

Up to 70% of yeast proteins are N-terminally acetylated, but in few cases is the function known. The NatB Nα-acetyltransferase is essential for ER-associated degradation of luminal proteins (ERAD-L). Der1, an ERAD-L cofactor of the Hrd1 ubiquitin ligase, is acetylated by NatB and is the only N-acetylation substrate crucial to ERAD-L.
Abstract
Two conserved ubiquitin ligases, Hrd1 and Doa10, mediate most endoplasmic reticulum–associated protein degradation (ERAD) in yeast. Degradation signals (degrons) recognized by these ubiquitin ligases remain poorly characterized. Doa10 recognizes the Deg1 degron from the MATα2 transcription factor. We previously found that deletion of the gene (NAT3) encoding the catalytic subunit of the NatB N-terminal acetyltransferase weakly stabilized a Deg1-fusion protein. By contrast, a recent analysis of several MATα2 derivatives suggested that N-terminal acetylation of these proteins by NatB was crucial for recognition by Doa10. We now analyze endogenous MATα2 degradation in cells lacking NatB and observe minimal perturbation relative to wild-type cells. However, NatB mutation strongly impairs degradation of ER-luminal Hrd1 substrates. This unexpected defect derives from a failure of Der1, a Hrd1 complex subunit, to be N-terminally acetylated in NatB mutant yeast. We retargeted Der1 to another acetyltransferase to show that it is the only ERAD factor requiring N-terminal acetylation. Preventing Der1 acetylation stimulates its proteolysis via the Hrd1 pathway, at least partially accounting for the ERAD defect observed in the absence of NatB. These results reveal an important role for N-terminal acetylation in controlling Hrd1 ligase activity toward a specific class of ERAD substrates.
INTRODUCTION
The eukaryotic ubiquitin-proteasome system (UPS) recognizes and degrades both naturally short-lived and aberrant proteins (Ravid and Hochstrasser, 2008; Schwartz and Ciechanover, 2009; Ulrich, 2012). Degradation of the latter, including those proteins that fail to fold or assemble properly, is part of cellular protein quality control. Most substrates of the UPS are first covalently modified with polymers of ubiquitin. Protein ubiquitylation requires the action of a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). The 26S proteasome recognizes polyubiquitylated proteins and cleaves them into short peptides with concomitant recycling of ubiquitin.
A prominent cellular site of protein quality control is the endoplasmic reticulum (ER), where most transmembrane (TM) and secreted proteins first insert into or cross the membrane and undergo additional folding, assembly, and posttranslational modification. Ubiquitin-dependent proteolysis initiated at this organelle is referred to as ER–associated degradation (ERAD; Carvalho et al., 2006; Hampton and Garza, 2009; Guerriero and Brodsky, 2012). In the baker's yeast Saccharomyces cerevisiae, the TM RING-domain proteins Doa10 and Hrd1 are the major E3s that target ER-associated proteins for proteolysis (Bays et al., 2001; Deak and Wolf, 2001; Swanson et al., 2001). Doa10 is believed to preferentially target proteins with degrons present on the cytoplasmic/nucleoplasmic side of the ER/nuclear membrane in a process termed ERAD-C (cytoplasm). By contrast, Hrd1 is believed to recognize predominantly proteins with ER luminal or intramembrane degrons—ERAD-L (lumen) and ERAD-M (membrane) substrates, respectively (Vashist and Ng, 2004; Sato et al., 2009). Both Doa10 and Hrd1 function with the E2 Ubc7 and the Ubc7 cofactor Cue1; Doa10 also requires a second E2, Ubc6 (Meusser et al., 2005).
The first identified Doa10 substrate was the MATα2 transcriptional repressor, the rapid degradation of which is essential for regulation of yeast mating-type switching (Swanson et al., 2001; Laney and Hochstrasser, 2003). The first ∼60 residues of MATα2 contain a degron, named Deg1, which is necessary and sufficient for Doa10-dependent degradation (Hochstrasser and Varshavsky, 1990; Deng and Hochstrasser, 2006; Chen et al., 1993; Johnson et al., 1998; Swanson et al., 2001; Ravid et al., 2006). Deg1 confers ubiquitin-dependent instability when fused to normally long-lived soluble or integral membrane proteins. Mutations of the hydrophobic face of a predicted amphipathic helix (Deg1 amino acids 14–32) strongly stabilize Deg1 fusion proteins; mutations both upstream and downstream of this region have partially stabilizing effects (Ho et al., 1994; Johnson et al., 1998; Xie et al., 2010).
We previously screened the S. cerevisiae gene deletion library for additional Doa10 pathway components (Ravid et al., 2006). We used a chimeric Deg1-Ura3 reporter construct, which confers growth to cells in the absence of uracil only when stabilized. Deletion of Doa10, Doa10 cofactors, and proteasome-related factors was found to support growth on medium lacking uracil. (A nup120Δ strain was also initially identified as a strong Deg1-Ura3 stabilizer, but the strain was found to have a cryptic mutation in DOA10.) Mutants allowing much slower growth on uracil-free medium were also identified; among these was a nat3Δ strain, which lacked the catalytic subunit of the heterodimeric NatB Nα-acetyltransferase (Nat3/Mdm20; Hollebeke et al., 2012). Direct biochemical analysis of the degradation of a Deg1–β-galactosidase fusion protein failed to confirm a proteolytic defect in nat3Δ cells. However, another Doa10 substrate, Ubc6, was weakly stabilized in the absence of Nat3 (Ravid et al., 2006). Deg1–β-galactosidase is expected to be N-terminally acetylated by NatB; Ubc6 is not.
Protein N-terminal acetylation is largely, but not entirely, determined by the residue following the initiator methionine (reviewed in Starheim et al., 2012; Hollebeke et al., 2012). S. cerevisiae cells express multiple Nα-acetyltransferases, with three—NatA (Ard1/Nat1), NatB (Nat3/Mdm20), and NatC (Mak3/Mak10/Mak31)—responsible for most protein Nα-acetylation. For NatA-mediated Nα-acetylation, the initiator Met residue must first be removed by a methionine aminopeptidase. NatB recognizes proteins with a second residue Glu, Asp, or Asn and occasionally Gln, and it Nα-acetylates the initiator Met residue. There are >900 predicted NatB substrates in yeast (Helbig et al., 2010).
A recent study suggested that loss of NatB impairs Deg1-dependent degradation to the same extent as deletion of DOA10 and that Doa10 binds directly to the acetylated N-terminus of MATα2-derived model substrates (Hwang et al., 2010). The MATα2 N-terminus is directly acetylated by NatB. These findings prompted us to reexamine the role of NatB in Doa10-mediated protein degradation. Whereas previous studies addressed the effect of NatB deficiency on sequence-modified derivatives of MATα2, we were particularly interested in the effects of NatB mutation on degradation of endogenous, full-length MATα2.
We report here that deletion of the NatB catalytic subunit results in little, if any, metabolic stabilization of endogenous MATα2. The same is true for MATα2 derivatives bearing just the Doa10-dependent Deg1 degron. NatB mutants display a mild induction of the ER unfolded-protein response (UPR), suggesting that mutation of NatB may broadly perturb ER protein homeostasis. In fact, we found that loss of NatB causes a striking defect in the degradation of ERAD-L (but not ERAD-M) substrates of Hrd1, including the classic model ERAD substrate CPY* (Hiller et al., 1996). The defect of nat3Δ cells in supporting CPY* degradation correlates with a failure of the Hrd1 holocomplex member, Der1, to be N-terminally acetylates. Der1 is specifically required for targeting ERAD-L substrates. We show that Der1 is acetylated in a NatB-dependent manner in vivo. Unacetylated wild-type (WT) Der1 is metabolically destabilized, and this enhanced degradation depends on Hrd1. Thus failure to acetylate the N-terminus of Der1 switches it from functioning as a Hrd1 cofactor to serving as a substrate. The ERAD-L defect of nat3Δ cells can be fully suppressed by overexpressing Der1. Moreover, by mutating the N-terminus of Der1 to make it a substrate of a different N-terminal acetyltransferase (NatA), we could render CPY* degradation NatA dependent. Loss of NatB does not cause a major change in either Der1 structure or topology. Taken together, our data indicate that N-terminal acetylation directly controls the function of Der1, a transmembrane protein critically involved in the degradation of ER luminal substrates of Hrd1.
RESULTS
NatB is not required for MATα2 degradation
Both our earlier genomic screen (Ravid et al., 2006) and the data of Hwang et al. (2010) suggested that the NatB Nα-acetyltransferase is important for maximal rates of Doa10-mediated degradation of substrates bearing the MATα2-derived Deg1 degron. MATα2 begins with a NatB consensus sequence (Met-Asn), and the majority of MATα2 is Nα-acetylated in vivo (Hwang et al., 2010). Binding experiments suggest a direct, albeit weak, interaction of Doa10 or Doa10 fragments with acetylated, but not unacetylated, MATα2-derived N-terminal peptides. However, previous random mutagenesis of Deg1 did not reveal any N-terminal mutations that significantly stabilized fusion proteins (Johnson et al., 1998), and attempts to validate the contribution of NatB to the degradation of such proteins suggested little if any role (Ravid et al., 2006).
To address this apparent discrepancy, we first analyzed degradation of Deg1-Ura3 in nat3Δ yeast. Deg1-Ura3 was the fusion protein used in our genomic screen that first suggested a potential role for NatB in Doa10 substrate degradation (Ravid et al., 2006). Deg1-Ura3–related fusions were also the principal substrates analyzed by Hwang et al. (2010). As shown in Figure 1A, Deg1-Flag-Ura3 was rapidly degraded in WT cells and strongly stabilized in doa10Δ cells. In contrast, deletion of NAT3 caused little stabilization of the substrate. The absence of a strong proteolytic defect in the absence of Nat3 is consistent with a failure of nat3Δ cells to stabilize the Deg1–β-galactosidase fusion protein or confer robust Deg1-Ura3–dependent growth on medium lacking uracil (Ravid et al., 2006).
FIGURE 1:
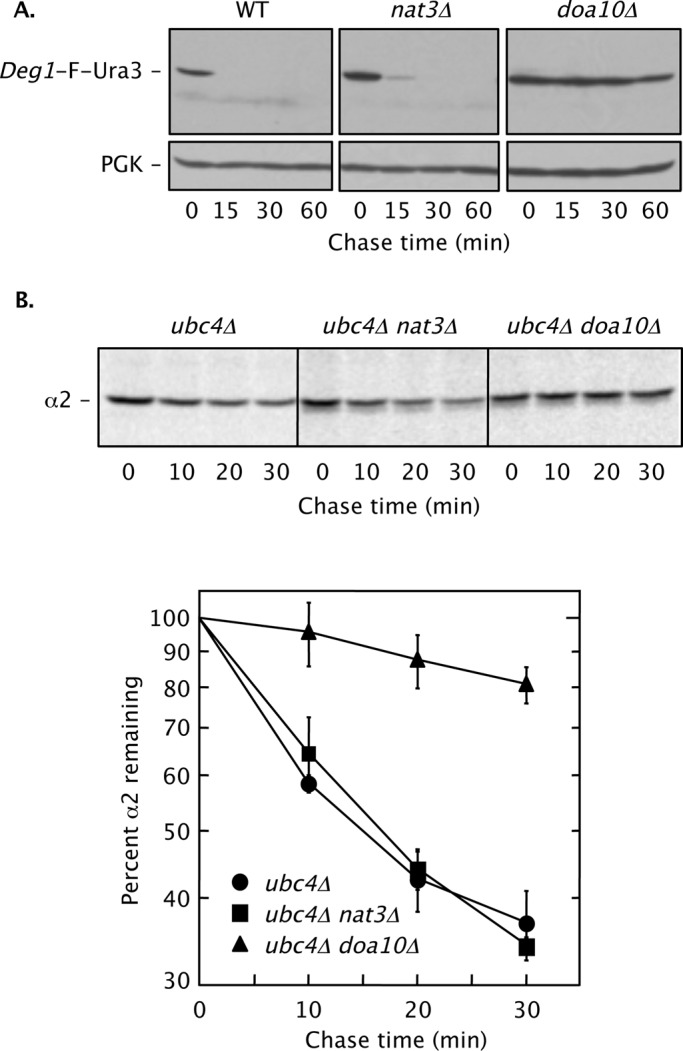
NatB is not essential for Doa10-dependent MATα2 degradation. (A) Cycloheximide chase/immunoblot analysis of Deg1-F-Ura3 degradation in WT (BY4741), nat3Δ (MHY7428), and doa10Δ (MHY3033) strains. Anti-FLAG (F) antibody was used for immunoblotting. Cell culture and incubation after cycloheximide addition were both conducted at 30°C. Deg1-F-Ura3 was expressed from the plasmid p415MET25-Deg1-FL-URA3. PGK, phosphoglycerate kinase (used as a loading control). (B) Pulse-chase analysis of native MATα2 degradation. Cell extracts were immunoprecipitated with anti-MATα2 antibody. Top, representative pulse-chase autoradiogram. Bottom, quantification of degradation kinetics. Each curve represents the average of four independent experiments. Error bars depict standard errors. Yeast strains used: ubc4Δ (MHY498), ubc4Δ nat3Δ (MHY6850), and ubc4Δ doa10Δ (MHY1648).
This and previous analyses were performed with ectopically expressed, modified versions of MATα2 or MATα2 fragments. We wished to know whether NatB-dependent Nα-acetylation is required for the degradation of endogenous, full-length MATα2. MATα2 is ubiquitylated by two principal mechanisms, one mediated by the Doa10 pathway, which includes Ubc6 and Ubc7, and the other by the Slx5/Slx8 pathway, which uses the Ubc4 E2 (Chen et al., 1993; Xie et al., 2010). To specifically measure Doa10-dependent MATα2 degradation, we analyzed degradation of chromosomally expressed, unmodified MATα2 in a ubc4Δ-strain background (Figure 1B). Degradation kinetics of MATα2 were experimentally indistinguishable in the presence or absence of Nat3. By contrast, MATα2 was strongly stabilized in a ubc4Δ doa10Δ strain.
We previously reported weak stabilization of Ubc6 bearing an internal hemagglutinin (HA) epitope tag in a nat3Δ strain (Ravid et al., 2006). Analysis of endogenous Ubc6 degradation confirmed a modest increase in half-life in the nat3Δ mutant, much milder than that seen in doa10Δ cells (Supplemental Figure S1A). Of note, Ubc6 begins with the Met-Ala dipeptide and is predicted to be a substrate of NatA (Ard1/Nat1) rather than NatB. Ubc6 is known to be Nα-acetylated after removal of the initiator Met by methionine aminopeptidase even in nat3Δ cells (Van Damme et al., 2012). Thus the inhibitory effect of the nat3Δ mutation on Ubc6 degradation is not likely due to a change in Ubc6 acetylation. Eliminating Ard1, the catalytic subunit of NatA, had no effect on Ubc6 degradation rate (unpublished data). We also measured the turnover of Ste6*, an integral ER membrane quality-control substrate of Doa10 (Huyer et al., 2004). Because its cytosolically disposed N-terminal sequence begins with Met-Asn, Ste6* is a candidate NatB target, although its acetylation status was not tested. Ste6* degradation was not affected by deletion of NAT3 (Supplemental Figure S1B).
We conclude that MATα2, an endogenous substrate of Doa10, is efficiently targeted to the Doa10 pathway in the absence of Nα-acetylation. Degradation of several additional Doa10 substrates predicted to be Nα-acetylated by NatB also appears to be largely if not entirely independent of NatB activity.
Weak induction of the UPR in a NatB mutant
The finding that Ubc6 degradation was weakly inhibited in nat3Δ cells despite not being a NatB substrate suggested that NatB might instead act on the Doa10 complex or other ERAD components. Doa10 itself is a predicted NatB substrate (N-terminus: Met-Asp), and NatB-dependent N-terminal acetylation of Doa10 was recently confirmed experimentally (Van Damme et al., 2012). With an N-terminal dipeptide Met-Glu, Cue1, the membrane-anchoring cofactor of the Ubc7 E2, is also a potential NatB substrate (Biederer et al., 1997). Reduced levels or activity due to N-terminal acetylation of either Doa10 or Cue1 might affect Ubc6 degradation, which is known to be uniquely sensitive to other mutations in Doa10 (Kreft and Hochstrasser, 2011). Doa10 levels appeared essentially unchanged when NAT3 was deleted (Supplemental Figure S2A). Cue1 levels actually increased in nat3Δ relative to WT cells (Figure 2A). Increased Cue1 abundance in nat3Δ cells correlated with a similar accumulation of Ubc7 (unpublished data), the levels of which depend on Cue1 (Ravid and Hochstrasser, 2007). Cue1 association with membranes was not detectably perturbed in the mutant (Supplemental Figure S2B). These data argue against a requirement for NatB-mediated acetylation in maintaining sufficient levels of these proteins. However, we cannot rule out subtle functional alterations to these factors when they are not Nα-acetylated, potentially accounting for the weak effect of the nat3Δ allele on Ubc6 degradation.
FIGURE 2:

NatB activity is important for degradation of CPY*, a Hrd1 ERAD substrate. (A) Loss of NatB causes increased levels of the Ubc7 cofactor Cue1 based on anti-Cue1 immunoblotting. PGK, loading control. (B) A mild UPR in cells lacking NatB. WT (MHY501), nat3Δ (MH6599), and doa10Δ hrd1Δ (MHY1703) strains were transformed with the pSZ1 UPRE-lacZ reporter plasmid (Travers et al., 2000), and at least three independent transformants were evaluated for β-galactosidase activity. Error bars represent SDs. (C) Cycloheximide-chase/anti-CPY immunoblot analysis of CPY* in prc1-1 (MHY1366), prc1-1 nat3Δ (MHY6920), and prc1-1 der1Δ (MHY7110) cells. PGK, loading control. Graph represents quantification of the cycloheximide-chase analyses at the top. CPY* levels were normalized to PGK at each time point. (D) KHN, another Hrd1-dependent luminal ER substrate, is also stabilized by loss of Nat3. Degradation was evaluated by cycloheximide-chase/anti-HA immunoblot analysis. (E) Turnover of 6myc-Hmg2, a membrane substrate of Hrd1, does not require NatB. Cycloheximide-chase was followed by anti-myc immunoblot analysis. Percentage of 6myc-Hmg2 remaining was normalized to PGK at each time point. Strains used were MHY7719 (WT), MHY7720 (nat3Δ), and MHY1661 (ubc7Δ).
Cue1 and Ubc7 accumulate during ER stress (Friedlander et al., 2000). The increase in their levels in the nat3Δ mutant might reflect accumulation of misfolded proteins in the ER and an induction of the ER UPR, although NatB was not previously implicated in the UPR (Jonikas et al., 2009). A more general protein stress response was suggested by a modest increase in the accumulation of bulk ubiquitin–protein conjugates in nat3Δ cells relative to WT cells (Supplemental Figure S2C). Using a UPRE-lacZ transcriptional reporter, we observed a ∼2.7-fold increase in β-galactosidase activity in nat3Δ cells (Figure 2B). The effect was not as great as that observed in a doa10Δ hrd1Δ double mutant, but it was comparable in magnitude to the induction previously seen in a hrd1Δ single mutant (Swanson et al., 2001).
An ERAD-L defect in nat3Δ cells
We therefore asked whether deleting NAT3 affected the Hrd1 pathway. Indeed, a striking stabilization of CPY*, an ERAD-L substrate, was observed in the absence of Nat3 (Figure 2C). The inhibition of CPY* degradation was comparable to that observed in der1Δ cells (or hrd1Δ cells; see Figure 7 later in this paper). The defect was suppressed by reintroduction of NAT3 on a plasmid (Figure 2C). Loss of Mdm20, the noncatalytic subunit of NatB, also strongly inhibited CPY* degradation (not shown). CPY* was similarly glycosylated in WT and nat3Δ cells, implying that its ER import and processing were not perturbed by mutation of NAT3 (Supplemental Figure S3A).
FIGURE 7:
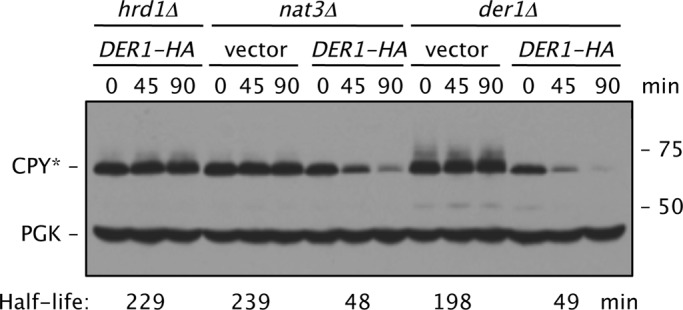
Increased Der1-HA levels suppress the CPY* degradation defect in nat3Δ cells. Cells all carried the prc1-1 allele and had the indicated additional chromosomal deletions; the strains were transformed with p415GPD-DER1-HA. The estimated CPY* half-lives were determined by quantitative immunoblotting using antibodies to HA and PGK and normalization to PGK levels. Strains used: prc1–1 hrd1Δ (MHY6855); prc1–1 nat3Δ (MHY6920); and prc1–1 der1Δ (MHY7110).
Additional Hrd1 substrates were also tested for Nat3 dependence of degradation. Degradation of another model ERAD-L substrate, KHN (Vashist and Ng, 2004), was impaired in the nat3Δ mutant (Figure 2D). By contrast, the ERAD-M substrate 6myc-Hmg2 (Hampton and Bhakta, 1997) was degraded efficiently in the absence of Nat3 (Figure 2E). These results suggest that NatB might be specifically required for the degradation of ERAD-L substrates of Hrd1.
Der1 function normally requires its acetylation by NatB
The Hrd1 holocomplex is a multisubunit membrane protein complex with components dedicated to the recognition or processing of specific substrate subclasses (Guerriero and Brodsky, 2012). Der1 is an integral membrane subunit specifically required for the degradation of ERAD-L substrates. The mechanistic role of Der1 is not known, although studies of the related mammalian Derlins have suggested that they participate in ERAD substrate retrotranslocation or release from the ER membrane after substrate traversal of the bilayer (Lilley and Ploegh, 2004; Ye et al., 2004; Greenblatt et al., 2011). Derlins are also related to the integral-membrane rhomboid proteases. The latter proteins induce a thinning and disruption of the membrane bilayer (Wang et al., 2007). Derlins might facilitate retrotranslocation in part by lowering the energy barrier for moving hydrophilic domains of luminal substrates across the membrane.
The N-terminus of Der1, which faces the cytosol (Hitt and Wolf, 2004), begins with the sequence Met-Asp. This matches the NatB consensus, although Der1 had not been identified as a NatB substrate in previous proteomic surveys (Helbig et al., 2010; Van Damme et al., 2012). To determine whether Der1 is indeed Nα-acetylated in vivo, we purified C-terminally FLAG-tagged Der1 protein from NAT3 and nat3Δ yeast cells (Figure 3A) and subjected them to liquid chromatography-tandem mass spectrometric (LC-MS/MS) analysis (Figure 3, B and C). Based on quantitative ion current measurements and MS/MS sequencing of N-terminal tryptic peptides, ∼93% of Der1 protein was Nα-acetylated; this modification was completely dependent on NatB (Figure 3, B and C; Table 1).
FIGURE 3:

Nα-acetylation of Der1 by NatB (Nat3/Mdm20). (A) Purified Der1-FLAG from hrd1Δ NAT3 (MHY3032) and hrd1Δ nat3Δ (MH7430) cells. Indicated bands from the Coomassie brilliant blue (CBB)–stained gel were excised, subjected to in-gel trypsin digestion, and evaluated by LC-MS/MS. (B) MS/MS sequencing of the N-terminal tryptic peptide of Der1 isolated from NAT3 (WT). Indicated in red are the y and b ions that matched the sequence of the fragmented N-terminal peptide. MS/MS spectra were searched using the Mascot algorithm. No Nα-acetylated peptide was identified by MS/MS for Der1-FLAG purified from the hrd1Δ nat3Δ (MHY 7430) strain (Table 1). (C) Normalized ion intensity peaks of the acetylated peptide are shown at the bottom using Progenesis LC-MS software (Nonlinear Dynamics, Durham, NC).
TABLE 1:
MS/MS analysis of Der1 N-terminal acetylation in NAT3 and nat3Δ cells.
| WT (MD) Der1 | |||||
|---|---|---|---|---|---|
| Peptide identified by mascot search | Charge state | m/z | MH+ | nat3ΔTIC max | WTTIC max |
| MDAVILNLLGDIPLVTR | 2 | 927.0326 | 1853.0652 | 6.18E+06 | 2.87E+05 |
| MDAVILNLLGDIPLVTR + Met oxidation | 2 | 935.0289 | 1869.0578 | 1.22E+06 | 2.38E+05 |
| MDAVILNLLGDIPLVTR + Met oxidation | 3 | 623.6899 | 1869.0697 | 6.76E+06 | 4.61E+05 |
| MDAVILNLLGDIPLVTR + acetylation | 2 | 948.0381 | 1895.0762 | Not seen | 1.11E+07 |
| MDAVILNLLGDIPLVTR + acetylation | 3 | 632.2623 | 1894.7869 | Not seen | 7.45E+05 |
| MDAVILNLLGDIPLVTR +acetylation + Met oxidation | 2 | 956.0353 | 1911.0706 | Not seen | 8.57E+05 |
| Total TIC (± Met oxidation and ± acetylation) | 1.42E+07 | 1.37E+07 | |||
| Total TIC for acetylated peptides (± Met oxidation) | 0.00E+00 | 1.27E+07 | |||
| Percentage of total peptides acetylated | 0 | 93 | |||
If the ERAD-L defects observed in NatB mutants were attributable solely to a failure to N-terminally acetylate Der1, it might be possible to switch ERAD-L dependence to a different Nα-acetyltransferase by altering the Der1 sequence to make it a substrate of that enzyme. Sequence comparisons of Der1 to its likely orthologues in a wide range of eukaryotes revealed that most are predicted to be Nα-acetylated. The second residue of many of these proteins is alanine, making them predicted substrates of NatA (rather than NatB).
We therefore mutated the second residue of Der1 from Asp to Ala (MA-Der1). The initiator Met should be cleaved from this construct, exposing an N-terminal Ala residue that is predicted to be Nα-acetylated by NatA. MA-Der1-FLAG was purified from WT cells and analyzed by LC-MS/MS. The N-terminal Met had indeed been removed, and ∼41% of the resultant Der1 protein with an N-terminal Ala was Nα-acetylated (Supplemental Figure S4 and Supplemental Table S1). The reduced levels of Nα-acetylation of the MA-Der1 compared with WT Der1 (MD-Der1) might be related to the recent observation that a cotranslationally ER-translocated NatA substrate was not acetylated, probably due to competition for binding between NatA and signal recognition particle (SRP; Forte et al., 2011). As will be shown later, Nα-acetylation of MA-Der1 is indeed NatA dependent.
Degradation of CPY* in the resulting MA-der1 strain was almost as rapid as in cells expressing the WT Der1 protein, indicating that MA-Der1 retains substantial function (Figure 4, A and B, and Supplemental Figure S3B). In a MA-der1 nat1 double mutant, which does not support Nα-acetylation of MA-Der1, ∼98% had the Met removed and no acetyl group (Supplemental Table S1; Arendt and Hochstrasser, 1997), CPY* degradation was strongly impaired. Of importance, CPY* degradation was unaffected in nat1 cells expressing wild-type Der1 (Figure 4A and Supplemental Figure S3B). Therefore disruption of NatA does not inhibit ERAD-L unless Der1 has been converted to a NatA substrate.
FIGURE 4:

N-terminal mutation of Der1 creates a novel Nα-acetyltransferase dependence. (A) WT Der1 (MD-Der1-HA) is sensitive to loss of NatB (Nat3/Mdm20) but not NatA (Ard1/Nat1). CPY* and Der1-HA degradation were measured by cycloheximide chase/immunoblotting in cells with the indicated genotypes (all strains were prc1-1 der1Δ and carried either the pRS314 plasmid [der1Δ] or p414DER1-HA plasmid). PGK, loading control. (B) Conversion of Der1 into a NatA substrate makes ERAD-L sensitive to loss of NatA. The MA-Der1 protein was functional in WT but not nat1 (or nat3Δ) cells. CPY* and MA-Der1-HA degradation was measured in cells with the indicated genotypes (all strains were prc1–1 der1Δ and had either the pRS314 plasmid [der1Δ nat1] or p414der1-D2A-HA plasmid). PGK, loading control. (C) Degradation of CPY* and endogenous Der1 measured by cycloheximide chase/immunoblot analysis in WT and nat3Δ cells. The same extracts were used for anti-CPY and anti-Der1 immunoblotting but were resolved on separate gels. pRS415DER1 is a low-copy (CEN) vector expressing DER1 from its own promoter. PGK, loading control.
Contrary to expectation, the MA-der1 allele failed to rescue ERAD-L in nat3Δ cells (Figure 4B). LC-MS/MS analysis of MA-Der1-FLAG purified from a nat3Δ mutant confirmed that the N-terminal Met had been cleaved (Supplemental Table S1). The percentage of Der1 acetylation determined from total ion current measurements in nat3Δ cells was only slightly decreased compared with WT (Table 1). However, when peptide amounts were normalized between the two strains, we observed an apparent ∼55% reduction in the abundance of acetylated Der1 (Supplemental Figure S4B). This partially reduced acetylation of MA-Der1 does not appear sufficient to account for the CPY* degradation defect in MA-der1 nat3Δ cells (see Discussion).
Taken together, the data argue for a specific requirement for Der1 Nα-acetylation for its function in ERAD-L.
Effects of mutating the N-terminus of Der1
To investigate how Nα-acetylation might affect Der1 function in ERAD-L, we analyzed the levels and stability of WT and N-terminally mutated Der1 proteins. WT Der1, either with or without a C-terminal HA epitope tag, exhibited a relatively long half-life (>2 h) in WT cells, although the HA tag caused a mild destabilization (Figures 4, A and C, and 5A). Of interest, WT Der1-HA was degraded approximately twice as fast in nat3Δ cells as in WT cells (Figure 5A).
FIGURE 5:
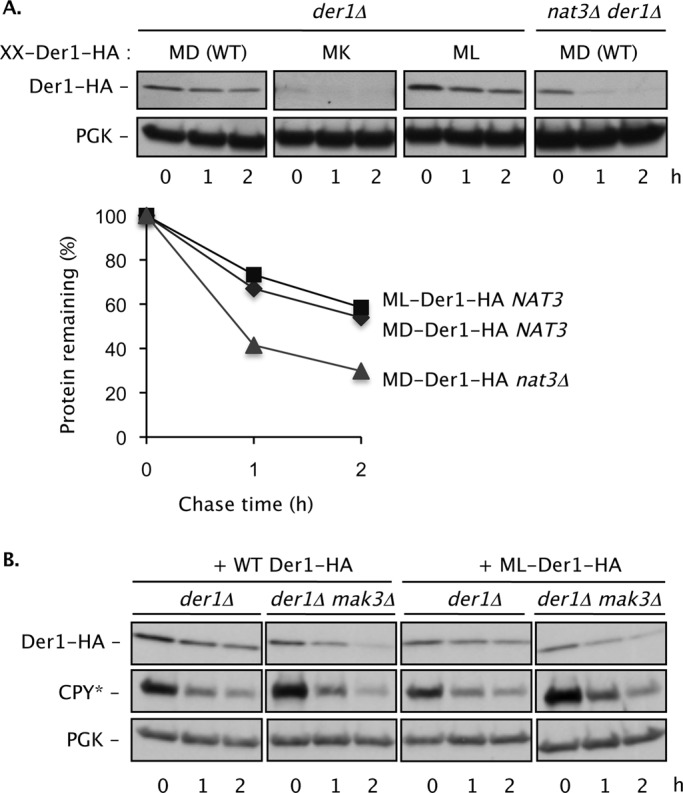
Der1 function and degradation rate is modulated by its N-terminal sequence. (A) A C-terminally HA-tagged WT Der1 that begins with the sequence Met-Asp (MD) is degraded slowly in NAT3 cells but is destabilized ∼2- to 2.5-fold in a nat3Δ strain. A mutant Der1 (MK) with a Lys at the second position is strongly destabilized (the anti-HA blot required prolonged exposure compared with the others), whereas the ML mutant is degraded at the same slow rate as WT Der1 (bottom). Der1 amounts for each time point in the plot were normalized to PGK levels. (B) The ML-Der1-HA protein is functional in ERAD-L even when not acetylated (mak3Δ cells). A low-copy plasmid expressing ML-Der1-HA from the DER1 promoter was transformed into cells of the indicated genotypes. PGK, loading control.
Mutation of the second residue of Der1 to residues predicted to alter its N-terminal processing also affected its steady-state levels, sometimes very strongly (Figures 4B and 5A). For example, when the second residue of Der1 was mutated from Asp to Lys (generating an N-terminus not predicted to be modified by any known yeast Nα-acetyltransferase), the resulting protein (MK-Der1-HA) could barely be detected in vivo due to rapid degradation (Figures 5A and 6). CPY* was strongly stabilized in these cells, consistent with depletion of Der1 (unpublished data). Conversely, when the N-terminus of Der1-HA was mutated in a manner not predicted to alter Nα-acetyltransferase specificity (ME-Der1), CPY* was rapidly degraded in a Nat3-dependent manner (Supplemental Figure S5, A and B). This implies that the ME-Der1 protein retained its function in ERAD-L but only when Nα-acetylated by NatB.
FIGURE 6:
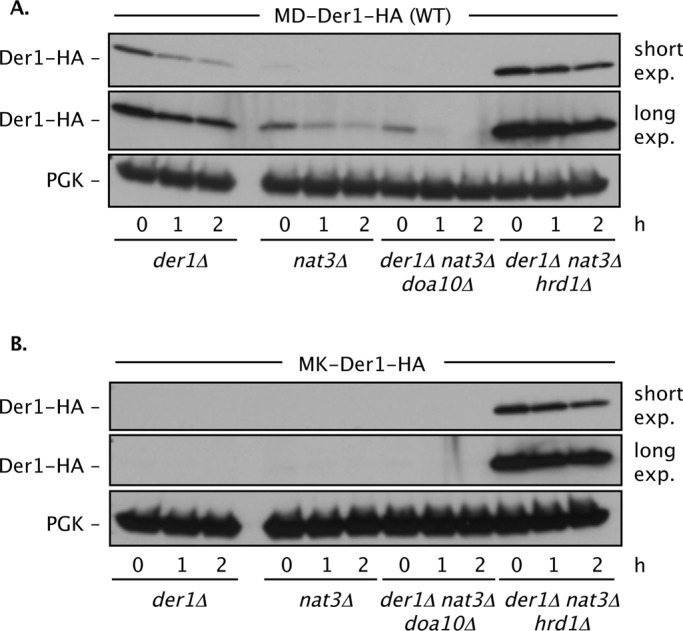
Nα-acetylation of Der1 regulates its degradation by the Hrd1 ligase. Degradation of WT MD-Der1 (A) and N-terminally mutated MK-Der1 (B) depends on Hrd1. Degradation of Der1-HA was followed by cycloheximide-chase assay and anti-HA immunoblotting. PGK, loading control. Strains used: prc1–1 der1Δ (MHY7110); prc1–1 der1Δ nat3Δ (MHY7111); der1Δ nat3Δ doa10Δ (MHY7834); and der1Δ nat3Δ hrd1Δ (MHY7839).
Finally, we also attempted to convert Der1 to a NatC (Mak3/Mak10/Mak31) substrate by mutating its second residue to Leu. The resulting ML-Der1 protein supported CPY* degradation in WT cells and rescued the CPY* degradation defect of nat3Δ cells (Figure 5B and Supplemental Figure S5C). This contrasts with the NatA substrate MA-Der1-HA, which was not functional in a nat3Δ mutant (Figure 4B). Surprisingly, ML-Der1 promoted CPY* degradation even in cells in which its predicted cognate Nα-acetyltransferase (NatC) was inactivated by MAK3 deletion (Supplemental Figure S5C). Therefore, when the N-terminus of Der1 is mutated to begin with the Met-Leu dipeptide, its function does not require Nα-acetylation.
Unacetylated Der1 becomes a substrate of Hrd1
Der1 was rapidly degraded in WT yeast when its second residue had been mutated to particular amino acids or when expressed in strains lacking specific Nα-acetyltransferase activities. Because the Der1 alterations affect its cytosolically disposed N-terminus, this enhanced turnover might be mediated by the Doa10 (ERAD-C) pathway. Alternatively, since Der1 is normally recruited to the Hrd1 complex, mutated Der1 might be ubiquitylated by Hrd1. We tested the degradation rates of WT and two N-terminally mutated forms of Der1 in doa10Δ and hrd1Δ mutants, as well as in mutants that combined these E3 gene deletions with deletion of NAT3 (Figure 6 and Supplemental Figure S6). In all cases, Der1 degradation was inhibited by loss of Hrd1 but not Doa10. Thus mutations that impair Der1 Nα-acetylation make it susceptible to ubiquitin-dependent degradation by the same ubiquitin ligase for which it normally serves as a cofactor.
The inference that under some circumstances Der1 can function when not acetylated was corroborated by the finding that the defect in CPY* degradation in nat3Δ cells was fully rescued by overexpression of WT Der1 even though it cannot be acetylated in this strain (Figure 7). The effect was specific, as Der1 overexpression did not suppress the hrd1Δ defect in ERAD-L (but did suppress the cognate der1Δ defect). We conclude that Der1 function is extremely sensitive to the structure of its N-terminus. In particular, at normal levels of expression, WT Der1 must be Nα-acetylated to function in ERAD-L.
Loss of acetylation does not cause gross changes in Der1 structure
How does the block to Nα-acetylation of Der1 impair its function and increase its Hrd1-mediated degradation? Acetylation could potentially affect Der1 conformation or folding, membrane insertion, or multimerization (Goder et al., 2008), its interaction with other Hrd1 complex components (Horn et al., 2009), or its association with substrates. The fact that unacetylated Der1 functions effectively in ERAD-L if overproduced suggests that unacetylated Der1 is not grossly misfolded and is still able to interact with the Hrd1 complex. Consistent with this, we find by coimmunoprecipitation analysis that Der1 continues to multimerize and associate with Usa1, its direct binding partner in the Hrd1 complex (Supplemental Figure S7).
We tested the possibility that Der1 Nα-acetylation affected its membrane topology, using Der1 derivatives with N-glycosylation consensus sequences inserted into the three predicted loops of the protein (after residues 45, 85, and 128; Hitt and Wolf, 2004). Residues at positions 45 and 128 had been found to be glycosylated and therefore inferred to be luminal in WT cells (Hitt and Wolf, 2004). By contrast, an N-glycosylation site at position 85 was not glycosylated and was thus inferred to be cytoplasmically disposed. We confirmed these findings in WT cells and observed the same results in nat3Δ cells, suggesting that at least the first three TM segments were inserted correctly in both strains (Supplemental Figure S8, A and B). Mammalian Derlin-1 was recently suggested to have a six-TM topology (Greenblatt et al., 2011). Our computational analysis suggests a similar topology for yeast Der1, among the most divergent of the Derlin family members (Supplemental Figure S8, B and C; Chen et al., 2010). It remains to be determined whether Der1 does indeed have six TM segments and whether N-terminal acetylation affects this topology.
We conclude that N-terminal acetylation of Der1, although essential for Der1 activity in ERAD-L, is unlikely to cause a major change in Der1 dimerization, affinity for the Hrd1 complex, or membrane topology.
DISCUSSION
Nα-acetylation is among the most common modifications of eukaryotic proteins. Some 50–70% of proteins in yeast and 70–90% of proteins in human cells are Nα-acetylated (Starheim et al., 2012). Several links between protein Nα-acetylation and the UPS have been proposed, including substrate acetylation that mediates direct recognition by the Doa10 ubiquitin ligase (Hwang et al., 2010). Here we show that multiple Doa10 substrates do not require Nα-acetylation for their degradation. Conversely, we find an unexpected link between Nα-acetylation and the other major ERAD ubiquitin ligase, Hrd1. In particular, Der1, a cofactor required specifically for Hrd1 substrates with ER luminal lesions (ERAD-L), is acetylated by NatB and normally requires this modification for its function. Failure to Nα-acetylate Der1 causes it to become a Hrd1 substrate.
N-terminal protein acetylation in the Doa10 pathway
Both we (Ravid et al., 2006) and Hwang et al. (2010) identified NatB as a potential factor in Doa10-mediated substrate degradation. However, unlike Hwang et al., our experiments uncovered only mild defects with tested substrates, including a variety of Deg1-bearing proteins. These observations hold for engineered Deg1 substrates analyzed in different strain backgrounds, with different cell-lysis protocols, and using either radioactive pulse-chase or translational-shutoff methods to measure degradation (Figure 1 and unpublished data). Most important, degradation of unmodified, endogenous MATα2 in cells lacking NatB was not significantly inhibited.
Recently an elegant optical reporter of in vivo protein degradation rates was used to screen yeast deletion strains for impaired turnover of different N-end–targeted substrates (Khmelinskii et al., 2012). One engineered reporter used an N-terminal degron beginning with Cys-Leu (the CL degron), which is recognized by the Doa10 pathway (Hwang et al., 2010). This substrate is predicted to be acetylated by NatA. Loss of Doa10, Ubc7, or Cue1 (and, separately, Ubc6, which was not in the library) caused the most severe defects in CL-degron-reporter degradation. These results exactly parallel our earlier screen using a Deg1-Ura3 reporter with the same library (Ravid et al., 2006). The ard1Δ strain, which lacks the NatA catalytic subunit, was not identified, and the nat1Δ stain (lacking the NatA noncatalytic subunit) was ranked 24 among the apparent stabilizers. Of note, mdm20Δ and nat3Δ mutations, which inactivate NatB, were identified among the 11 strongest stabilizers of the CL-degron reporter even though the reporter is not a NatB substrate.
These data suggest that NatB may contribute to the function of components of the UPS. For example, proteasomal subunits Rpt3, Rpn11, and Pre1/β4 are all confirmed targets of NatB (Kimura et al., 2003). This might help to explain the enhanced levels of ubiquitin–protein conjugates in nat3Δ cells (Supplemental Figure S2C). A proteolysis-promoting role for NatB through its modification of UPS components, rather than of substrates, could account for the weak Ubc6 stabilization in nat3Δ cells since Ubc6 is not acetylated by NatB (Van Damme et al., 2012).
Role of Der1 Nα-acetylation in ERAD-L
In contrast to the weak effects of Nα-acetylation on the Doa10 pathway, Nα-acetylation of the Der1 subunit of the Hrd1 complex is essential to ERAD-L. NatB is dispensable for ERAD-M, which does not require Der1. Several results suggest that Der1 is the only Nα-acetylation target of functional relevance to ERAD-L. First, Der1 overexpression is sufficient to suppress completely the nat3Δ defect in CPY* degradation. Second, altering the N-terminal sequences of Der1 changes ERAD-L Nα-acetyltransferase dependence. For instance, changing the second residue of Der1 from Asp to Leu renders CPY* degradation NatB independent (Supplemental Figure S5C). Mutating the second residue to Ala results in removal of the N-terminal methionine, NatA-mediated Nα-acetylation and an accompanying novel dependence of ERAD-L on NatA (Figure 4B and Supplemental Figure S3B). That NatA is not normally required for ERAD-L indicates there is no other protein in the Hrd1 pathway that must be modified by this enzyme. In addition, ERAD-L exhibited no detectable dependence on NatC. These results imply that other than Der1 modification by NatB, Nα-acetylation by any of the principal yeast acetyltransferases is not essential for ERAD-L, although it remains possible that degradation of certain substrates is more sensitive to other Nα-acetylation events.
Altering the second residue of Der1 to a leucine should cause Der1 to be acetylated by NatC; however, ML-Der1 function in ERAD-L is independent of NatC. Nα-acetylation of Der1 may normally help to create a relatively hydrophobic binding surface at its N-terminus. Replacement of an acidic residue (Asp) with a large aliphatic one (Leu) might serve the same function and obviate the requirement for acetylation.
Surprisingly, ERAD-L in MA-der1 cells was still sensitive to NatB inactivation (Figure 4). Nα-acetylation of MA-Der1 remained partially dependent on NatB (Supplemental Figure S4B). This may reflect altered SRP binding to the ribosome-nascent-chain complex (RNC), which is mediated by the emerging hydrophobic N-terminal segment of nascent proteins (Zhang et al., 2012). In cells lacking NatB, SRP might bind more tightly to the RNC-translating mutant MA-Der1 protein, limiting access of NatA (Forte et al., 2011). The strong ERAD-L defect in MA-der1 nat3Δ cells can be viewed as a synthetic genetic deficiency. Partially defective mutant MA-der1 cells may become sensitized to defects in other Hrd1 pathway components, such as the Ubc7-binding Cue1 cofactor, which is also a NatB substrate.
Mutations in NatB or in Der1 itself enhance rates of Der1 degradation, and this degradation depends on Hrd1. This suggests that Hrd1 ubiquitylates Der1 when the N-terminus of Der1 is aberrant. Mutant Der1-Hrd1 binding may nevertheless be mediated by the same interactions with the Hrd1 complex normally required for Der1 function. A potential precedent for this is the Ubc6 E2, which also exhibits susceptibility to ubiquitylation by its cognate ubiquitin ligase, Doa10 (Kreft and Hochstrasser, 2011). In this case, data suggest a second binding site in the Doa10 complex where Ubc6 is ubiquitylated by a mechanism requiring another Ubc6 molecule.
General functions of Nα-acetylation
Surprisingly few proteins have been shown to rely on their Nα-acetylation for their function despite the common occurrence of this modification. One problem in assessing effects of Nα-acetylation on a specific protein is that it usually requires mutating that protein's N-terminal region. As seen with Der1, a wide range of effects on the mutated substrate can be engendered, and these are difficult to predict. To our knowledge, the change in Der1 Nα-acetyltransferase dependence to NatA by mutating Der1 is the first successful execution of such a switch in functional dependence to a different Nα-acetyltransferase (Starheim et al., 2012).
It is likely that a general role for Nα-acetylation is to modulate the affinity or relative orientation of proteins toward one another. A clear example of such binding modulation is the Nα-acetylation of the Ubc12E2 and its interaction with its cognate E3 ligase (Scott et al., 2011). A binding site of the acetylated N-terminus of Der1 in the Hrd1 complex remains to be identified. Usa1 is an obvious candidate (Horn et al., 2009), although we could not detect a change in Der1-Usa1 binding by coimmunoprecipitation from cell extracts. Alternatively, Nα-acetylation of Der1 might conformationally alter or orient the cofactor relative to the Hrd1 complex in a way that prevents it from becoming ubiquitylated and degraded while maximizing its substrate retrotranslocation and/or ubiquitylation activities. A final possibility is that the hydrophobic N-terminal segment of Der1 interacts directly with the membrane, and Nα-acetylation enhances or helps to properly position this interaction.
MATERIALS AND METHODS
Yeast and bacterial methods
Yeast rich (yeast extract/peptone/dextrose [YPD]) and minimal (SD) media were prepared as described previously, and standard methods were used for genetic manipulation of yeast (Guthrie and Fink, 2002). Standard techniques were used for recombinant DNA work in Escherichia coli.
Yeast strain constructions
S. cerevisiae strains used in this study are listed in Supplemental Table S2. Deletion strains in the BY4741 genetic background were retrieved from the Saccharomyces Gene Deletion Project collection (Open Biosystems, Huntsville, AL); in some cases these were crossed to congenic strains for creation of double mutants. Mutant strains were also generated in the MHY501 background (Chen et al., 1993), as indicated in Supplemental Table S2. For gene knockouts, marker genes from the pFA6a series (European Saccharomyces cerevisiae Archive for Functional Analysis, Institute for Molecular Biosciences, Johann Wolfgang Goethe-University Frankfurt, Frankfurt, Germany) were PCR amplified with appropriate flanking primers; in some cases, the knockout alleles from the Saccharomyces deletion collection were used as PCR templates. The PCR fragments were used for yeast transformation, and correct single-site integration was verified by colony PCR and marker segregation analysis. The nat3Δ allele was also tracked by the slower, temperature-sensitive growth it conferred on cells. In crosses involving the prc1-1 allele, which encodes CPY*, the CPY* protein was distinguished from WT CPY by its distinct glycosylation pattern, which was monitored by anti-CPY immunoblotting.
Plasmid constructions
All plasmids described in the present study (Supplemental Table S3) were constructed using standard approaches. Previously described plasmids are also listed in Supplemental Table S3. The pSZ1 plasmid (Friedlander et al., 2000) was used for quantitative measurement of the UPR using ortho-nitrophenyl-β-galactoside as substrate (Chen et al., 1993). The DER1 open reading frame (ORF) was PCR amplified from yeast genomic DNA with primers containing XbaI and XhoI restriction sites in the 5′ and 3′ ends, respectively. The PCR fragment was initially cloned into a p415MET25 vector (Mumberg et al., 1994). MK, MS, and ML variants of Der1 were also generated by PCR amplification in which the 5′-primer contained the respective mutations in the second codon. All HA- and FLAG-tagged Der1-encoding constructs were also made by PCR amplification with the 3′-primer containing the tag sequence fused in-frame to the terminal DER1 ORF followed by a stop codon and an XhoI site.
For plasmids in which Der1 was expressed under its natural promoter, the MET25 promoter from p415MET25-DER1 was replaced with a 500–base pair sequence upstream of the DER1 ORF; the sequence was amplified from yeast genomic DNA using 5′ and 3′ primers with SacI and XbaI sites, respectively. Overexpression of Der1 proteins was done from p416GPD or p415GPD vectors (Mumberg et al., 1995) after subcloning the DER1 ORFs into these vectors by XbaI/XhoI digestion. The der1-D2A mutant alleles (encoding MA-Der1) were generated by QuikChange mutagenesis using either the p415-DER1 or p415-DER1-HA plasmid as template. Subsequently, for overexpression of MA-der1-FLAG for protein modification analysis, a der1-D2A-FLAG fragment was generated by PCR using 5′-primer with the D2A mutation and an XbaI restriction site and the 3′-primer encoding the FLAG sequence followed by an XhoI site. This fragment was then cloned into a p416GPD plasmid.
The p416NAT3 plasmid (pNAT3) was constructed by PCR amplification from yeast genomic DNA. The amplified BamHI–XhoI fragment included the NAT3 ORF flanked by 500 base pairs of upstream and 300 base pairs of downstream sequence. The PCR product was cloned into pRS416. To make p415MET25-Deg1-FLAG-URA3, the Deg1-FLAG-URA3 ORF was amplified as a PstI–XhoI fragment using as template the p416GPD-Deg1-FLAG-URA3 vector and then inserted into p415MET25. All PCR-amplified DNA fragments were fully sequenced.
Purification and mass-spectrometric analysis of Der1 proteins
Purification of FLAG-tagged Der1 proteins.
WT Der1-FLAG was expressed from a p416GPD plasmid in hrd1Δ NAT3 (MHY3032) and hrd1Δ nat3Δ (MHY7430) cells. Similarly, the MA-Der1-FLAG mutant protein was expressed using a p416GPD vector in hrd1Δ NAT3 (MHY3032), hrd1Δ nat3Δ (MHY7430), and hrd1Δ nat1-4 (MHY7860) cells. Yeast cultures were grown in 1 l of selective medium at 30°C until OD600 was ∼1.2. Each culture was split into 4 × 250-ml tubes and centrifuged at 4000 relative centrifugal force (rcf) for 10 min. Each pellet was washed with 25 ml of dH2O, transferred to a 50-ml conical tube, and centrifuged again. Cell pellets from each culture were ground in liquid nitrogen and then resuspended in 100 ml of wash buffer (20 mM Na 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.5, 200 mM NaCl, and 10% glycerol containing Complete Protease Inhibitor tablets [Roche, Indianapolis, IN], 1 mM phenylmethylsulfonyl fluoride, and 20 μg/ml pepstatin A). A 25-ml amount of each extract was carefully layered on 10 ml of cushion buffer (1 M sucrose, 20 mM Na-HEPES, pH 7.5) and centrifuged for 15 min at 3000 rcf (4°C); 25 ml of supernatant (top phase) from each tube (∼100 ml total) were transferred to separate tubes and rotated at 4°C in the presence of 1% Triton X-100 for 1 h. Solubilized extracts were then added to 600 μl of FLAG-M2 resin (50% slurry; Sigma-Aldrich, St. Louis, MO) and rotated for 14–16 h at 4°C. The resin was subsequently washed three times in batch with 6 ml of high-salt wash buffer (wash buffer containing 500 mM NaCl and 1% Triton X-100). A fourth wash with 6 ml of high-salt wash buffer was performed in batch containing 0.1% Triton X-100 instead of 1%. Protein was eluted with 3X-FLAG peptide (200 μg/ml). A 2× Laemmli sample buffer was added to the final eluate, and samples were run on 12% SDS–polyacrylamide gels.
In-gel protein digestion.
Analyses in this subsection were performed by the Keck Protein Microchemistry Facility at Yale University (New Haven, CT) on a fee basis. Proteins excised from the foregoing gels were subjected to in situ enzymatic digestion. Gel plugs were washed with 250 μl of 50% acetonitrile/50% water for 5 min, followed by 250 μl of 50 mM ammonium bicarbonate/50% acetonitrile/50% water for 30 min. A final wash was done using 10 mM ammonium bicarbonate/50% acetonitrile/50% water for 30 min. After washing, the gel plugs were dried in a SpeedVac and rehydrated with 0.1 μg of modified trypsin (Promega, Madison, WI) per (approximately) 15 mm3 of gel in 15 μl of 10 mM ammonium bicarbonate. Samples were digested at 37°C for 16 h. Details regarding LC-MS/MS analysis can be found in the Supplementary Methods.
Pulse-chase and cycloheximide-chase/immunoblot analyses.
Pulse-chase analysis was performed as described (Chen et al., 1993; Rubenstein et al., 2012). Yeast cells were labeled with ∼20 μCi of TRAN 35S-LABEL (MP Biomedicals, Solon, OH) per OD600 unit of cells at 30°C for 5–6 min in SD medium lacking methionine and cysteine. Chases with excess unlabeled Met and Cys were performed in the absence of cycloheximide. Immunoprecipitation of MATα2 was performed with anti-MATα2 antibodies (Laney and Hochstrasser, 2003) and agarose–protein A (RepliGen, Waltham, MA). Immunoprecipitated proteins were separated by SDS–PAGE and analyzed by autoradiography with a Storm 860 PhosphorImager system and ImageQuant 5.2 software (Molecular Dynamics, Sunnyvale, CA).
Cycloheximide-chase/immunoblot assays followed Ravid et al. (2006). Protein degradation after immunoblotting was quantified using a G:Box system (Syngene, Frederick, MD). The following mouse monoclonal antibodies were used: anti–HA 16B12 (Covance, Berkeley, CA); anti–yeast 3-phosphoglycerate kinase (PGK; Molecular Probes, Eugene, OR); and anti–yeast CPY (Nava Segev, University of Illinois, Chicago, IL). Rabbit polyclonal antibodies used were anti-Der1 (Horn et al., 2009), anti-Doa10 (Kreft et al., 2006), anti-Cue1, and anti-ubiquitin (Dako, Carpinteria, CA). Primary antibody incubations were followed by incubation with peroxidase-coupled anti–immunoglobulin G, which was visualized by enhanced chemiluminescence (GE Healthcare, Piscataway, NJ).
Supplementary Material
Acknowledgments
We thank Chris Hickey and Robb Tomko Jr. for helpful comments on the manuscript; Randy Hampton, Ernst Jarosch, Davis Ng, Thomas Sommer, Alex Varshavsky, and Dieter H. Wolf for yeast strains and/or plasmids; and Ernst Jarosch and Nava Segev for anti-Der1 and anti-CPY antibodies, respectively. This work was funded by National Institutes of Health Grant GM046904 to M.H. Support is also acknowledged from National Institutes of Health Grant T32GM7223 (D.Z.) and National Institutes of Health National Research Service Award postdoctoral fellowships to D.J.A. and E.M.R.
Abbreviations used:
- CPY
carboxypeptidase Y
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- GPD
glycerol-3-phosphate dehydrogenase
- HA
hemagglutinin
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- ORF
open reading frame
- PGK
phosphoglycerate kinase
- SD
synthetic dextrose
- SRP
signal recognition particle
- TM
transmembrane
- UPR
unfolded protein response
- UPRE
unfolded protein response element
- UPS
ubiquitin-proteasome system
- YPD
yeast extract–peptone–dextrose
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-11-0838) on January 30, 2013.
Present addresses: *Protein Sciences Corp., 1000 Research Parkway, Meriden, CT 06450;
†Department of Biology, Ball State University, 2111 W. Riverside Ave., Muncie, IN 47306.
REFERENCES
- Arendt CS, Hochstrasser M. Identification of the yeast 20S proteasome catalytic centers and subunit interactions required for active-site formation. Proc Natl Acad Sci USA. 1997;94:7156–7161. doi: 10.1073/pnas.94.14.7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER- associated degradation. Nat Cell Biol. 2001;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278:1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MATα2 repressor. Cell. 1993;74:357–369. doi: 10.1016/0092-8674(93)90426-q. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak PM, Wolf DH. Membrane topology and function of Der3/Hrd1p as a ubiquitin-protein ligase (E3) involved in endoplasmic reticulum degradation. J Biol Chem. 2001;276:10663–10669. doi: 10.1074/jbc.M008608200. [DOI] [PubMed] [Google Scholar]
- Deng M, Hochstrasser M. Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature. 2006;443:827–831. doi: 10.1038/nature05170. [DOI] [PubMed] [Google Scholar]
- Forte GM, Pool MR, Stirling CJ. N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol. 2011;9:e1001073. doi: 10.1371/journal.pbio.1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2:379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- Goder V, Carvalho P, Rapoport TA. The ER-associated degradation component Der1p and its homolog Dfm1p are contained in complexes with distinct cofactors of the ATPase Cdc48p. FEBS Lett. 2008;582:1575–1580. doi: 10.1016/j.febslet.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, Olzmann JA, Kopito RR. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat Struct Mol Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to Yeast Genetics and Molecular and Cell Biology. San Diego, CA: Academic Press; 2002. [Google Scholar]
- Hampton RY, Bhakta H. Ubiquitin-mediated regulation of 3-hydroxy-3-methylglutaryl-CoA reductase. Proc Natl Acad Sci USA. 1997;94:12944–12948. doi: 10.1073/pnas.94.24.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Garza RM. Protein quality control as a strategy for cellular regulation: lessons from ubiquitin-mediated regulation of the sterol pathway. Chem Rev. 2009;109:1561–1574. doi: 10.1021/cr800544v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig AO, Rosati S, Pijnappel PW, van Breukelen B, Timmers MH, Mohammed S, Slijper M, Heck AJ. Perturbation of the yeast N-acetyltransferase NatB induces elevation of protein phosphorylation levels. BMC Genomics. 2010;11:685. doi: 10.1186/1471-2164-11-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- Hitt R, Wolf DH. Der1p, a protein required for degradation of malfolded soluble proteins of the endoplasmic reticulum: topology and Der1-like proteins. FEMS Yeast Res. 2004;4:721–729. doi: 10.1016/j.femsyr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Ho Y-C, Adamson JG, Hodges RS, Smith M. Heterodimerization of the yeast MATa1 and MATα2 proteins is mediated by two leucine zipper-like coiled-coil motifs. EMBO J. 1994;13:1403–1413. doi: 10.1002/j.1460-2075.1994.tb06394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M, Varshavsky A. In vivo degradation of a transcriptional regulator: the yeast α2 repressor. Cell. 1990;61:697–708. doi: 10.1016/0092-8674(90)90481-s. [DOI] [PubMed] [Google Scholar]
- Hollebeke J, Van Damme P, Gevaert K. N-terminal acetylation and other functions of Nalpha-acetyltransferases. Biol Chem. 2012;393:291–298. doi: 10.1515/hsz-2011-0228. [DOI] [PubMed] [Google Scholar]
- Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell. 2009;36:782–793. doi: 10.1016/j.molcel.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science. 2010;327:973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PR, Swanson R, Rakhilina L, Hochstrasser M. Degradation signal masking by heterodimerization of MATα2 and MATa1 blocks their mutual destruction by the ubiquitin-proteasome pathway. Cell. 1998;94:217–227. doi: 10.1016/s0092-8674(00)81421-x. [DOI] [PubMed] [Google Scholar]
- Jonikas MC, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khmelinskii A, et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol. 2012;30:708–714. doi: 10.1038/nbt.2281. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Saeki Y, Yokosawa H, Polevoda B, Sherman F, Hirano H. N-Terminal modifications of the 19S regulatory particle subunits of the yeast proteasome. Arch Biochem Biophys. 2003;409:341–348. doi: 10.1016/s0003-9861(02)00639-2. [DOI] [PubMed] [Google Scholar]
- Kreft SG, Hochstrasser M. An unusual transmembrane helix in the endoplasmic reticulum ubiquitin ligase doa10 modulates degradation of its cognate e2 enzyme. J Biol Chem. 2011;286:20163–20174. doi: 10.1074/jbc.M110.196360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreft SG, Wang L, Hochstrasser M. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI) J Biol Chem. 2006;281:4646–4653. doi: 10.1074/jbc.M512215200. [DOI] [PubMed] [Google Scholar]
- Laney JD, Hochstrasser M. Ubiquitin-dependent degradation of the yeast Matα2 repressor enables a switch in developmental state. Genes Dev. 2003;17:2259–2270. doi: 10.1101/gad.1115703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994;22:5767–5768. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- Ravid T, Hochstrasser M. Autoregulation of an E2 enzyme by ubiquitin-chain assembly on its catalytic residue. Nat Cell Biol. 2007;9:422–427. doi: 10.1038/ncb1558. [DOI] [PubMed] [Google Scholar]
- Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–690. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein EM, Kreft SG, Greenblatt W, Swanson R, Hochstrasser M. Aberrant substrate engagement of the ER translocon triggers degradation by the Hrd1 ubiquitin ligase. J Cell Biol. 2012;197:761–773. doi: 10.1083/jcb.201203061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starheim KK, Gevaert K, Arnesen T. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem Sci. 2012;37:152–161. doi: 10.1016/j.tibs.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001;15:2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Ulrich HD. Ubiquitin, SUMO, phosphate: how a trio of posttranslational modifiers governs protein fate. Mol Cell. 2012;47:335–337. doi: 10.1016/j.molcel.2012.07.016. [DOI] [PubMed] [Google Scholar]
- Van Damme P, et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc Natl Acad Sci USA. 2012;109:12449–12454. doi: 10.1073/pnas.1210303109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Maegawa S, Akiyama Y, Ha Y. The role of L1 loop in the mechanism of rhomboid intramembrane protease GlpG. J Mol Biol. 2007;374:1104–1113. doi: 10.1016/j.jmb.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Rubenstein EM, Matt T, Hochstrasser M. SUMO-independent in vivo activity of a SUMO-targeted ubiquitin ligase toward a short-lived transcription factor. Genes Dev. 2010;24:893–903. doi: 10.1101/gad.1906510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Berndt U, Golz H, Tais A, Oellerer S, Wolfle T, Fitzke E, Rospert S. NAC functions as a modulator of SRP during the early steps of protein targeting to the endoplasmic reticulum. Mol Biol Cell. 2012;23:3027–3040. doi: 10.1091/mbc.E12-02-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.