C, D) Role of S885 in GEF-H1 activation toward RhoA. LLC-PK1 cells were transfected with GFP-GEF-H1WT, GFP-GEF-H1S885A, or GFP-GEF-H1T678D (labeled as TD) or GFP-GEF-H1T678D/S885A (labeled as TD/SA) as indicated. Activated GFP-GEF-H1 was precipitated using RhoA(G17A) and detected by Western blotting with anti-GFP, as described earlier. In C, p38 in the cell lysates was also detected. Note that the transfected FLAG-tagged p38 is visualized as an additional, higher band (see arrows). Throughout the figure representative blots of three independent experiments are shown.
S885 in GEF-H1 is required for TNF-α–induced GEF-H1 activation toward RhoA
Previous studies implicated the S885 site in regulation of GEF-H1–induced RhoA activation (Zenke et al., 2004; Callow et al., 2005; Birkenfeld et al., 2007; Meiri et al., 2009, 2012; Yamahashi et al., 2011). Our data described so far suggest that the S885 site could affect RhoA activation indirectly through regulation of ERK activation and subsequent T678 phosphorylation. However, it is conceivable that S885 phosphorylation is also a direct regulator of activity of GEF-H1 toward RhoA. To test this possibility, we first asked whether absence of the S885 phosphorylation site affects TNF-α–induced GEF-H1 activation toward RhoA. As shown in Figure 7C GFP-GEF-H1S885A showed no TNF-α–induced enhanced association with GST-RhoA(G17A). To further substantiate a potential direct effect of S885 on RhoA activation, we induced RhoA-specific GEF-H1 activation by overexpressing FLAG-p38. As described earlier, active p38 induces TACE-dependent ERK activation even when GEF-H1 is silenced (Figures 2D and 5F). As expected, GEF-H1wt was activated by coexpression of p38 (Figure 8C). In contrast, activation of GFP-GEF-H1S885A by p38 was much reduced (Figure 8C). This finding suggests that S885 phosphorylation might directly regulate activation of GEF-H1 toward RhoA. An alternative possibility, however, is that effective phosphorylation of T678 (and thus activation toward RhoA) requires S885 phosphorylation even in the presence of active ERK. To test this possibility we generated a GEF-H1 molecule with a phosphomimetic mutation at T678 (GEF-H1T678D). As expected, this mutant showed enhanced precipitation with RhoA(G17A) compared with WT (Figure 8D), verifying that phosphorylation of this site mediates activation of GEF-H1 toward RhoA. Elimination of the S885 site by introducing an S885A mutation did not seem to alter this enhanced activity, as indicated by comparable precipitation of the T678D single and the T678D/S885A double mutants by RhoA(G17A). Taken together, our data suggest that S885 phosphorylation regulates TNF-α–induced GEF-H1 activation toward RhoA possibly through both direct and indirect effect(s) (see Discussion).
TNF-α enhances epithelial wound healing through TACE, ERK, and GEF-H1
We earlier showed that TNF-α accelerates migration of tubular epithelial cells in a wound-healing assay (Szaszi et al., 2012). Of importance, Rho-family small GTPases play key roles in cell migration, and GEF-H1 was also implicated in this process (Nalbant et al., 2009; Tsapara et al., 2010; Spiering and Hodgson, 2011; Tonami et al., 2011; Cheng et al., 2012). Therefore we next asked whether TNF-α–induced activation of GEF-H1 plays a role in enhanced epithelial cell migration in TNF-α–treated cells. We used an electric cell-substrate impedance sensing (ECIS)–based wound healing assay that allows real-time quantification of cell migration–dependent repopulation of an injured area within the epithelial layer (Keese et al., 2004; Szaszi et al., 2012). In ECIS, cells grown on gold electrodes are continuously exposed to small, biologically inert AC currents to determine the impedance and the capacitance (C) of the system (Wegener et al., 2000). The value of C measured at 32 kHz reflects confluence of the cells on the electrode and can be used to follow wound healing. LLC-PK1 cells were plated on the electrodes and monitored using ECIS. The value of C measured at 32 kHz drops as the cells grow to confluence (Figure 9, A, C, and D) and reaches a minimum around 1 nF when confluence is reached. After confluence was reached, a wound was generated in the layer by exposing the cells to an elevated current pulse. Cell death and the lifting up of the cells from the surface is indicated by the immediate increase in C, which is followed by a gradual decrease of C as cells migrate into the wounded area to regenerate the intact monolayer (Figure 9). Of importance, TNF-α added at the time of wounding enhanced the regeneration of the monolayer, as indicated by ∼25% reduction in the half–recovery time (see quantification in Figure 9, B and E). To test whether GEF-H1 is required for the TNF-α–induced stimulation of wound healing, we transfected cells with NR or GEF-H1-specific siRNA. Silencing of GEF-H1 reduced basal (unstimulated) wound healing, in accordance with the previously reported role of GEF-H1 in cell migration (Figure 9, A and B). Of importance, TNF-α failed to enhance wound healing when GEF-H1 was silenced. Similar to GEF-H1 silencing, inhibition of TACE using TAPI-1 (Figure 9, C and E) and ERK using PD98059 (Figure 9, D and E) slowed wound healing and prevented stimulation by TNF-α. Taken together, these data suggest that GEF-H1 is key in mediating TNF-α–induced enhanced wound healing, likely through both TACE regulation and RhoA activation.
FIGURE 9:

TNF-α enhances epithelial migration through GEF-H1, TACE, and ERK. (A, B) GEF-H1 mediates TNF-α–induced enhanced migration. LLC-PK1 cells were transfected with NR or GEF-H1-specific siRNA and 24 h later plated into wells of an ECIS 8W1E array and grown with continuous measurement of C at 32 kHz until confluence was reached (indicated by C reaching its minimum value). Next a wound was generated by applying an elevated voltage pulse. Where indicated, before wounding the cells were treated with 20 ng/ml TNF-α. Recovery of the layer was monitored by measuring C at 32 kHz. Typical recovery curves are shown. The graph in B shows the half–recovery times for each condition, calculated as described in Materials and Methods. Values for the controls were taken as unity, and the other conditions were expressed as fold changes. Mean ± SEM of n = 4 independent experiments performed in duplicate. Note that although the trend was detectable in all experiments, the combined data do not reach statistical significance. (C–E) TACE and ERK are required for TNF-α–induced enhanced migration. Wound-healing assays using LLC-PK1 cells were performed as described. Where indicated, before wounding the cells were treated with 20 ng/ml TNF-α with or without 10 μM TAPI-1 or PD98059. In E, half–recovery times are shown (mean ± SEM of n = 4 independent experiments, performed in duplicate).
DISCUSSION
TNF-α–induced EGFR transactivation in the tubular epithelium mediates ERK and RhoA activation, required for cellular responses, including junction remodeling and proliferation (Kakiashvili et al., 2011). The aim of this work was to explore mechanisms of TNF-α–induced EGFR transactivation. Our major findings are the following: 1) GEF-H1 and Rac are central regulators of TACE and are essential for TNF-α–induced, p38-mediated activation of the TACE/EGFR/ERK pathway. 2) GEF-H1 mediates both TNF-α–induced Rac and RhoA activation but through different mechanisms. EGFR- and ERK-dependent phosphorylation of T678 is necessary only for GEF-H1 activation toward RhoA, whereas phosphorylation at the S885 site is necessary for activation toward both Rac and RhoA. Of interest, Rac and RhoA are activated in a hierarchical manner because GEF-H1–stimulated Rac activation is a prerequisite for ERK-mediated GEF-H1 phosphorylation, which in turn is necessary for RhoA activation. Figure 10 summarizes the proposed mechanism of TNF-α–induced signaling toward Rac and RhoA. 3) TNF-α enhances epithelial migration in a wound-healing assay through GEF-H1, TACE, and ERK.
FIGURE 10:
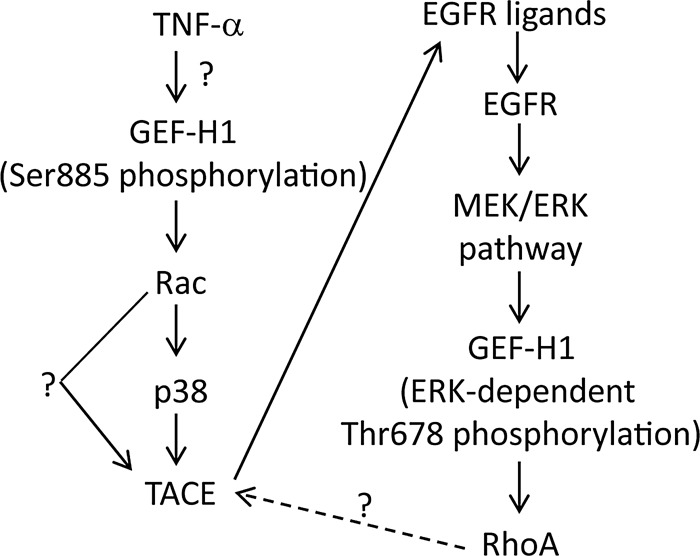
The proposed mechanism of TNF-α–induced TACE and subsequent EGFR/ERK/GEF-H1/RhoA activation.
Many stimuli were shown to transactivate the EGFR through ADAM family enzymes that release EGFR ligands, including HB-EGF, transforming growth factor-α, and amphiregulin (Liebmann, 2011). Here we show that TNF-α activates ADAM17/TACE in tubular cells. Because the substrates of TACE include pro-TNF-α and the TNF receptors (Black et al., 1997; Moss et al., 1997; reviewed in Wajant et al., 2003), TNF-α–induced activation of this enzyme could represent a significant feedback step. TACE is believed to be regulated by translocation to the membrane, where it cleaves substrates (Schlondorff et al., 2000; Soond et al., 2005), and through phosphorylation by ERK, PDK1, Src and p38 (Diaz-Rodriguez et al., 2002; Soond et al., 2005; Zhang et al., 2006; Xu and Derynck, 2010; Scott et al., 2011). Here we show that GEF-H1 and Rac regulate TACE through p38. Rac is also a major regulator of NADPH oxidase (Miyano and Sumimoto, 2007) and could potentially affect TACE through TNF-α–induced reactive oxygen species generation; however, this remains to be tested. Of interest, we found that RhoA silencing also reduced TACE activity. Although this could be partly due to reduced GEF-H1 expression caused by RhoA silencing, p38 activation under these conditions was only slightly decreased (unpublished data), suggesting that Rac and RhoA might regulate TACE through different mechanisms. RhoA might exert its effect through the cytoskeleton or by regulating translocation of the enzyme. In colonic epithelial cells TACE conveys TNF-α–induced survival signals (Hilliard et al., 2011). Of interest, TACE activation in these cells is MEK dependent but p38 independent (Liebmann, 2011). The exchange factor mediating TNF-α–induced Rac activation might also be cell type dependent: in fibroblasts, TNF-α–induced Rac and cdc42 activation were shown to be mediated by Vav (Kant et al., 2011).
GEF-H1 is activated by physical stimuli, including mechanical force and hyperosmolarity (Birukova et al., 2010; Waheed et al., 2010; Guilluy et al., 2011; Heck et al., 2012; Nie et al., 2012; Ly et al., 2013), and its overexpression leads to cell transformation (Mizuarai et al., 2006) and promotes migration (Nalbant et al., 2009; Liao et al., 2012). When the results are taken together, it is conceivable that GEF-H1 is a central signaling hub for EGFR transactivation induced by a variety of stimuli. Such a role of GEF-H1 warrants further exploration.
TNF-α traditionally was viewed as a proinjury cytokine, but a more complex picture is starting to emerge. In many epithelial cells TNF-α promotes survival and proliferation, possibly due to transactivation of ErbB family receptors (Argast et al., 2004; Yamaoka et al., 2008; Hilliard et al., 2011; Kakiashvili et al., 2011). Here we show that TNF-α enhances epithelial migration in a wound-healing assay. Of importance, this effect requires GEF-H1, as well as TACE and ERK. Thus, GEF-H1 might affect cell migration both as a regulator of TACE and EGFR transactivation and through ERK-dependent RhoA activation. In line with a central role of the EGFR, its deletion in the proximal tubules was shown to delay recovery from acute kidney injury (Chen et al., 2012a). Of interest, however, EGFR overactivation can also contribute to nephropathies. Angiotensin II, a well-established fibrogenic factor, exerts some of its effects through TACE and EGFR (Chen et al., 2006; Shah and Catt, 2006), and sustained EGFR activation enhanced expression of transforming growth factor-β1 (TGFβ1), a major inducer of epithelial–mesenchymal transition (EMT) and fibrosis (Chen et al., 2012b). Of importance, RhoA and GEF-H1 were also shown to regulate the expression of smooth muscle actin, a hallmark of EMT (Masszi et al., 2003; Fan et al., 2007; Tsapara et al., 2010; Ly et al., 2013).
An important finding of this study is that GEF-H1 mediates both TNF-α–induced RhoA and Rac activation. Although most of the recent studies focus on RhoA activation by GEF-H1, earlier it was also shown to exert Rac-GEF activity (Ren et al., 1998). Further, Tonami et al. (2011) recently showed that knockdown of calpain-6 resulted in GEF-H1–dependent Rac activation. Our study provides the first example of a signaling pathway in which GEF-H1 can act as an activator of both Rac and RhoA, depending on its phosphorylation state. EGFR- and ERK-dependent phosphorylation of T678, required for GEF-H1–mediated RhoA activation, is not needed for Rac activation. In contrast, surprisingly, Rac is upstream from the T678 phosphorylation. S885 phosphorylation is a prerequisite for both TNF-α–induced Rac and RhoA activation. Mass spectrometry analysis, as well as Western blotting with a phospho-S885–specific antibody, revealed that S885 is phosphorylated both in unstimulated and TNF-α–stimulated cells, with a trend for enhanced S885 phosphorylation in TNF-α–treated cells. Of importance, a nonphosphorylatable mutant of S885 no longer showed enhanced association with the nucleotide-free Rac(G15A) upon TNF-α stimulation. This mutant was also not activated toward RhoA by TNF-α and showed reduced activation upon p38-induced stimulation of the ERK pathway, which is independent of Rac. These data imply that the S885 site might have a direct role in GEF-H1 activation toward RhoA. Of interest, we found that introducing an S885A mutation into an active GEF-H1 containing a phosphomimetic mutation at T678 (GEF-H1T678D/S885A) did not reduce its activity toward RhoA. Thus, it is very likely that the natural phosphorylation of T678 depends upon the S885 site not only because of the demonstrated indirect effect (through ERK), but also through an additional (direct) effect. Our future work will address this.
When the results are taken together, the S885 site seems to play a central role in GEF-H1 activation. Indeed, this site was shown to be phosphorylated by many kinases, including PAK1, PAK4, Aurora A, Par1b/MARK2, and PKA (Zenke et al., 2004; Callow et al., 2005; Birkenfeld et al., 2007; Meiri et al., 2009; Yamahashi et al., 2011). It was also suggested to regulate binding to microtubules (Zenke et al., 2004; Callow et al., 2005). In line with our present findings, Callow et al. (2005) showed that expression of a phosphorylation-incompetent mutant of the S810 site in the short splice variant GEF-H1M (analogous to S885 in the full protein) reduced the abundance of stress fibers in fibroblasts. Further, a phosphomimetic S885D mutant showed enhanced RhoA activation (Birkenfeld et al., 2007). However, S885 phosphorylation is likely not the only switch turning on the protein. S959 (Birkenfeld et al., 2007), S143, and S3 (Callow et al., 2005; Yoshimura and Miki, 2011) were also implicated in GEF-H1 regulation. Of interest, single S885 mutants seem to show opposite effects to those of double mutants of S885 and S959, suggesting a collaboration between these sites (Birkenfeld et al., 2007; Yamahashi et al., 2011). Overall it is likely that differential single or double phosphorylation/dephosphorylation of these sites and T678 can fine-tune the RhoA and Rac exchange activities of GEF-H1. The potential role of other serine sites and kinases targeting them in TNF-α–induced GEF-H1 activation remains to be established.
Finally, our study demonstrates a hierarchical relationship between TNF-α–induced Rac and RhoA activation in tubular cells. This concept is in line with the pioneering studies of Alan Hall and his group showing that active Rac in fibroblasts stimulates RhoA (Ridley et al., 1992; Nobes and Hall, 1995). Our findings provide a possible mechanism for a hierarchy between Rac and RhoA: GEF-H1-dependent Rac activation regulates the RhoA exchange activity of GEF-H1 by controlling its ERK-mediated T678 phosphorylation (Figure 10). In many cells Rac and RhoA activities were reported as mutually antagonistic or spatially restricted (e.g., Rac in the front, RhoA in the back of migrating cells). However, a more complex picture is emerging, suggesting that activation of the two GTPases can spatiotemporally coexist, for example, at the lamellipodium (Kurokawa et al., 2005; Pertz et al., 2006, 2011). Such context-dependent fine tuning requires tight pathway-specific control of regulators. This work provides a prominent example for a mechanism that can achieve differential regulation, coordination, and coupling of activities of a single GEF toward Rac and Rho, likely contributing to complex functions such as epithelial sheet migration. Understanding such mechanisms could help in the development of strategies to selectively affect Rac- or RhoA-specific activation of GEFs.
MATERIALS AND METHODS
Chemicals and antibodies
PD98059, SB203580, AG1478, TAPI-1, and calyculin A were from EMD Biosciences (Mississauga, Canada). TNF-α was from Sigma-Aldrich (St. Louis, MO). Bovine serum albumin (BSA) was from BioShop Canada (Burlington, Canada). The Complete Mini Protease inhibitor and PhosSTOP Phosphatase Inhibitor tablets were from Roche Diagnostics (Laval, Canada).
Antibodies against the following proteins were used: RhoA, GEF-H1 (55B6), Rac1/2/3, p38, phospho-p38 (Thr-180/Tyr-182), GFP, EGFR, phospho-EGFR (Y845), and IκBα from Cell Signaling Technology (Danvers, MA); TACE and phospho–S885-GEF-H1 (ab94348) from Abcam (Cambridge, MA); phospho-p44/42 mitogen-activated protein kinase (MAPK; ERK1/2; Thr-202/Tyr-204), p44/42 MAPK (ERK1/2), and p65 NFκB from Santa Cruz Biotechnology (Santa Cruz, CA); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from EMD Biosciences; HA tag from Covance (Princeton, NJ); and tubulin from Sigma-Aldrich. Peroxidase- and Cy3-labeled secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). HA-tag antibody coupled to agarose beads was from Santa Cruz Biotechnology. 4′,6-diamidino-2-phenylindole (DAPI) nucleic acid stain was from Invitrogen (Burlington, Canada).
Cells and cell treatment
LLC-PK1, a kidney proximal tubule epithelial cell line (clones 101 and 4; Kakiashvili et al., 2009) and NRK-52-E cell lines (American Type Culture Collection, Manassas, VA) were used. Cells were maintained in low-glucose (for LLC-PK1) or high-glucose (NRK) DMEM supplemented with 10% fetal bovine serum and 1% antibiotic suspension (penicillin and streptomycin; Invitrogen) in an atmosphere containing 5% CO2. Confluent cells were serum depleted for at least 3 h in DMEM before the experiments.
The Na+ medium used for the experiments exploring the effects of depolarization contained 130 mM NaCl, 3 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM glucose, and 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4), and the K+ medium contained 130 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM glucose, and 20 mM HEPES (pH 7.4).
Vectors
The vectors used were kind gifts from the following investigators: cDNAs encoding for the GST-RBD portion of Rhotekin, the GST-PBD portion of Pak, GST-RhoA(G17A), and GST-Rac(G15A) (Garcia-Mata et al., 2006) from K. Burridge (University of North Carolina, Chapel Hill, NC); active pCMV-FLAG p38-α (Flag-p38) from R. J. Davis (University of Massachusetts, Worcester, MA; Raingeaud et al., 1995); myc-RacT17N, a dominant-negative Rac (DN-Rac), and pCMV5-HA3-WT-GEF-H1 (HA-GEF-H1) were from G. Bokoch (Scripps Institute, La Jolla, CA; Zhang et al., 1995; Zenke et al., 2004); and HA-ERK2 and GFP-tagged wild-type and T678A mutant GEF-H1 were from M. Kohno (Nagasaki University, Nagasaki, Japan; Fujishiro et al., 2008). The GEF-H1 point mutants GEF-H1S885A and GEF-H1T678D were generated from the WT-GFP-GEF-H1 construct using PCR-based mutagenesis with the following primers: for GEF-H1S885A, 5′-GTGGATCCTCGGCGGCGCGCCCTCCCCGCAGGCGATG-3′ and 5′-CATCGCCTGCGGGGAGGGCGCGCCGCCGAGGATCCAC-3′; and for GEF-H1T678D, 5′-AACTGCTCTTGGATCCCCGAGAGCCAGCC-3′ and 5′-GGCTGGCTCTCGGGGATCCAAGAGCAGTTC-3′. The GEF-H1S885A/T678D double mutant was prepared by introducing the S-to-A mutation into GEF-H1T678D.
Gene silencing using siRNA
The following porcine sequences were targeted by the siRNAs.
GEF-H1: #1, AACAAGAGCATCACAGCCAAG (Kakiashvili et al., 2009; Waheed et al., 2010), and #2, AACGGGCATCTCTTCACCACC (porcine specific).
Rac 1/2: #1, AAATACCTGGAGTGCTCGGCG, and #2, UCGAGAAACUGAAGGAGAA.
TACE: #1, GGUGAAAGGCACUACAAUAUU, and #2, UAUUGUAGUGCCUUUCACCUU.
RhoA, AAAGCAGGTAGAGTTGGCTTT (Ly et al., 2013).
The siRNAs were obtained from Applied Biosystems/Ambion (Austin, TX) or ThermoScientific/Dharmacon (Lafayette, CO). All experiments using Rac, GEF-H1, and TACE silencing in LLC-PK1 cells were performed with two different siRNAs, and the data obtained were pooled. TACE in NRK cells was silenced using a predesigned and validated ON-TARGETplus siRNA from ThermoScientific/Dharmacon. Cells were transfected with 100 nM siRNA oligonucleotide using the Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer's instructions. Control cells were transfected with 100 nM Silencer siRNA negative control # 2 (NR siRNA; Applied Biosystems/Ambion).
For the porcine-specific shRNA plasmid, two complementary oligonucleotides were generated: the porcine GEF-H1–specific sequence GCTATACCAACGGGCATCT and the hairpin loop sequence TTCAAGAGA and restriction site overhangs to allow directional cloning into the BamH1 and Xho1 sites of the pRNAT-CMV3.2 expression vector (GenScript, Piscataway, NJ). The two strands were annealed and ligated to the cut and purified vector. Positive clones were purified and sequenced. Empty pRNAT vector was used for control.
Transient transfection
LLC-PK1 cells were transiently transfected with DNA vectors using FuGENE 6 (Roche Molecular Biochemicals, Indianapolis, IN; or Promega, Madison, WI) or jetPRIME transfection reagent (Polyplus-Transfection, New York, NY), according to the manufacturers’ instructions. Unless otherwise indicated, experiments were performed 48 h after transfection. The levels of the silenced proteins were routinely checked by Western blotting.
The following DNA concentrations were used for transfecting 10-cm dishes using FuGENE 6: 2 μg of HA-ERK with or without 5 μg of DN-Rac or active p38; or 6 μg of WT or mutant GFP-GEF-H1; or 5 μg of HA-GEF-H1. For expression of DNA vectors along with silencing of endogenous GEF-H1, two different protocols were used, which allowed efficient silencing and protein expression without significant cell toxicity. In a sequential transfection protocol, LLC-PK1 cells were transfected with the porcine-specific GEF-H1 siRNA #2 using Lipofectamine RNAiMAX, as described. Twenty-four hours later, cells were transfected with human GFP-GEF-H1S885A or GFP-GEF-H1T678A along with HA-ERK2 using FuGENE 6, as described, and experiments were performed 1 d later. In some experiments a cotransfection protocol was followed. The jetPRIME transfection reagent was used to cotransfect siRNA and DNA vectors, as well as the shRNA vector and other DNA-based vectors. The following shRNA and DNA concentrations were used for 6-cm dishes: 3 μg of empty pRNAT vector or GEF-H1–specific shRNA along with 1.0 μg of HA-ERK-2.
MMP activity assay
MMP activity was measured as in Ge et al. (2009). LLC-PK1 or NRK cells were grown to confluence, treated as indicated in the respective figure legends, and lysed using a buffer containing 150 mM NaCl, 50 mM Tris-HCl, 0.1% SDS, 1% sodium deoxycholate, 1% Nonidet P40, and 1% Triton X-100, pH 7.4, supplemented with Mini Protease Inhibitor Tablet (Roche Diagnostics). The lysates were clarified by centrifugation at 16,000 × g for 20 min, and the supernatants were collected. TACE activity was followed using the Fluorigenic Peptide Substrate III (Mca-Pro-Leu-Ala-Gln-Ala-Val-Dpa-Arg-Ser-Ser-Ser-Arg-NH2; R&D Systems, Minneapolis, MN). This peptide contains the TACE-specific cleavage site Ala–Val and becomes fluorescent after cleavage. Samples containing 10 μg of protein from each supernatant were incubated with the fluorigenic substrate for 3 h at room temperature in an assay buffer (50 mM Tris-HCl, pH 7.4, 25 mM NaCl, 4% glycerol, and protease inhibitors), and the fluorescence was measured using a SpectraMax M5e plate reader (Molecular Devices, Sunnyvale, CA) at 320-nm excitation and 405-nm emission wavelengths. Samples incubated for 3 h with the fluorigenic peptide without cell lysates were used to determine the background, which was subtracted from all values.
Preparation of GST-fusion proteins
Preparation of GST-RBD (amino acids 7–89 of Rhotekin) and GST-PDB (p21-binding domain of PAK1), GST-RhoA(G17A), and GST-Rac(G15A) has been described (Di Ciano-Oliveira et al., 2003; Waheed et al., 2012). Protein bound to the beads was estimated by SDS–PAGE, followed by Coomassie blue staining, and the beads were kept at 4°C for immediate use or stored frozen in the presence of glycerol.
Rac and Rho activity assays
Active (GTP-bound) Rac and RhoA were captured using GST-PBD or GST-RBD, respectively, as described (Sebe et al., 2008; Kakiashvili et al., 2009). Briefly, confluent LLC-PK1 cells grown on 6- or 10-cm dishes were treated as indicated in the respective figure legends. Cells were lysed with ice-cold buffer. The Rac assay buffer contained 25 mM HEPES (pH 7.5), 150 mM NaCl, 1% NP-40, 10% glycerol, 10 mM MgCl2, 1 mM EDTA, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), 25 mM NaF, and protease inhibitors. The RhoA assay buffer contained 100 mM NaCl, 50 mM Tris base (pH 7.6), 20 mM NaF, 10 mM MgCl2, 1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS, 1 mM Na3VO4, and protease inhibitors. After centrifugation, aliquots for determination of total Rac or RhoA were removed. The remaining supernatants were incubated at 4°C for 45 min with 20–25 μg of GST-RBD or GST-PBD beads, followed by extensive washing. Total cell lysates and the RBD- or PDB-captured proteins were analyzed by Western blotting using Rac1/2/3 or RhoA antibody. Results were quantified by densitometry.
Affinity precipitation of activated GEFs
Active GEFs were affinity precipitated from cell lysates using the Rac(G15A) or RhoA(G17A) mutant, which cannot bind nucleotide and therefore has high affinity for activated GEFs (Garcia-Mata et al., 2006), as in our earlier work (Kakiashvili et al., 2009; Waheed et al., 2010). This method is described in a video protocol (Waheed et al., 2012). GEF-H1 in the precipitates was detected by Western blotting. Precipitation with glutathione–Sepharose beads containing no fusion proteins resulted in no GEF-H1 precipitation (Kakiashvili et al., 2009). GEF-H1 in total cell lysates was also detected for each sample (total GEF-H1). Precipitated (active) and total GEF-H1 were quantified by densitometry.
Immunoprecipitation
To assess phosphorylation of HA-ERK2, we transfected LLC-PK1 cells in 10-cm dishes with HA-ERK2 with or without cotransfections, as described for the specific experiments. Forty-eight hours later the cells were serum depleted and treated as indicated in the corresponding figure legends. Cells were lysed with the lysis buffer used for preparing Western blotting samples, and HA-tagged ERK was precipitated using 20 μl of HA antibody coupled to agarose beads for 1 h at 4°C. The precipitates were washed and eluted in sample buffer, then subjected to Western blot analysis using anti–phospho-ERK and anti-HA. Control experiments in which lysates from nontransfected cells were used verified the specificity of the immunoprecipitation. For exploring the phosphorylation of HA-GEF-H1 the lysis buffer was also supplemented with 10 nM calyculin (Kakiashvili et al., 2009). HA-GEF-H1 transfection and precipitation was done as for HA-ERK. Phosphorylation of S885 of the precipitated protein was tested using anti–phospho-S885 GEF-H1.
Western blotting
After treatment, cells were lysed on ice with cold lysis buffer containing 100 mM NaCl, 30 mM HEPES (pH 7.5), 20 mM NaF, 1 mM ethylene glycol tetraacetic acid, and 1% Triton X-100, supplemented with 1 mM Na3VO4, 1 mM PMSF, and Mini Protease Inhibitor Tablet (Roche Diagnostic). For the detection of phosphoproteins the lysis buffer was also supplemented with PhosSTOP phosphatase inhibitor (Roche Diagnostic). Protein concentration was determined by the bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL) with BSA used as standard. SDS–PAGE and Western blotting was performed as in Kakiashvili et al. (2009). Blots were blocked in Tris-buffered saline containing 3% BSA and incubated with the primary antibody overnight. Antibody binding was visualized with the corresponding peroxidase-conjugated secondary antibodies and the enhanced chemiluminescence method (kit from GE Healthcare Lifesciences, Piscataway, NJ). Where indicated, blots were stripped and reprobed to demonstrate equal loading or detect levels of down-regulated proteins. Because the phospho-ERK (pERK) antibody proved difficult to strip, those blots were first developed using total ERK antibody, followed by reprobing with pERK. Data were quantified using densitometry.
Densitometry
Films with nonsaturated exposures were scanned and densitometry analysis performed using a GS-800 calibrated densitometer and the “band analysis” option of Quantity One software (Bio-Rad, Hercules, CA) as described previously (Waheed et al., 2010). In each assay the amount of the investigated protein species was normalized to the appropriate control (e.g., active RhoA to total RhoA, active GEF-H1 to total GEF-H1, pERK to total ERK protein, etc). Because the basal levels of many investigated proteins were often either undetectable or just slightly above the background, to achieve accurate and stringent comparison, signals were expressed relative to the response detected in stimulated cells, taken as 100%, as described in the figure legends.
ECIS-based wound-healing assay
Wound healing was quantified using the ECIS Ztheta system (Applied Biophysics, Troy, NY), as in Szaszi et al. (2012). LLC-PK1 cells were plated in wells of an 8W1E array (2 × 105 cells/well in 400 μl of culture medium). In experiments in which GEF-H1 was silenced, LLC-PK1 cells were transfected with the NR or GEF-H1–specific siRNA using Lipofectamine RNAiMAX, as described, and 24 h later the cells were trypsinized, counted, and plated on the electrode. In all experiments, after plating on the electrode, the cells were grown for 20–24 h to reach confluence, as indicated by the drop in C measured at 32 kHz. Next a wound was generated in the monolayer by applying a 3-mA, 32-kHz voltage pulse for 20 s, and recovery of the layer was monitored by measuring C at 32 kHz. To quantify and compare wound healing, the half–recovery time was calculated for each curve, as in Szaszi et al. (2012). Briefly, the difference in the C values at the last time point before wounding and the first time point after wounding was calculated and taken as the total wounding (100%). Next the recovery percentage was calculated at each time point from the highest C value (taken as 0% recovery) and plotted against the time from wounding (taken as 0 h). The 50% recovery time for each curve was determined, and expressed as fold from the control, taken as 1.
Immunofluorescence microscopy
Confluent cells grown on coverslips were treated as indicated in the corresponding figure legend and fixed with 4% paraformaldehyde. Immunofluorescence staining was carried out as in Kakiashvili et al. (2009). Briefly, after permeabilization with 0.1% Triton X-100, the coverslips were blocked with 3% BSA in phosphate-buffered saline. Next cells were incubated with anti-p65 NFκB (1:100). Bound antibody was detected using the corresponding fluorescent secondary antibody (1:1000), which also contained DAPI to counterstain nuclei. All samples were viewed using an Olympus IX81 microscope (Melville, NY) coupled to an Evolution QEi Monochrome camera (Media Cybernetics, Bethesda, MD).
Statistical analysis
All shown blots are representatives of at least three similar experiments. Data are presented as mean ± SE of the number of experiments indicated (n). Statistical significance was assessed by one-way analysis of variance with Newman–Keuls posttesting or Student's t test, as appropriate, using Prism (GraphPad Software, La Jolla, CA). For clarity on the figures only the most important significant differences are indicated: *p < 0.05, **p < 0.01, ***p < 0.001, ns, nonsignificant.
Supplementary Material
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research (MOP-97774 to K.S. and MOP-106625 to A.K.) and the Kidney Foundation of Canada and a Grant Miller Cancer Research grant from the University of Toronto. K.S. was a recipient of a KRESCENT New Investigator Award (a joint award of the Kidney Foundation of Canada, Canadian Nephrology Society, and Canadian Institute of Health Research) and an Early Researcher Award from the Ontario Ministry of Research and Innovation. F.W. was supported by a Li Ka Shing scholarship and a University of Toronto open scholarship. We thank the coordinators of the Li Ka Shing Knowledge Institute Core Facility for their help.
Abbreviations used:
- ADAM
a disintegrin and metalloproteinase
- BSA
bovine serum albumin
- ECIS
electric cell-substrate impedance sensing
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- EMT
epithelial–mesenchymal transition
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- NR
nonrelated
- RBD
Rho-binding domain
- TACE
TNF-α convertase enzyme
- TGF
transforming growth factor
- TNF-α
tumor necrosis factor-α
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-09-0661) on February 6, 2013.
*These authors contributed equally to this work.
REFERENCES
- Argast GM, Campbell JS, Brooling JT, Fausto N. Epidermal growth factor receptor transactivation mediates tumor necrosis factor-induced hepatocyte replication. J Biol Chem. 2004;279:34530–34536. doi: 10.1074/jbc.M405703200. [DOI] [PubMed] [Google Scholar]
- Benais-Pont G, Punn A, Flores-Maldonado C, Eckert J, Raposo G, Fleming TP, Cereijido M, Balda MS, Matter K. Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J Cell Biol. 2003;160:729–740. doi: 10.1083/jcb.200211047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12:699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld J, Nalbant P, Yoon SH, Bokoch GM. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis. Trends Cell Biol. 2008;18:210–219. doi: 10.1016/j.tcb.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Fu P, Xing J, Yakubov B, Cokic I, Birukov KG. Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2010;298:L837–L848. doi: 10.1152/ajplung.00263.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Callow MG, Zozulya S, Gishizky ML, Jallal B, Smeal T. PAK4 mediates morphological changes through the regulation of GEF-H1. J Cell Sci. 2005;118:1861–1872. doi: 10.1242/jcs.02313. [DOI] [PubMed] [Google Scholar]
- Chen J, Chen JK, Harris RC. Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int. 2012a;82:45–52. doi: 10.1038/ki.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC. EGFR signaling promotes TGFbeta-dependent renal fibrosis. J Am Soc Nephrol. 2012b;23:215–224. doi: 10.1681/ASN.2011070645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chen JK, Neilson EG, Harris RC. Role of EGF receptor activation in angiotensin II-induced renal epithelial cell hypertrophy. J Am Soc Nephrol. 2006;17:1615–1623. doi: 10.1681/ASN.2005111163. [DOI] [PubMed] [Google Scholar]
- Cheng IK, Tsang BC, Lai KP, Ching AK, Chan AW, To KF, Lai PB, Wong N. GEF-H1 over-expression in hepatocellular carcinoma promotes cell motility via activation of RhoA signaling. J Pathol. 2012;228:575–585. doi: 10.1002/path.4084. [DOI] [PubMed] [Google Scholar]
- Citi S, Spadaro D, Schneider Y, Stutz J, Pulimeno P. Regulation of small GTPases at epithelial cell-cell junctions. Mol Membr Biol. 2011;28:427–444. doi: 10.3109/09687688.2011.603101. [DOI] [PubMed] [Google Scholar]
- Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 2007;18:335–343. doi: 10.1016/j.cytogfr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Di Ciano-Oliveira C, Sirokmany G, Szaszi K, Arthur WT, Masszi A, Peterson M, Rotstein OD, Kapus A. Hyperosmotic stress activates Rho: differential involvement in Rho kinase-dependent MLC phosphorylation and NKCC activation. Am J Physiol Cell Physiol. 2003;285:C555–C566. doi: 10.1152/ajpcell.00086.2003. [DOI] [PubMed] [Google Scholar]
- Diaz-Rodriguez E, Montero JC, Esparis-Ogando A, Yuste L, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Mol Biol Cell. 2002;13:2031–2044. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens JR, Mahimkar RM, Black RA. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem Biophys Res Commun. 2003;308:331–338. doi: 10.1016/s0006-291x(03)01381-0. [DOI] [PubMed] [Google Scholar]
- Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258–289. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol Biol Cell. 2007;18:1083–1097. doi: 10.1091/mbc.E06-07-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujishiro SH, Tanimura S, Mure S, Kashimoto Y, Watanabe K, Kohno M. ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem Biophys Res Commun. 2008;368:162–167. doi: 10.1016/j.bbrc.2008.01.066. [DOI] [PubMed] [Google Scholar]
- Gao Y, Xing J, Streuli M, Leto TL, Zheng Y. Trp(56) of rac1 specifies interaction with a subset of guanine nucleotide exchange factors. J Biol Chem. 2001;276:47530–47541. doi: 10.1074/jbc.M108865200. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 2006;406:425–437. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- Ge L, et al. Sheddase activity of tumor necrosis factor-alpha converting enzyme is increased and prognostically valuable in head and neck cancer. Cancer Epidemiol Biomarkers Prev. 2009;18:2913–2922. doi: 10.1158/1055-9965.EPI-08-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45:146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooz M, Gooz P, Luttrell LM, Raymond JR. 5-HT2A receptor induces ERK phosphorylation and proliferation through ADAM-17 tumor necrosis factor-alpha-converting enzyme (TACE) activation and heparin-bound epidermal growth factor-like growth factor (HB-EGF) shedding in mesangial cells. J Biol Chem. 2006;281:21004–21012. doi: 10.1074/jbc.M512096200. [DOI] [PubMed] [Google Scholar]
- Guilluy C, Swaminathan V, Garcia-Mata R, O'Brien ET, Superfine R, Burridge K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol. 2011;13:722–727. doi: 10.1038/ncb2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck JN, Ponik SM, Garcia-Mendoza MG, Pehlke CA, Inman DR, Eliceiri KW, Keely PJ. Microtubules regulate GEF-H1 in response to extracellular matrix stiffness. Mol Biol Cell. 2012 doi: 10.1091/mbc.E11-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashiyama S, Iwabuki H, Morimoto C, Hieda M, Inoue H, Matsushita N. Membrane-anchored growth factors, the epidermal growth factor family: beyond receptor ligands. Cancer Sci. 2008;99:214–220. doi: 10.1111/j.1349-7006.2007.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard VC, Frey MR, Dempsey PJ, Peek RM, Jr, Polk DB. TNF-α converting enzyme-mediated ErbB4 transactivation by TNF promotes colonic epithelial cell survival. Am J Physiol Gastrointest Liver Physiol. 2011;301:G338–G346. doi: 10.1152/ajpgi.00057.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Kakiashvili E, Dan Q, Vandermeer M, Zhang Y, Waheed F, Pham M, Szaszi K. The epidermal growth factor receptor mediates tumor necrosis factor-alpha-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium. J Biol Chem. 2011;286:9268–9279. doi: 10.1074/jbc.M110.179903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakiashvili E, Speight P, Waheed F, Seth R, Lodyga M, Tanimura S, Kohno M, Rotstein OD, Kapus A, Szaszi K. GEF-H1 mediates tumor necrosis factor-α-induced Rho activation and myosin phosphorylation: role in the regulation of tubular paracellular permeability. J Biol Chem. 2009;284:11454–11466. doi: 10.1074/jbc.M805933200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MG, Guo Y, Huganir RL. AMPA receptor and GEF-H1/Lfc complex regulates dendritic spine development through RhoA signaling cascade. Proc Natl Acad Sci USA. 2009;106:3549–3554. doi: 10.1073/pnas.0812861106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant S, Swat W, Zhang S, Zhang ZY, Neel BG, Flavell RA, Davis RJ. TNF-stimulated MAP kinase activation mediated by a Rho family GTPase signaling pathway. Genes Dev. 2011;25:2069–2078. doi: 10.1101/gad.17224711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapus A, Szaszi K. Coupling between apical and paracellular transport processes. Biochem Cell Biol. 2006;84:870–880. doi: 10.1139/o06-202. [DOI] [PubMed] [Google Scholar]
- Keese CR, Wegener J, Walker SR, Giaever I. Electrical wound-healing assay for cells in vitro. Proc Natl Acad Sci USA. 2004;101:1554–1559. doi: 10.1073/pnas.0307588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killock DJ, Ivetic A. The cytoplasmic domains of TNFalpha-converting enzyme (TACE/ADAM17) and L-selectin are regulated differently by p38 MAPK and PKC to promote ectodomain shedding. Biochem J. 2010;428:293–304. doi: 10.1042/BJ20091611. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Nakamura T, Aoki K, Matsuda M. Mechanism and role of localized activation of Rho-family GTPases in growth factor-stimulated fibroblasts and neuronal cells. Biochem Soc Trans. 2005;33:631–634. doi: 10.1042/BST0330631. [DOI] [PubMed] [Google Scholar]
- Leonard M, Ryan MP, Watson AJ, Schramek H, Healy E. Role of MAP kinase pathways in mediating IL-6 production in human primary mesangial and proximal tubular cells. Kidney Int. 1999;56:1366–1377. doi: 10.1046/j.1523-1755.1999.00664.x. [DOI] [PubMed] [Google Scholar]
- Liao YC, Ruan JW, Lua I, Li MH, Chen WL, Wang JR, Kao RH, Chen JH. Overexpressed hPTTG1 promotes breast cancer cell invasion and metastasis by regulating GEF-H1/RhoA signalling. Oncogene. 2012;31:3086–3097. doi: 10.1038/onc.2011.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebmann C. EGF receptor activation by GPCRs: an universal pathway reveals different versions. Mol Cell Endocrinol. 2011;331:222–231. doi: 10.1016/j.mce.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Ly DL, Waheed F, Lodyga M, Speight P, Masszi A, Nakano H, Hersom M, Pedersen SF, Szaszi K, Kapus A. Hyperosmotic stress regulates the distribution and stability of myocardin-related transcription factor, a key modulator of the cytoskeleton. Am J Physiol Cell Physiol. 2013;304:C115–C127. doi: 10.1152/ajpcell.00290.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretzky T, Zhou W, Huang XY, Blobel CP. A transforming Src mutant increases the bioavailability of EGFR ligands via stimulation of the cell-surface metalloproteinase ADAM17. Oncogene. 2011;30:611–618. doi: 10.1038/onc.2010.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Martin N, Dan Q, Amoozadeh Y, Waheed F, McMorrow T, Ryan MP, Szaszi K. RhoA and Rho kinase mediate cyclosporine A and sirolimus-induced barrier tightening in renal proximal tubular cells. Int J Biochem Cell Biol. 2012;44:178–188. doi: 10.1016/j.biocel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Masszi A, Di Ciano C, Sirokmany G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–F924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- Meiri D, Greeve MA, Brunet A, Finan D, Wells CD, LaRose J, Rottapel R. Modulation of Rho guanine exchange factor Lfc activity by protein kinase A-mediated phosphorylation. Mol Cell Biol. 2009;29:5963–5973. doi: 10.1128/MCB.01268-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiri D, et al. Mechanistic insight into the microtubule and actin cytoskeleton coupling through dynein-dependent RhoGEF inhibition. Mol Cell. 2012;45:642–655. doi: 10.1016/j.molcel.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Miyano K, Sumimoto H. Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie. 2007;89:1133–1144. doi: 10.1016/j.biochi.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Mizuarai S, Yamanaka K, Kotani H. Mutant p53 induces the GEF-H1 oncogene, a guanine nucleotide exchange factor-H1 for RhoA, resulting in accelerated cell proliferation in tumor cells. Cancer Res. 2006;66:6319–6326. doi: 10.1158/0008-5472.CAN-05-4629. [DOI] [PubMed] [Google Scholar]
- Moss ML, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Nalbant P, Chang YC, Birkenfeld J, Chang ZF, Bokoch GM. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol Biol Cell. 2009;20:4070–4082. doi: 10.1091/mbc.E09-01-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M, Aijaz S, Leefa Chong San IV, Balda MS, Matter K. The Y-box factor ZONAB/DbpA associates with GEF-H1/Lfc and mediates Rho-stimulated transcription. EMBO Rep. 2009;10:1125–1131. doi: 10.1038/embor.2009.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M, Balda MS, Matter K. Stress- and Rho-activated ZO-1-associated nucleic acid binding protein binding to p21 mRNA mediates stabilization, translation, and cell survival. Proc Natl Acad Sci USA. 2012;109:10897–10902. doi: 10.1073/pnas.1118822109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Pascher A, Klupp J. Biologics in the treatment of transplant rejection and ischemia/reperfusion injury: new applications for TNFalpha inhibitors. BioDrugs. 2005;19:211–231. doi: 10.2165/00063030-200519040-00002. [DOI] [PubMed] [Google Scholar]
- Pertz O. Spatio-temporal Rho GTPase signaling—where are we now. J Cell Sci. 2011;123:1841–1850. doi: 10.1242/jcs.064345. [DOI] [PubMed] [Google Scholar]
- Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Ren Y, Li R, Zheng Y, Busch H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J Biol Chem. 1998;273:34954–34960. doi: 10.1074/jbc.273.52.34954. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Samarin S, Nusrat A. Regulation of epithelial apical junctional complex by Rho family GTPases. Front Biosci. 2009;14:1129–1142. doi: 10.2741/3298. [DOI] [PubMed] [Google Scholar]
- Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE) Biochem J. 2000;347:131–138. [PMC free article] [PubMed] [Google Scholar]
- Scott AJ, O'Dea KP, O'Callaghan D, Williams L, Dokpesi JO, Tatton L, Handy JM, Hogg PJ, Takata M. Reactive oxygen species and p38 mitogen-activated protein kinase mediate tumor necrosis factor alpha-converting enzyme (TACE/ADAM-17) activation in primary human monocytes. J Biol Chem. 2011;286:35466–35476. doi: 10.1074/jbc.M111.277434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebe A, Masszi A, Zulys M, Yeung T, Speight P, Rotstein OD, Nakano H, Mucsi I, Szaszi K, Kapus A. Rac, PAK and p38 regulate cell contact-dependent nuclear translocation of myocardin-related transcription factor. FEBS Lett. 2008;582:291–298. doi: 10.1016/j.febslet.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah BH, Catt KJ. TACE-dependent EGF receptor activation in angiotensin-II-induced kidney disease. Trends Pharmacol Sci. 2006;27:235–237. doi: 10.1016/j.tips.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Soond SM, Everson B, Riches DW, Murphy G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci. 2005;118:2371–2380. doi: 10.1242/jcs.02357. [DOI] [PubMed] [Google Scholar]
- Spiering D, Hodgson L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr. 2011;5:170–180. doi: 10.4161/cam.5.2.14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaszi K, Vandermeer M, Amoozadeh Y. Epithelial wound healing and the effects of cytokines investigated by ECIS. In: WG Jiang., editor. Electric Cell-Substrate Impedance Sensing and Cancer Metastasis. Dordrecht, Netherlands: Springer; 2012. pp. 131–175. [Google Scholar]
- Tonami K, Kurihara Y, Arima S, Nishiyama K, Uchijima Y, Asano T, Sorimachi H, Kurihara H. Calpain-6, a microtubule-stabilizing protein, regulates Rac1 activity and cell motility through interaction with GEF-H1. J Cell Sci. 2011;124:1214–1223. doi: 10.1242/jcs.072561. [DOI] [PubMed] [Google Scholar]
- Tsapara A, Luthert P, Greenwood J, Hill CS, Matter K, Balda MS. The RhoA activator GEF-H1/Lfc as a TGF-β target gene and effector that regulates α-smooth muscle actin expression and cell migration. Mol Biol Cell. 2010;21:860–870. doi: 10.1091/mbc.E09-07-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielhauer V, Mayadas TN. Functions of TNF and its receptors in renal disease: distinct roles in inflammatory tissue injury and immune regulation. Semin Nephrol. 2007;27:286–308. doi: 10.1016/j.semnephrol.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Waheed F, Speight P, Dan Q, Garcia-Mata R, Szaszi K. Affinity precipitation of active Rho-GEFs using a GST-tagged mutant Rho protein (GST-RhoA(G17A)) from epithelial cell lysates. J Vis Exp. 2012;61:e3932. doi: 10.3791/3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed F, Speight P, Kawai G, Dan Q, Kapus A, Szaszi K. Extracellular signal-regulated kinase and GEF-H1 mediate depolarization-induced Rho activation and paracellular permeability increase. Am J Physiol Cell Physiol. 2010;298:C1376–C1387. doi: 10.1152/ajpcell.00408.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- Wegener J, Keese CR, Giaever I. Electric cell-substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp Cell Res. 2000;259:158–166. doi: 10.1006/excr.2000.4919. [DOI] [PubMed] [Google Scholar]
- Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell. 2010;37:551–566. doi: 10.1016/j.molcel.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamahashi Y, Saito Y, Murata-Kamiya N, Hatakeyama M. Polarity-regulating kinase partitioning-defective 1b (PAR1b) phosphorylates guanine nucleotide exchange factor H1 (GEF-H1) to regulate RhoA-dependent actin cytoskeletal reorganization. J Biol Chem. 2011;286:44576–44584. doi: 10.1074/jbc.M111.267021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka T, Yan F, Cao H, Hobbs SS, Dise RS, Tong W, Polk DB. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci USA. 2008;105:11772–11777. doi: 10.1073/pnas.0801463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Miki H. Dynamic regulation of GEF-H1 localization at microtubules by Par1b/MARK2. Biochem Biophys Res Commun. 2011;408:322–328. doi: 10.1016/j.bbrc.2011.04.032. [DOI] [PubMed] [Google Scholar]
- Zenke FT, Krendel M, DerMardirossian C, King CC, Bohl BP, Bokoch GM. p21-activated kinase 1 phosphorylates and regulates 14–3-3 binding to GEF-H1, a microtubule-localized Rho exchange factor. J Biol Chem. 2004;279:18392–18400. doi: 10.1074/jbc.M400084200. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Thomas SM, Lui VW, Xi S, Siegfried JM, Fan H, Smithgall TE, Mills GB, Grandis JR. Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci USA. 2006;103:6901–6906. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, Bokoch GM. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.