

The morphogenesis checkpoint stabilizes the mitotic inhibitor Swe1p and prevents mitosis following stresses that affect bud formation. It is shown that, following some stresses, Swe1p stabilization is an indirect effect of cyclin-dependent kinase inhibition.
Abstract
Saccharomyces cerevisiae cells exposed to a variety of physiological stresses transiently delay bud emergence or bud growth. To maintain coordination between bud formation and the cell cycle in such circumstances, the morphogenesis checkpoint delays nuclear division via the mitosis-inhibitory Wee1-family kinase, Swe1p. Swe1p is degraded during G2 in unstressed cells but is stabilized and accumulates following stress. Degradation of Swe1p is preceded by its recruitment to the septin scaffold at the mother-bud neck, mediated by the Swe1p-binding protein Hsl7p. Following osmotic shock or actin depolymerization, Swe1p is stabilized, and previous studies suggested that this was because Hsl7p was no longer recruited to the septin scaffold following stress. However, we now show that Hsl7p is in fact recruited to the septin scaffold in stressed cells. Using a cyclin-dependent kinase (CDK) mutant that is immune to checkpoint-mediated inhibition, we show that Swe1p stabilization following stress is an indirect effect of CDK inhibition. These findings demonstrate the physiological importance of a positive-feedback loop in which Swe1p activity inhibits the CDK, which then ceases to target Swe1p for degradation. They also highlight the difficulty in disentangling direct checkpoint pathways from the effects of positive-feedback loops active at the G2/M transition.
INTRODUCTION
Cell cycle checkpoints are surveillance pathways that monitor the progress of key events and act to delay cell cycle progression in the face of problems (Hartwell and Weinert, 1989; Morgan, 2007). The best-known checkpoints monitor DNA damage, DNA replication, and bipolar alignment of chromosomes during mitosis (Morgan, 2007). In budding yeast, a “morphogenesis checkpoint” also monitors aspects of bud formation and delays nuclear division until a bud has been formed (Lew, 2003). The precise aspects of morphogenesis that might be monitored by the checkpoint remain controversial and may include bud emergence (Theesfeld et al., 2003; McNulty and Lew, 2005), bud growth (Anastasia et al., 2012), septin organization (Barral et al., 1999), and actin organization (McMillan et al., 1998).
The cell cycle delay triggered by the morphogenesis checkpoint involves inhibition of mitosis-promoting cyclin-dependent kinase (CDK) complexes via tyrosine phosphorylation of the CDK Cdc28p at Tyr-19 (Lew and Reed, 1995; Sia et al., 1996). Tyr-19 phosphorylation is controlled by the balance between the activities of the Wee1-family kinase Swe1p and the Cdc25-family phosphatase Mih1p (Russell et al., 1989; Booher et al., 1993). Following stress, Swe1p degradation is temporarily halted, and its abundance increases (Sia et al., 1998). Mih1p phosphorylation is also regulated by stress (Anastasia et al., 2012), perhaps causing its inhibition (Harrison et al., 2001). In combination, Swe1p stabilization and Mih1p inhibition promote a delay in nuclear division that is thought to maintain coordination between budding and the nuclear cycle (Lew, 2003).
Swe1p degradation in unstressed cells proceeds via several sequential modifications: initial phosphorylation by Cdc28p primes Swe1p for further phosphorylation by the Polo-family kinase Cdc5p, leading to ubiquitination (possibly by the ubiquitin ligases Dma1p and Dma2p) and degradation by the proteasome (Sakchaisri et al., 2004; Asano et al., 2005; Raspelli et al., 2011). Interestingly, Swe1p degradation appears to be coupled to its localization: after bud emergence, Swe1p accumulates at the mother-bud neck, and mutations that interfere with Swe1p neck targeting also block its degradation (Longtine et al., 2000; McMillan et al., 2002; Theesfeld et al., 2003; Crutchley et al., 2009). At least one such mutation can be bypassed by artificial tethering of Swe1p to the neck (King et al., 2012), directly implicating Swe1p neck targeting as an important step in Swe1p degradation.
Swe1p is recruited to the neck by interaction with Hsl7p, which in turn binds to the checkpoint kinase Hsl1p, which binds to septins at the mother-bud neck (Lew, 2003). The septins are a family of conserved filament-forming proteins that polymerize to form a stable scaffold beneath the plasma membrane at the neck (Gladfelter et al., 2001). Localization of Swe1p to the septin scaffold may provide enhanced access to the kinases (Clb2p/Cdc28p and Cdc5p) that target it for degradation, as both of those kinases are themselves enriched at the neck (Song et al., 2000; Bailly et al., 2003).
Septins assemble into a ring at the future bud site, and then expand to form an hourglass-shaped collar at the neck following bud emergence (Haarer and Pringle, 1987; Kim et al., 1991). Hsl1p and Hsl7p both localize to the collar (Barral et al., 1999; Shulewitz et al., 1999; Longtine et al., 2000), but when bud emergence is blocked by depolymerizing F-actin with latrunculin (Lat), the septin ring recruits only Hsl1p, and not Hsl7p, so Swe1p also fails to localize (Theesfeld et al., 2003). Even after a bud has formed, osmotic shock transiently displaces Hsl7p from the neck (Clotet et al., 2006), and temperature shift transiently displaces Swe1p from the neck (Longtine et al., 2000). Thus an appealing “localization hypothesis” is that stresses stabilize Swe1p by displacing it from the septin scaffold.
The localization hypothesis predicts that, if Swe1p displacement from the neck were blocked, then Swe1p would continue to be degraded, even following stress. We set out to test this prediction. Our findings imply that, at least in budded cells, stresses do not stabilize Swe1p by displacing it from the neck. Rather, stresses empower Swe1p to inhibit Cdc28p, and it is the Cdc28p inhibition that indirectly leads to Swe1p stabilization. Thus Swe1p stabilization is due to a feedback loop and is not the primary cause of checkpoint-mediated cell cycle delay.
RESULTS
We recently reported that a septin-Swe1p fusion protein is targeted to the neck independent of the normally required targeting factors Hsl1p and Hsl7p, and is phosphorylated and degraded with normal timing during the cell cycle (King et al., 2012). Furthermore, septin tethering bypassed the need for Hsl1p-Hsl7p interaction in Swe1p degradation (King et al., 2012). We set out to test whether septin tethering would similarly force Swe1p degradation to continue after osmotic shock, a stress reported to displace Hsl7p from the neck (Clotet et al., 2006).
To assess Swe1p and septin-Swe1p degradation, we performed single-cycle synchrony experiments in which cells released from pheromone-induced G1 arrest were rearrested in G1 following a single cell cycle. This protocol allowed us to follow the fate of Swe1p that is made in a transcriptional pulse in late G1/S phase (Sia et al., 1996), without confounding effects from the lack of perfect synchrony as cells enter a second cell cycle. For direct comparison of the behavior of septin-tethered Swe1p with that of wild-type Swe1p, both proteins were tagged with the myc epitope and expressed from the SWE1 promoter in the same cells. To avoid potential indirect effects due to Cdc28p inhibition following stress, we replaced wild-type CDC28 with the Swe1p-resistant CDC28E12K mutant. It might seem more natural to use the nonphosphorylatable CDC28Y19F mutant, but previous work showed that CDC28Y19F was still susceptible to Swe1p inhibition via binding, whereas the CDC28E12K mutant is more (though still not completely) resistant to Swe1p (McMillan et al., 1999b). Thus, in these cells, Cdc28p should remain active, even following stresses that would normally trigger Swe1p-mediated inhibition of Cdc28p.
Swe1p degradation following osmotic shock
Both Swe1p and septin-Swe1p accumulated and then disappeared as cells progressed through the cell cycle (Figure 1, A and B). Remarkably, osmotic shock had no effect on this profile for either protein (Figure 1, C and D). This was surprising because degradation of wild-type Swe1p was previously shown to stop following comparable osmotic shock treatments (Sia et al., 1998). To assess whether degradation of wild-type Swe1p might be indirectly affected by the presence of the septin-tethered Swe1p, we repeated the experiment in cells containing only wild-type Swe1p. Again, osmotic shock failed to block or even detectably delay Swe1p degradation (Figure 1, E and G), although the altered budding profile indicated that the salt-shocked cells were experiencing stress (Figure 1, F and H).
FIGURE 1:

Degradation of Swe1p is not affected by osmotic shock in the absence of feedback via Cdc28p. (A) Swe1p and septin-Swe1p levels rise and fall in parallel as cells traverse the cell cycle. Cells containing CDC28E12K to prevent Swe1p-mediated Cdc28p inhibition (DLY12321) were arrested in G1 with pheromone and released into fresh media at 30°C to traverse the cell cycle. Pheromone was reintroduced 60 min after release to rearrest cells at the next G1. Samples were taken at 15-min intervals and the levels of myc-tagged Swe1p and septin-Swe1p were analyzed by Western blotting with anti-myc (Cdc11p: loading control), and quantitated (graph). (B) Synchrony parameters (budding and nuclear division) for the experiment in (A) were scored for >200 cells per sample. (C) Swe1p and septin-Swe1p levels are unaffected by osmotic shock. Cells were treated as in (A), except that 0.4 M NaCl was added at 45 min. (D) Synchrony parameters for the experiment in (C) show that osmotic stress transiently perturbed bud formation and nuclear division. (E) A single-cycle synchrony experiment as in (A) was performed with strain DLY12034, which does not express septin-Swe1p. (F) Synchrony parameters for (E). (G) Cells were treated as in (E), except that 0.4 M NaCl was added at 45 min. (H) Synchrony parameters for (G).
In cells with wild-type CDC28, Cdc28p inhibition following stress leads to delayed repression of the SWE1 promoter (Sia et al., 1996) and continued Swe1p synthesis, as well as a transient block of Swe1p degradation (Sia et al., 1998), which combine to promote further Swe1p accumulation for a time following osmotic shock. We reproduced the previous finding that Swe1p levels continue to accumulate following osmotic shock in cells with wild-type CDC28 (Figure 2). The key difference between the experiment of Figure 1 and that of Figure 2 is the presence of the CDC28E12K mutant in Figure 1. These findings imply that stabilization of Swe1p in budded cells exposed to osmotic shock is an indirect consequence of Cdc28p inhibition: when such inhibition is (mostly) abrogated, osmotic shock does not block Swe1p degradation.
FIGURE 2:
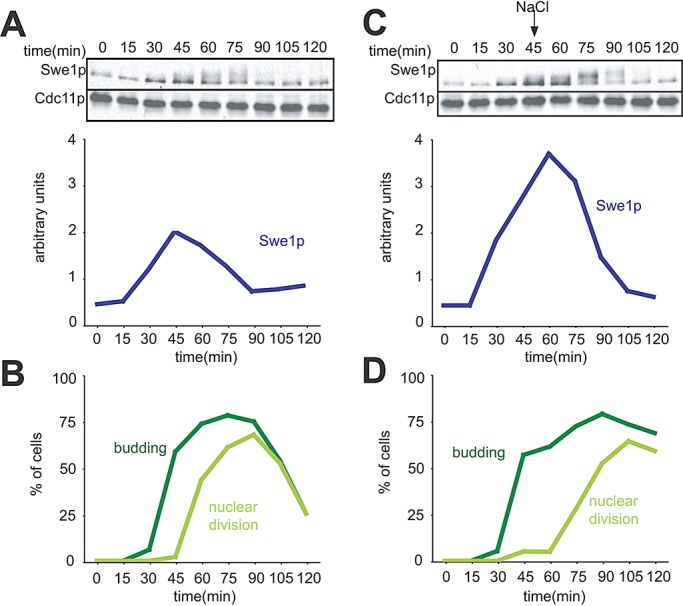
Swe1p accumulates following osmotic shock in cells with wild-type Cdc28p. (A) A single-cycle synchrony experiment was performed using cells containing wild-type CDC28 (DLY5330). The amount of Swe1p was quantified as in Figure 1 and is plotted in arbitrary units for comparison with the parallel stress experiment in (C). (B) Synchrony parameters (budding and nuclear division) for the experiment in (A). (C) Swe1p accumulates following osmotic shock. Cells were treated as in (A), except that 0.4 M NaCl was added at 45 min. (D) Synchrony parameters for the experiment in (C).
Swe1p degradation requires assembled septins
The finding that Swe1p degradation continues even following stress in CDC28E12K mutants suggests that Swe1p stabilization is not a primary effect of stress but rather an indirect effect of Cdc28p inhibition. Combined with the previous finding that osmotic shock displaces Hsl7p from the neck (Clotet et al., 2006), our results raised the possibility that Swe1p degradation is occurring without need for Swe1p to visit the neck. Although previous studies showed that Swe1p promotes a G2 delay in septin mutants (Barral et al., 1999; Longtine et al., 2000), Swe1p degradation has not been directly examined in septin mutants, and it seemed possible that, as with osmotic shock, septin disassembly might affect Swe1p indirectly via inhibition of Cdc28p. To address this question, we asked whether septin disassembly would affect Swe1p degradation in CDC28E12K mutants. We performed single-cycle synchrony experiments as above, but used cdc12-6 mutants in which the septin scaffold is temperature-sensitive (Haarer and Pringle, 1987; Ford and Pringle, 1991; Kim et al., 1991). Unlike in cells exposed to osmotic shock, Swe1p was stabilized upon shift of the septin mutant to restrictive temperature, even in cells with CDC28E12K (Figure 3). The slow decline in Swe1p abundance seen in the septin mutant may reflect residual slow degradation or simply dilution, as the cells continue to grow without making more Swe1p. Thus septin assembly is needed for rapid Swe1p degradation whether or not Cdc28p is inhibited.
FIGURE 3:
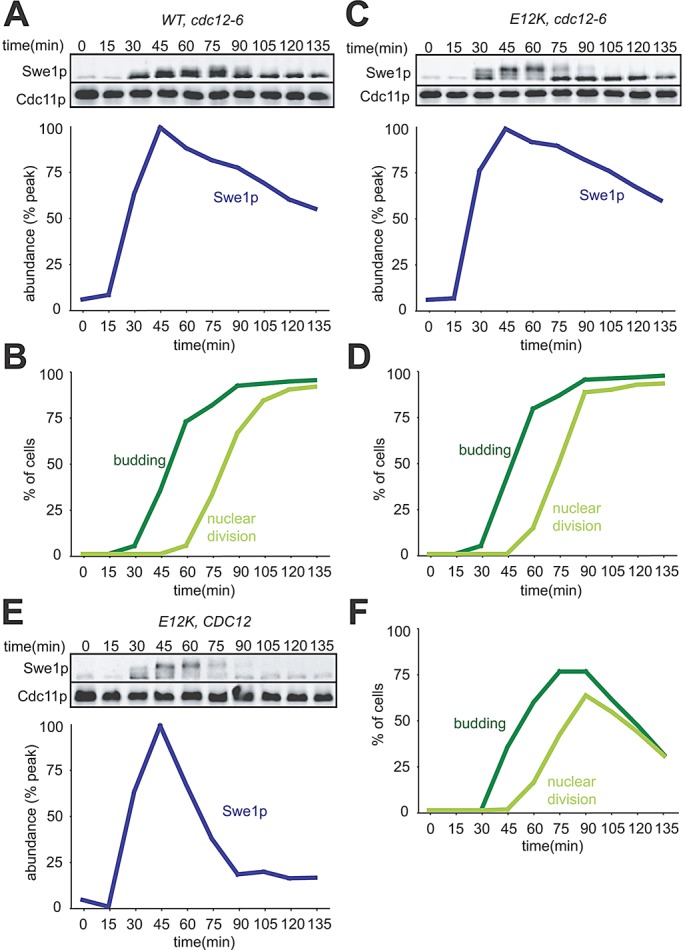
Swe1p is stable in cells lacking assembled septins, regardless of feedback. (A) A single-cycle synchrony experiment was performed using temperature-sensitive cdc12-6 mutant cells (DLY14556). Cells were grown and arrested in pheromone at 18°C, then released into fresh media at 33°C, and Swe1p-myc levels were quantified as in Figure 1. (B) Synchrony parameters for (A). (C) Swe1p is stable regardless of feedback in septin-mutant cells. cdc12-6 CDC28E12K cells were treated as in (A). (D) Synchrony parameters for (C). (E) CDC28E12K cells (with wild-type septins) were treated as in (A): Swe1p accumulates to lower levels and is degraded more rapidly when septins are intact. (F) Synchrony parameters for (E).
Reexamination of Hsl7p localization in cells exposed to osmotic shock
Our results present an apparent conundrum: if Swe1p degradation requires its recruitment to the septin cortex by Hsl7p (McMillan et al., 1999a; Longtine et al., 2000; Figure 3), then displacement of Hsl7p (and hence Swe1p) from the septins following stress (Clotet et al., 2006) should result in Swe1p stabilization. Yet Swe1p in CDC28E12K mutants is not stabilized following salt shock (Figure 1). To resolve this conundrum, we reassessed whether or not Hsl7p is displaced from the septins following salt shock. To that end, we used a green fluorescent protein (GFP)-Hsl7p fusion that was functional by the criterion that it rescued the hsl7Δ mih1Δ synthetic lethality (McMillan et al., 1999a).
Cells expressing GFP-Hsl7p were mixed with cells expressing Hog1p-GFP in a microfluidics chamber. Hog1p is a mitogen-activated protein kinase activated by osmotic stress, and Hog1p concentrates in the nucleus following stress (Brewster et al., 1993; Ferrigno et al., 1998). After cells were switched to media containing 0.4 M NaCl, Hog1p-GFP rapidly accumulated in the nucleus (Figure 4 and Supplemental Video S1: eight out of eight cells accumulated nuclear Hog1p), confirming that the cells in the chamber experienced osmotic stress. However, in neighboring cells in the same chamber, GFP-Hsl7p remained at the mother-bud neck and was not displaced following stress (Figure 4 and Video S1: 17 out of 17 cells retained Hsl7p at the neck for >30 min). The continued recruitment of Hsl7p to the neck following osmotic shock can explain the continuing septin-dependent degradation of Swe1p following osmotic shock in CDC28E12K mutants.
FIGURE 4:
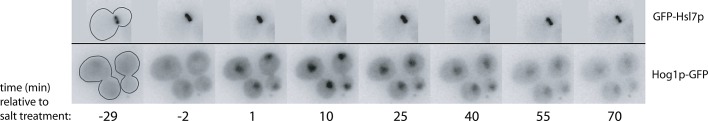
Hsl7p does not leave the septin collar following osmotic shock. Cells marked with GFP-Hsl7p (DLY15839) or Hog1p-GFP (DLY15123) were mixed and imaged in a microfluidics chamber. After 30 min, the culture medium was replaced with fresh medium containing 0.4 M NaCl. Hog1p-GFP (bottom row) rapidly translocated into the nucleus following the shock and gradually returned to the cytoplasm over the subsequent 90 min as cells adapted to the stress. GFP-Hsl7p (top row) remained at the bud neck throughout the 90 min following the shock. Representative cells of each genotype are shown at selected times. For complete movies see Supplemental Video 1.
Swe1p degradation following actin depolymerization
The experiments discussed above indicate that, rather than directly stabilizing Swe1p, osmotic shock leads to Swe1p stabilization indirectly via Cdc28p inhibition. This raised the question of whether other stresses would similarly regulate Swe1p. One well-characterized stress that leads to Swe1p-dependent cell cycle delay is actin depolymerization caused by Lat (Ayscough et al., 1997; McMillan et al., 1998). In budded cells, Hsl7p remained at the neck following Lat treatment (Longtine et al., 2000). However, immunofluorescence experiments indicated that, if budding was prevented by Lat treatment, then Hsl7p was not recruited to the septin ring (Theesfeld et al., 2003). Thus, as proposed for osmotic shock, a block of budding would be expected to stabilize Swe1p by preventing its Hsl7p-mediated recruitment to the septin ring.
We examined Swe1p degradation in single-cycle synchrony experiments similar to those described above. Lat was added 30 min after release from G1 arrest, at which point only ∼20% of cells had formed visible buds (Figure 5, B and D). In cells containing wild-type CDC28, Swe1p levels remained elevated following exposure to Lat, as expected (Figure 5A). However, in cells containing CDC28E12K, Swe1p degradation (Figure 5C) was similar to that seen in untreated cells (Figure 1E). We have shown that Swe1p profiles for untreated cells are highly reproducible (King et al., 2012). Thus, as with osmotic shock, Swe1p stabilization upon actin depolymerization is to a large degree an indirect consequence of Cdc28p inhibition.
FIGURE 5:
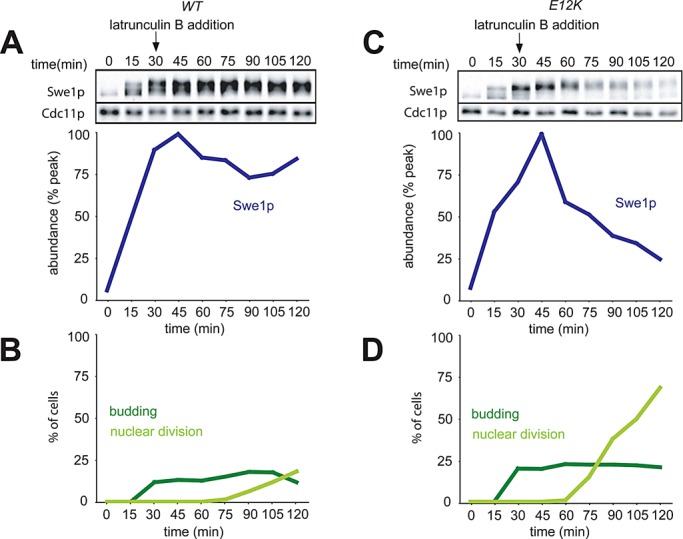
Degradation of Swe1p in cells treated with Lat. (A) Accumulation of Swe1p in wild-type cells following treatment with Lat B. A single-cycle synchrony experiment was performed with cells containing wild-type Cdc28p (DLY5330), and 100 μM Lat B was added at 30 min. Swe1p-myc levels were quantified as in Figure 1. (B) Synchrony parameters for (A). (C) Swe1p accumulation is due to feedback via Cdc28. CDC28E12K cells (DLY12034) were treated as in (A). (D) Synchrony parameters for (C).
In our other experiments, the degree of Swe1p phosphorylation, as inferred by slower gel mobility in the Western blots, was consistent with the expectation that active Cdc28p phosphorylates Swe1p, and Cdc28p inactivation during mitotic exit is rapidly followed by Swe1p dephosphorylation. However, in Lat-treated cells, Swe1p phosphorylation remained high, even after nuclear division (Figure 5). We do not know why that should be the case, but two (nonexclusive possibilities) may account for this surprising observation: 1) Lat treatment may delay Cdc28p inactivation (perhaps via the spindle orientation checkpoint), or 2) Lat treatment may impact the activity of the phosphatases that dephosphorylate Swe1p.
Reexamination of Hsl7p localization in cells exposed to Lat
As with osmotic shock, the degradation of Swe1p in Lat-treated cells raised the conundrum of how such degradation could proceed in the absence of Hsl7p-mediated recruitment of Swe1p to the septins. One possibility was suggested by the previous finding that, in cells released from pheromone arrest, recruitment of Hsl7p to the septin ring could occur even following Lat treatment, if the local cell geometry near the septin ring was tubular rather than flat (i.e., if the septin ring formed within the mating projection; Theesfeld et al., 2003). Thus it seemed possible that Hsl7p was indeed recruited to the septin rings, even in the cells treated with Lat. Using live-cell microscopy of cells expressing Cdc3p-mCherry (a septin) and GFP-Hsl7p, we imaged cells that were released from G1 arrest and then, ∼10 min later, placed on agar slabs either with or without Lat to block budding. In the control cells, septin rings formed and recruited Hsl7p after ∼15 min, approximately coincident with bud emergence (Figure 6, A and C). On Lat treatment, Hsl7p recruitment was delayed (∼30 min after initiating a septin ring), but not blocked, despite the apparent absence of a bud (Figure 6, B and C). Interestingly, in many cells, Hsl7p was recruited to septin rings, even though they did not form within the mating projection (Figure 6B). These findings suggest that Hsl7p recruitment to the septin ring is dependent on some event (possibly septin “maturation”: see Discussion) that is normally coincident with bud emergence and is delayed, but not blocked, upon Lat treatment.
FIGURE 6:

Hsl7p can be recruited to septin rings even in unbudded cells. Cells marked with both GFP-Hsl7p and the septin Cdc3p-mcherry (DLY15772) were arrested in G1 with pheromone and released into fresh media. Ten minutes later, the cells were placed on agar slabs for imaging. (A) Control cells assembled septin rings, budded, and recruited Hsl7p to the neck. (B and D). Cells placed on slabs containing 100 μM Lat B assembled septin rings but failed to form visible buds. Nevertheless, Hsl7p was recruited to the septin ring (often after a delay). Most cells in this experiment formed septin rings away from the mating projection (B) but some formed rings within the projection (D). Hsl7p was recruited in both cases. Selected frames from the indicated times after plating are shown and marked with arrows indicating initial septin ring assembly (Cdc3p-mCherry) or initial Hsl7p recruitment (GFP-Hsl7p). Prior to its recruitment to the septins, Hsl7p is localized to the spindle pole body (Cid et al., 2001), which appears as a dot. (C) Quantification of the interval between initial septin ring assembly and initial Hsl7p recruitment. Each circle is one cell, and the line marks the average interval. For this analysis, we scored only cells in which septin rings formed away from the mating projection (as in A and B) because, when rings formed within the projection, it was difficult to determine exactly when the ring assembled due to the presence of septins at the base of the projection. The Lat-induced delay was statistically significant (p < 0.001) as assessed by the Student's t test.
Delay of Swe1p degradation upon earlier exposure to Lat
As Hsl7p recruitment to the septin ring is delayed following exposure of cells to Lat, we would expect that degradation of Swe1p might also be delayed, yet Swe1p degradation proceeded without a noticeable delay in our previous experiment (Figure 5). A possible explanation is that Hsl7p recruitment is necessary but not rate-limiting for Swe1p degradation. Alternatively, exposure to Lat at 30 min following release from G1 arrest (as in Figure 5) may not have affected Hsl7p recruitment. To distinguish between these possibilities, we repeated the experiment of Figure 4 but added Lat just 15 min after release from G1 arrest. In this case, degradation of Swe1p in cells carrying CDC28E12K was delayed, compared with the experiment in which Lat was added at 30 min (Figure 7). That some Swe1p degradation still occurred was suggested by comparing Swe1p levels with those in the septin-mutant situation (Figure 7E). We conclude that, if Lat is added early enough to delay Hsl7p recruitment to the septin ring, then Swe1p degradation is also delayed. However, Lat exposure after that time does not stabilize Swe1p in CDC28E12K mutants. Thus the Swe1p stabilization observed in wild-type cells upon later exposure to Lat is an indirect effect of Swe1p-mediated inhibition of Cdc28p.
FIGURE 7:

Delayed Swe1p degradation upon earlier treatment with Lat. (A–D) The experiment of Figure 5 was repeated with the same cells, except that Lat was added 15 min following release, instead of 30 min following release, from G1 arrest. In this case, we did not detect any bud formation, and Swe1p degradation was delayed even in the cells with CDC28E12K (C). (E) The Swe1p abundance from (C) was compared with that from Figure 3C (cdc12-6, Swe1p stable) and that from Figure 5C (30 min Lat addition: Swe1p degraded).
DISCUSSION
Feedback control of Swe1p abundance following stress
Swe1p degradation requires Clb1-4/Cdc28p activity (Sia et al., 1998; Asano et al., 2005). In turn, Swe1p inhibits Clb1-2/Cdc28p and, to a lesser extent, Clb3-4/Cdc28p (Keaton et al., 2007). This sets up a potential positive-feedback loop in which Swe1p promotes its own stabilization by inhibiting Cdc28p (Figure 8). In this study, we show that at least two stresses (osmotic shock and actin depolymerization) stabilize Swe1p via this feedback loop.
FIGURE 8:
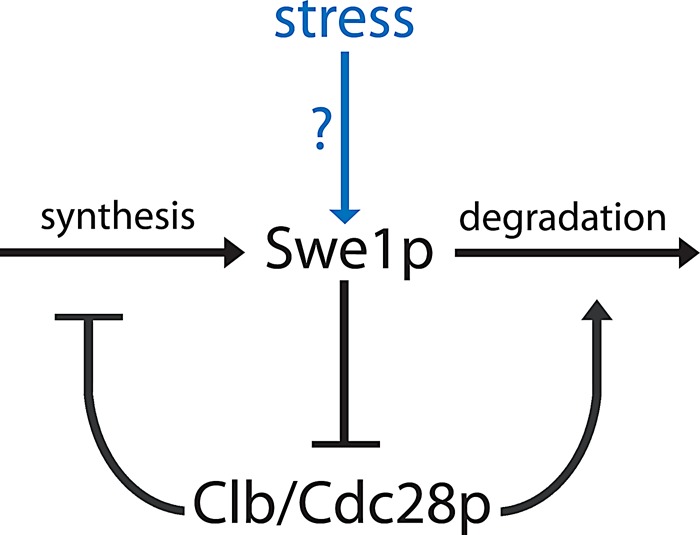
Feedback between Swe1p and Cdc28p controls Swe1p abundance following stress. Swe1p inhibits Cdc28p, which in turn antagonizes Swe1p by promoting its transcriptional repression and its degradation. In cells with mature septin rings, stresses due to osmotic shock or Lat do not directly stabilize Swe1p. Rather, they somehow promote Swe1p's ability to inhibit Cdc28p, leading to subsequent stabilization and accumulation of Swe1p via feedback.
Like Swe1p degradation, timely repression of SWE1 transcription requires Clb/Cdc28p activity, so Swe1p promotes its own continued transcription via a parallel feedback loop (Sia et al., 1996; Figure 8). We find that in budded cells, stress-mediated Swe1p accumulation can be entirely accounted for by feedback because CDC28E12K cells in which Swe1p cannot inhibit Cdc28p do not up-regulate Swe1p abundance in response to stress. The situation is a bit more complicated for unbudded cells, as discussed below.
Swe1p degradation requires septin ring assembly
We found that septin disassembly stabilized Swe1p, regardless of feedback. Thus unbudded cells would only be able to degrade Swe1p once they had assembled a septin ring capable of recruiting the Swe1p degradation machinery. We confirmed previous studies showing that there is a delay between assembly of a detectable septin ring and recruitment of Hsl7p to the ring (Theesfeld et al., 2003). When budding was prevented by actin depolymerization, Hsl7p recruitment was further delayed, and there was a concomitant delay in Swe1p degradation.
What is the basis for the delay in Hsl7p recruitment? We previously suggested that Hsl7p recruitment was responsive to a reorganization of the septins upon bud formation, triggered by the change in the local curvature of the plasma membrane (Theesfeld et al., 2003; Keaton and Lew, 2006). This model was attractive in that the geometrical change in cell shape during bud emergence would enable Hsl7p recruitment, licensing cells to degrade Swe1p. More recently, we discovered that septin ring assembly and collar formation are not clearly separable events; rather, septin proteins are gradually and continuously recruited to a growing septin ring over a period of several minutes spanning bud emergence (Chen et al., 2011). Moreover, Hsl7p can be recruited to the septin ring (albeit after a significant delay) even in unbudded cells. Thus Hsl7p recruitment appears to involve some “maturation” of the septin ring that is delayed by the absence of polymerized actin. The nature of that maturation remains to be determined.
We found that Hsl7p remains at the septin ring following osmotic shock. The basis for the apparent discrepancy between our results and those previously reported (Clotet et al., 2006) is unknown, but we believe that our treatment protocol (exposing cells in a microfluidics device to high-salt media) provides fewer opportunities for additional stresses connected to transferring the cells to microscope slides. It also remains possible that the discrepancy is due to strain background differences. However, it is unlikely that the cells in our experiments did not experience sufficient stress because cells in the same chamber displayed dramatic nuclear accumulation of Hog1p, and we did not detect any displacement of Hsl7p from the neck, even following exposure to 0.8 M NaCl (unpublished data).
We found that Hsl7p could be recruited to septin rings in unbudded cells (after a delay), even if the rings were not within the mating projection. This was also somewhat surprising, given previous findings that only unbudded rings within the projection could recruit Hsl7p (Theesfeld et al., 2003). It remains possible that we missed the presence of small “bumps” near the septin rings that may have sufficiently altered local cell geometry to allow shape-dependent septin maturation. Alternatively, it could be that the Hsl7p recruitment to rings outside the projection does not survive fixation/immunofluorescence protocols and was therefore missed in previous work. In any case, together with the Swe1p degradation data, our findings support the hypothesis that Hsl7p recruitment to the septin ring is a prerequisite for cells to degrade Swe1p.
Implications for other systems
Positive feedback appears to be a very common, if not universal, feature of the G2/M transition (Dunphy, 1994; Morgan, 2007). In that context, it is far from straightforward to determine how checkpoints restrain cell cycle progression, as disentangling direct effects of the checkpoint from indirect effects due to the feedback loops is difficult. For example, in mammalian cells, checkpoint kinases activated by DNA damage are able to phosphorylate Cdc25C at a regulatory site (Ser-216) that keeps it in the inactive, G2 state (Peng et al., 1997). This finding suggested the attractive model that checkpoint-mediated G2 arrest follows from direct checkpoint-induced phosphorylation of that site. However, several other kinases also phosphorylate Ser-216, and it remains unclear how quantitatively important or effective the additional checkpoint-induced phosphorylation might be. Dephosphorylation of Ser-216 occurs concomitant with entry into mitosis, and active mitotic CDK can promote Ser-216 dephosphorylation in a positive-feedback loop (Margolis et al., 2006). Thus continuing Cdc25 Ser-216 phosphorylation during a checkpoint arrest might simply reflect indirect feedback regulation, rather than direct checkpoint regulation of that regulatory site.
One way to determine whether a given checkpoint effect on a CDK regulator is direct or indirect is to render the CDK immune to checkpoint signals and to ask whether that bypasses the checkpoint effect on the regulator. Direct checkpoint effects would occur regardless of CDK status, whereas feedback effects would disappear if CDK regulation were disabled. Applying that strategy to the yeast morphogenesis checkpoint, we conclude that our findings suggest a distinction between two types of checkpoint triggers. Stresses (or mutations) that block assembly of a “mature” septin scaffold directly stabilize Swe1p, regardless of feedback. However, stresses that depolymerize actin or halt bud growth after septin maturation stabilize Swe1p indirectly via feedback. The direct effects of such stresses that cause G2 arrest remain to be determined.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strains are in the BF264-15DU (ade1, his2, leu2-3112, trp1-1a, ura3Δns [ Richardson et al., 1989]) background and are listed in Table 1. The following alleles have been described previously: SWE1myc:HIS2 (McMillan et al., 1999a); CDC3-GFP-SWE1-myc:TRP1 (King et al., 2012); CDC3-mCherry:URA3 (Fang et al., 2010); CDC28E12K (McMillan et al., 1999b); and cdc12-6:LEU2 (Longtine et al., 2000).
TABLE 1:
Yeast strains.
| Strain | Relevant genotype |
|---|---|
| DLY5330 | a bar1 SWE1myc:HIS2 |
| DLY12034 | a bar1 CDC28E12K SWE1myc:HIS2 |
| DLY12321 | a bar1 CDC28E12K SWE1myc:HIS2 CDC3-GFP-SWE1-myc:TRP1 |
| DLY14556 | a bar1 SWE1myc:HIS2 cdc12-6:LEU2 |
| DLY14559 | a bar1 CDC28E12K SWE1myc:HIS2 cdc12-6:LEU2 |
| DLY15123 | a HOG1-GFP::kanR |
| DLY15772 | a bar1 CDC28E12K GFP-HSL7 CDC3-mCherry:URA3 |
| DLY15839 | a bar1 GFP-HSL7 |
HOG1-GFP was generated by the PCR-based C-terminal tagging method (Longtine et al., 1998) using pFA6a-GFP-KanMX6 as template.
To create GFP-HSL7, we began with YCplac111-HSL7 (pDLB1545), which was constructed by amplifying the HSL7 open reading frame plus 606 base pairs of the 5′ untranslated region (UTR) and 398 base pairs of the 3′ UTR out of the genome, and cloning it into YCplac111 cut with HindIII and EcoRI. Next a PCR-amplified GFP fragment with NdeI sites at both ends and no stop codon was cloned into the NdeI site at the N-terminus of Hsl7p in pDLB1545, yielding YCplac111-GFP-HSL7 (pDLB3176). An integrating version of this plasmid was made by swapping the plasmid backbone with that of YIplac128 using AatII/SacI sites, yielding YIplac128-GFP-HSL7 (pDLB3591). A GFP-HSL7 fragment containing 564 base pairs of 5’ and 398 base pairs of 3’ HSL7 sequence was excised from this plasmid using HindIII and SacI and transformed into a strain containing hsl7::URA3. Replacement of hsl7::URA3 with GFP-HSL7 was selected on plates containing 5-fluoroorotic acid, which kills cells that have URA3. Expression of GFP-Hsl7p was confirmed by fluorescence microscopy.
Media, growth conditions, and cell cycle synchrony
Yeast strains were grown in YEPD (1% yeast extract 2% Bacto Peptone, 2% dextrose, 0.012% adenine, and 0.01% uracil) or synthetic complete media at 30°C. For α-factor arrest–release–rearrest experiments, exponentially growing cells were treated with 100 ng/ml α-factor (Research Genetics, Huntsville, AL) for 3 h, harvested by centrifugation, and released into fresh YEPD. After 60 min, fresh 100 ng/ml α-factor was added, so cells would arrest in the subsequent G1 phase. For the experiment of Figure 3, cells were grown and arrested at 18°C, and shifted to 33°C upon release from pheromone arrest. Samples for Western blotting and nuclear staining were taken every 15 min.
Osmotic shock and Lat B treatment
For osmotic shock experiments, NaCl was added to 0.4 M from a 5 M stock at the indicated time. Treatment with 100 μM Lat B (Enzo Life Sciences, Farmingdale, NY) occurred either 15 or 30 min after pheromone release, as indicated. Control cells were treated with 1% dimethyl sulfoxide instead. For imaging (Figure 6), 100 μM Lat B was added to the agarose slabs on the microscope slides.
Nuclear staining and microscopy
For visualization of nuclear DNA, cells were fixed in 70% ethanol overnight at 4°C. The cells were then harvested by centrifugation, resuspended in 0.2 μg/ml 4′,6′-diamidino-2-phenylindole in phosphate-buffered saline (PBS), centrifuged again, and resuspended in mounting medium (90% glycerol, 9.2 mM p-phenylenediamine [Sigma-Aldrich, St. Louis, MO] in PBS). Cells were scored for budding and nuclear division on an Axioskop apparatus with a 100× objective equipped with epifluorescence and differential interference contrast optics (Zeiss, Thornwood, NY).
Live-cell microscopy
Cells were grown at 30°C in synthetic media with dextrose and imaged at room temperature.
For Figure 6, cells were pheromone-arrested and released, mounted on slabs solidified with 2% agarose (Denville Scientific, Metuchen, NJ), and sealed with Vaseline (Unilever, London, UK). Imaging was performed using an Andor XD revolution spinning-disk confocal microscope (Andor Technology, Belfast, UK) with a 100×/1.4 Plan-Apochromat oil-immersion objective and an Andor Ixon3 EM-CCD camera controlled by MetaMorph software (Molecular Devices, Silicon Valley, CA). Both the GFP (488-nm laser) and mCherry (561-nm laser) channels were captured using a gain setting of 200 and a 50-ms exposure time. Time-lapse imaging was set up to take 30 Z-steps of 250 nm each every 1.5 min.
Images were deconvolved using Huygens Essential Software (Scientific Volume Imaging, Hilversum, Netherlands), using the classic maximum likelihood estimation and predicted point-spread function with a background value set constant across all images from the same time lapse and a signal-to-noise ratio of 10. Deconvolved images were compiled in MetaMorph (Molecular Devices, Silicon Valley, CA). GFP and mCherry localization was scored visually using these images.
Microfluidics
Cells for Figure 4 were introduced into a microfluidics device (Y04C; CellASIC, Hayward, CA) in which the flow of fresh media was controlled by the ONIX microfluid perfusion system (CellASIC) at a flow pressure of 4 lb/in2. Images were captured using a DeltaVision Elite deconvolution microscope with a 100×/1.4 numerical aperture oil-immersion objective (Applied Precision, Issaquah, Washington) equipped with an Evolve EM-CCD camera (Photometrics, Tucson, AZ) using 5% excitation light, 200-ms exposure and 25 Z-steps of 250 nm each every 3 min. Shown are maximum projections of the Z-stacks. Image processing was carried out using ImageJ (National Institutes of Health [NIH], Bethesda, MD).
Western blotting
Cell lysis was performed by the trichloroacetic acid method (Keaton, 2008). Samples were subjected to SDS–PAGE on a 6% polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane (Pall, East Hills, NY) and blocked for 1 h with PBS containing 3% non-fat dry milk (Kroger, Cincinnati, OH). Antibodies were diluted in blocking buffer supplemented with 0.1% Tween-20 (Bio-Rad, Hercules, CA), and membranes were probed overnight with 1:250 mouse anti-myc 9E10 and 1:50,000 rabbit anti-Cdc11 (Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were washed three times with PBS/0.1% Tween-20 and probed with 1:7500 goat anti-mouse IRdye800 (Rockland Immunochemicals, Gilbertsville, PA) or 1:7500 goat anti-rabbit Alexa Fluor 680 (Invitrogen, Carlsbad, CA) for 1 h in blocking buffer containing 0.1% Tween 20 and 0.01% SDS. Blots were then washed as above and visualized using an Odyssey scanner (Li-Cor Biosciences, Lincoln, NE). Fluorescence was quantified using Odyssey software (Li-Cor Biosciences), and the intensities of the myc bands were normalized to the Cdc11 loading controls.
Supplementary Material
Acknowledgments
We thank Nicolas Buchler, Sam Johnson, and the Duke Light Microscopy facility for help with the microscopy. This work was supported by NIH grant GM53050 to D.J.L.
Abbreviations used:
- CDK
cyclin-dependent kinase
- GFP
green fluorescent protein
- Lat
latrunculin
- PBS
phosphate-buffered saline
- UTR
untranslated region
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-11-0812) on February 6, 2013.
REFERENCES
- Anastasia SD, Nguyen DL, Thai V, Meloy M, MacDonough T, Kellogg DR. A link between mitotic entry and membrane growth suggests a novel model for cell size control. J Cell Biol. 2012;197:89–104. doi: 10.1083/jcb.201108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Park JE, Sakchaisri K, Yu LR, Song S, Supavilai P, Veenstra TD, Lee KS. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. EMBO J. 2005;24:2194–2204. doi: 10.1038/sj.emboj.7600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough KR, Stryker J, Pokala N, Sanders M, Crews P, Drubin DG. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J Cell Biol. 1997;137:399–416. doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly E, Cabantous S, Sondaz D, Bernadac A, Simon MN. Differential cellular localization among mitotic cyclins from Saccharomyces cerevisiae: a new role for the axial budding protein Bud3 in targeting Clb2 to the mother-bud neck. J Cell Sci. 2003;116:4119–4130. doi: 10.1242/jcs.00706. [DOI] [PubMed] [Google Scholar]
- Barral Y, Parra M, Bidlingmaier S, Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13:176–187. doi: 10.1101/gad.13.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Deshaies RJ, Kirschner MW. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993;259:1760–1763. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- Chen H, Howell AS, Robeson A, Lew DJ. Dynamics of septin ring and collar formation in Saccharomyces cerevisiae. Biol Chem. 2011;392:689–697. doi: 10.1515/BC.2011.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Shulewitz MJ, McDonald KL, Thorner J. Dynamic localization of the Swe1 regulator Hsl7 during the Saccharomyces cerevisiae cell cycle. Mol Biol Cell. 2001;12:1645–1669. doi: 10.1091/mbc.12.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clotet J, Escote X, Adrover MA, Yaakov G, Gari E, Aldea M, de Nadal E, Posas F. Phosphorylation of Hsl1 by Hog1 leads to a G(2) arrest essential for cell survival at high osmolarity. EMBO J. 2006;25:2338–2346. doi: 10.1038/sj.emboj.7601095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutchley J, King KM, Keaton MA, Szkotnicki L, Orlando DA, Zyla TR, Bardes ES, Lew DJ. Molecular dissection of the checkpoint kinase Hsl1p. Mol Biol Cell. 2009;20:1926–1936. doi: 10.1091/mbc.E08-08-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy WG. The decision to enter mitosis. Trends Cell Biol. 1994;4:202–207. doi: 10.1016/0962-8924(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Fang X, Luo J, Nishihama R, Wloka C, Dravis C, Travaglia M, Iwase M, Vallen EA, Bi E. Biphasic targeting and cleavage furrow ingression directed by the tail of a myosin II. J Cell Biol. 2010;191:1333–1350. doi: 10.1083/jcb.201005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrigno P, Posas F, Koepp D, Saito H, Silver PA. Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin β homologs NMD5 and XPO1. EMBO J. 1998;17:5606–5614. doi: 10.1093/emboj/17.19.5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford SK, Pringle JR. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC11 gene product and the timing of events at the budding site. Dev Genet. 1991;12:281–292. doi: 10.1002/dvg.1020120405. [DOI] [PubMed] [Google Scholar]
- Gladfelter AS, Pringle JR, Lew DJ. The septin cortex at the yeast mother-bud neck. Curr Opin Microbiol. 2001;4:681–689. doi: 10.1016/s1369-5274(01)00269-7. [DOI] [PubMed] [Google Scholar]
- Haarer BK, Pringle JR. Immunofluorescence localization of the Saccharomyces cerevisiae CDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol Cell Biol. 1987;7:3678–3687. doi: 10.1128/mcb.7.10.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JC, Bardes ES, Ohya Y, Lew DJ. A role for the Pkc1p/Mpk1p kinase cascade in the morphogenesis checkpoint. Nat Cell Biol. 2001;3:417–420. doi: 10.1038/35070104. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Keaton M, Szkotnicki L, Marquitz AR, Harrison JC, Zyla T, Lew DJ. Nucleocytoplasmic shuttling of G2/M regulators in yeast. Mol Biol Cell. 2008;19:4006–4018. doi: 10.1091/mbc.E08-03-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaton MA, Bardes ES, Marquitz AR, Freel CD, Zyla TR, Rudolph J, Lew DJ. Differential susceptibility of yeast S and M phase CDK complexes to inhibitory tyrosine phosphorylation. Curr Biol. 2007;17:1181–1189. doi: 10.1016/j.cub.2007.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaton MA, Lew DJ. Eavesdropping on the cytoskeleton: progress and controversy in the yeast morphogenesis checkpoint. Curr Opin Microbiol. 2006;9:540–546. doi: 10.1016/j.mib.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Kim HB, Haarer BK, Pringle JR. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC3 gene product and the timing of events at the budding site. J Cell Biol. 1991;112:535–544. doi: 10.1083/jcb.112.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K, Jin M, Lew D. The roles of Hsl1p and Hsl7p in Swe1p degradation: beyond septin-tethering. Eukaryot Cell. 2012;11:1496–1502. doi: 10.1128/EC.00196-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ. The morphogenesis checkpoint: how yeast cells watch their figures. Curr Opin Cell Biol. 2003;15:648–653. doi: 10.1016/j.ceb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J Cell Biol. 1995;129:739–749. doi: 10.1083/jcb.129.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, III, DeMarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Longtine MS, Theesfeld CL, McMillan JN, Weaver E, Pringle JR, Lew DJ. Septin-dependent assembly of a cell-cycle-regulatory module in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:4049–4061. doi: 10.1128/mcb.20.11.4049-4061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis SS, Perry JA, Weitzel DH, Freel CD, Yoshida M, Haystead TA, Kornbluth S. A role for PP1 in the Cdc2/cyclin B-mediated positive feedback activation of Cdc25. Mol Biol Cell. 2006;17:1779–1789. doi: 10.1091/mbc.E05-08-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Longtine MS, Sia RAL, Theesfeld CL, Bardes ESG, Pringle JR, Lew DJ. The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol Cell Biol. 1999a;19:6929–6939. doi: 10.1128/mcb.19.10.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Bardes ESG, Lew DJ. Phosphorylation-independent inhibition of Cdc28p by the tyrosine kinase Swe1p in the morphogenesis checkpoint. Mol Cell Biol. 1999b;19:5981–5990. doi: 10.1128/mcb.19.9.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Lew DJ. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol. 1998;142:1487–1499. doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Theesfeld CL, Harrison JC, Bardes ES, Lew DJ. Determinants of Swe1p degradation in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:3560–3575. doi: 10.1091/mbc.E02-05-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty JJ, Lew DJ. Swe1p responds to cytoskeletal perturbation, not bud size, in S. cerevisiae. Curr Biol. 2005;15:2190–2198. doi: 10.1016/j.cub.2005.11.039. [DOI] [PubMed] [Google Scholar]
- Morgan DO. The Cell Cycle: Principles of Control. Sunderland, MA: New Science Press/Sinauer; 2007. [Google Scholar]
- Peng C-Y, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- Raspelli E, Cassani C, Lucchini G, Fraschini R. Budding yeast Dma1 and Dma2 participate in regulation of Swe1 levels and localization. Mol Biol Cell. 2011;22:2185–2197. doi: 10.1091/mbc.E11-02-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson HE, Wittenberg C, Cross F, Reed SI. An essential G1 function for cyclin-like proteins in yeast. Cell. 1989;59:1127–1133. doi: 10.1016/0092-8674(89)90768-x. [DOI] [PubMed] [Google Scholar]
- Russell P, Moreno S, Reed SI. Conservation of mitotic controls in fission and budding yeast. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- Sakchaisri K, Asano S, Yu LR, Shulewitz MJ, Park CJ, Park JE, Cho YW, Veenstra TD, Thorner J, Lee KS. Coupling morphogenesis to mitotic entry. Proc Natl Acad Sci USA. 2004;101:4124–4129. doi: 10.1073/pnas.0400641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulewitz MJ, Inouye CJ, Thorner J. Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7123–7137. doi: 10.1128/mcb.19.10.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia RAL, Bardes ESG, Lew DJ. Control of Swe1p degradation by the morphogenesis checkpoint. EMBO J. 1998;17:6678–6688. doi: 10.1093/emboj/17.22.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia RAL, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell. 1996;7:1657–1666. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Grenfell TZ, Garfield S, Erikson RL, Lee KS. Essential function of the polo box of Cdc5 in subcellular localization and induction of cytokinetic structures. Mol Cell Biol. 2000;20:286–298. doi: 10.1128/mcb.20.1.286-298.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theesfeld CL, Zyla TR, Bardes EG, Lew DJ. A monitor for bud emergence in the yeast morphogenesis checkpoint. Mol Biol Cell. 2003;14:3280–3291. doi: 10.1091/mbc.E03-03-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.