

Centromeres are epigenetically defined by CENP-A nucleosomes. SNAP tagging is used to determine the composition of the heritable centromeric chromatin core. Assembly during G1 and stable maintenance at centromeres are restricted to CENP-A and H4. The CATD is the protein domain of CENP-A that is responsible for both features.
Abstract
Centromeres are the site of kinetochore formation during mitosis. Centromere protein A (CENP-A), the centromere-specific histone H3 variant, is essential for the epigenetic maintenance of centromere position. Previously we showed that newly synthesized CENP-A is targeted to centromeres exclusively during early G1 phase and is subsequently maintained across mitotic divisions. Using SNAP-based fluorescent pulse labeling, we now demonstrate that cell cycle–restricted chromatin assembly at centromeres is unique to CENP-A nucleosomes and does not involve assembly of other H3 variants. Strikingly, stable retention is restricted to the CENP-A/H4 core of the nucleosome, which we find to outlast general chromatin across several cell divisions. We further show that cell cycle timing of CENP-A assembly is independent of centromeric DNA sequences and instead is mediated by the CENP-A targeting domain. Unexpectedly, this domain also induces stable transmission of centromeric nucleosomes, independent of the CENP-A deposition factor HJURP. This demonstrates that intrinsic properties of the CENP-A protein direct its cell cycle–restricted assembly and induces quantitative mitotic transmission of the CENP-A/H4 nucleosome core, ensuring long-term stability and epigenetic maintenance of centromere position.
INTRODUCTION
Centromeres are the chromosomal loci for kinetochore formation during mitosis and thus form the site of interaction between DNA and the mitotic spindle (Cheeseman and Desai, 2008). As a result, centromeres are essential for proper chromosome segregation and prevention of aneuploidy. Although human centromeres are usually assembled on α-satellite (alphoid) DNA, specific sequences are neither necessary nor sufficient to stably maintain a centromere. Evidence for this comes primarily from the existence of neocentromeres, in which a specific centromere has repositioned to, and is stably maintained upon, a naive locus that differs in DNA sequence context and is not normally associated with centromere activity (Voullaire et al., 1993; Amor et al., 2004; Marshall et al., 2008). This has led to the proposal that centromeres are specified in a sequence-independent, epigenetic manner.
Whereas the vast majority of genomic DNA is packed by the canonical histones (H2A, H2B, H3.1, and H4), specific histone H3 variants package subsets of the genome. Among these, the H3.3 variant is mainly found at sites of active transcription (Ahmad and Henikoff, 2002), whereas centromere protein A (CENP-A) replaces H3.1 specifically in centromeric nucleosomes (Yoda et al., 2000; Foltz et al., 2006) and is required for the localization of nearly all other centromeric proteins (Foltz et al., 2006; Liu et al., 2006). Consistent with a role in epigenetic maintenance of centromere identity, CENP-A is a stable component of centromeric chromatin (Pearson et al., 2004; Schuh et al., 2007; Hemmerich et al., 2008) and is transmitted at centromeres during successive cell divisions (Jansen et al., 2007). In addition, it was recently shown in Drosophila S2 cells that targeting of CENP-ACID to ectopic loci for a short period of time is sufficient to initiate a sustainable epigenetic feedback loop, which recruits and maintains functional kinetochores for several subsequent cell division cycles (Mendiburo et al., 2011). Taken together, these findings strongly suggest that CENP-A plays a key role in epigenetic memory of centromere position and function.
Consistent with a critical role in centromere specification, assembly of CENP-A is tightly regulated and restricted to a specific stage in the cell cycle in order to maintain proper CENP-A levels. In metazoans, assembly of CENP-A is uncoupled from the S phase and depends on passage through mitosis (Jansen et al., 2007; Schuh et al., 2007; Bernad et al., 2011; Mellone et al., 2011; Moree et al., 2011). We previously showed that G1 phase–restricted assembly of CENP-A in human and chicken cells is directly coupled to cell cycle progression as a result of inhibitory action of Cdk1 and Cdk2 in S phase, G2, and mitosis (Silva et al., 2012). Although we have a basic understanding of the mechanism of cell cycle coupling of centromeric chromatin assembly, how this assembly is restricted to centromeres and how CENP-A chromatin is stably maintained are unclear.
In this study we determine whether centromeric chromatin assembly during G1 represents a general phase of nucleosome turnover or is a unique feature of CENP-A nucleosomes. In addition, we determine whether the previously reported stable maintenance of CENP-A (Jansen et al., 2007) is a feature of centromeric chromatin in general or is an intrinsic property of CENP-A–containing nucleosomes or even subnucleosomal complexes thereof. Using SNAP-tag–based fluorescent pulse labeling (Jansen et al., 2007; Bodor et al., 2012; Silva et al., 2012), we made the striking finding that CENP-A nucleosome assembly is the major form of nascent chromatin assembly in G1. This results in the formation of nucleosomes with a uniquely high in vivo stability of the CENP-A/H4 nucleosome core, a property induced in-cis by residues encoded by the CENP-A protein.
RESULTS
G1-phase histone assembly is restricted to CENP-A and H4
We previously used SNAP labeling to demonstrate that incorporation of nascent CENP-A is restricted to a brief window during early G1 phase (Jansen et al., 2007). SNAP is a self-labeling suicide enzyme that covalently and irreversibly reacts with benzylguanine or (fluorescent) derivatives thereof (Keppler et al., 2003, 2004). Sequential SNAP labeling steps allow for differential analysis of protein pools synthesized at distinct periods of time (Bodor et al., 2012). Timing of CENP-A assembly can be a consequence of an intrinsic property of this particular protein or result from a general wave of histone exchange at centromeres during G1. To determine whether a G1 assembly pathway exists for other histones, we used cells stably expressing SNAP-tagged versions of a variety of histone proteins. These include two other histone H3 family members—the canonical H3.1 and the replacement variant H3.3—as well as H4, the direct binding partner of all H3 variants, and H2B, a member of the more dynamic H2A/H2B histone subcomplex (Kimura and Cook, 2001). Direct pulse labeling of the total steady-state pool of SNAP-tagged histone showed signal in all cells, as expected (Supplemental Figure S1, A and B). To determine the pattern of assembly of nascent histones, we performed SNAP-based quench-chase-pulse experiments (Figure 1A; Bodor et al., 2012). To visualize stable chromatin assembly of nascent protein, we preextracted cells before fixation and imaging (Ray-Gallet et al., 2011). As anticipated, due to cell cycle–regulated assembly, nascent CENP-A–SNAP is found at centromeres in only a subset of cells (Figure 1B; Jansen et al., 2007). Similarly, nascent H3.1-SNAP is found in a subset of the population (Figure 1B), owing to its strict replication-coupled assembly (Ray-Gallet et al., 2011). Of interest, distinct subnuclear patterns of H3.1-SNAP staining can be observed, indicative of differential patterns of DNA synthesis throughout S phase (Figure 1B; Ray-Gallet et al., 2011). These results emphasize the power of SNAP-based pulse-chase assays, as they reveal strikingly different patterns of localization of the same protein synthesized and deposited into chromatin at different times during the cell cycle. Our H3.1-SNAP cell line therefore provides a powerful and accessible tool for marking S-phase progression without the need for an inducible expression system. In contrast, H3.3 (Ray-Gallet et al., 2002, 2011) and H2B (Kimura and Cook, 2001) are assembled throughout the cell cycle, and, consequently, nascent protein can be observed in all cells analyzed (Figure 1B).
FIGURE 1:

H4, but not H3.1, H3.3, or H2B, is coassembled with CENP-A in G1 phase. (A) Outline of quench-chase-pulse labeling strategy, allowing visualization of a newly synthesized pool of SNAP, followed by Triton-based preextraction. (B) Results of A for indicated histone–SNAP fusion proteins. Enlargement to the right shows rescaled images to indicate colocalization of newly synthesized H4-SNAP with centromeres (marked by CENP-C). Enlargements below show single–focal plane images to indicate specific subnuclear assembly patterns. Blue, green, and red arrows show G1, early S, and mid/late S phase cells, respectively. (C) Outline of quench-chase-pulse experiment on synchronized cells. (D) Results of C for SNAP-tagged histone proteins. CENP-C staining indicates centromere positions. Enlargement shows colocalization of newly synthesized H4-SNAP with centromeres.
It is intriguing that nascent H4-SNAP reveals a unique differential pattern of assembly, different from all other histone proteins analyzed. Whereas all cells display assembly throughout chromatin, consistent with a role as partner of H3.1 in S phase or H3.3 throughout the cell cycle, preferential assembly at discrete foci is observed in a subset of cells (Figure 1B). This pool of nascent H4 specifically colocalizes with centromeres, as marked by CENP-C (Figure 1B, enlargement), suggesting that histone H4 has a distinct phase of centromeric assembly.
CENP-A and H4 are coassembled during G1 phase
Prenucleosomal CENP-A forms a complex with H4 and HJURP, the CENP-A–specific histone chaperone (Dunleavy et al., 2009; Foltz et al., 2009; Shuaib et al., 2010; Hu et al., 2011). In addition, the CENP-A/H4 interface forms a highly rigid structure in nucleosomes (Black et al., 2007a), as well as in prenucleosomal (CENP-A/H4)2 tetramers (Black et al., 2004) and CENP-A/H4/HJURP trimers (Bassett et al., 2012). Thus, we reasoned that centromere-specific assembly of H4 results from coassembly with CENP-A during G1 phase in vivo. To test this directly, we labeled nascent pools of SNAP-tagged CENP-A, H3.1, H3.3, H2B, and H4 in cells synchronized in G2 phase of the cell cycle and analyzed assembly in the subsequent G1 phase (Figure 1C). Only CENP-A and H4-SNAP are assembled at centromere foci, indicating that centromeric assembly of H4 is contemporaneous with CENP-A (Figure 1D).
Of importance, however, whereas nascent CENP-A–SNAP and H4-SNAP colocalize at centromeres, newly synthesized H3.1-, H3.3-, and H2B-SNAP remain diffusely localized (Figure 1D). This indicates that these histones are not preferentially assembled at centromeres at this stage. This does not exclude the possibility that H2B is part of the centromeric nucleosome or that any of these histones are incorporated into centromeric chromatin at this time, albeit at a rate that is similar to the genome overall. This result, however, does demonstrate that the centromere is not a specialized chromatin domain that undergoes major nucleosome turnover events during G1 phase. Instead, CENP-A and H4 form a subnucleosomal core, which is specifically assembled at centromeres during G1 phase.
To validate that centromeric enrichment of H4 is not a consequence of the SNAP labeling procedure, we created a polyclonal HeLa cell line expressing H4–yellow fluorescent protein (YFP). Whereas endogenous pools of H4 are oscillating along the cell cycle, peaking in S phase (Marzluff and Duronio, 2002), the YFP-tagged H4 transgene, like our SNAP-tagged H4, is expressed at a constitutive level. Consequently, the relative levels of tagged versus endogenous H4 are higher in G1 phase than in S phase. For this reason we expect that tagged H4 can be detected at centromeres, despite genome-wide assembly in S phase, even without pulse-chase labeling. Indeed, when cells express low levels of H4-YFP, centromeric enrichment of this fusion protein can be detected over general chromatin (Supplemental Figure S1C), corroborating our observations with the SNAP tag.
Next we determined whether centromeric H4 assembly depends on G1-phase entry. For this, we labeled nascent proteins either in an asynchronous population of cells or in cells that were prevented from exiting mitosis by addition of nocodazole (Figure 2A). As expected, after an 8-h synthesis period, both CENP-A–SNAP and H4-SNAP readily assembled at centromeres in a subset of unperturbed cells. None of these cells stained positive for cyclin B (Figure 2, B and C), indicating that no centromere assembly occurred in late S, G2, or M phase. Consistent with this, virtually no cells assembled CENP-A or H4 at centromeres when entry into G1 was prevented by addition of nocodazole in asynchronous cells (Figure 2, B and C) or in a G2-synchronized population (Supplemental Figure S2, E and F). However, nocodazole treatment or consequent mitotic arrest does not irreversibly prevent assembly, as release into G1 by nocodazole washout promptly resulted in centromere targeting of CENP-A–SNAP and H4-SNAP, exclusively in cyclin B–negative cells (Figure 2, B and C). Analysis of cells synchronized at different stages along the cell cycle confirm that enrichment of H4-SNAP at centromere foci is only observed when cells carrying a nascent labeled pool of H4 cycle through G1 phase (Supplemental Figure S2), indicating that assembly of this histone at centromeres is uniquely restricted to G1 phase. We conclude that CENP-A and H4 assemble contemporaneously in a manner dependent on mitotic exit. In addition, because G1 assembly of H4 is largely restricted to centromeres, our data strongly suggest that other forms of nascent nucleosome assembly (i.e., H3.1/H4, H3.2/H4, or H3.3/H4 nucleosomes) throughout the rest of the genome represent a minority of assembly at this stage of the cell cycle. Thus, although it represents at most only ∼2% the total number of all nucleosomes (Black et al., 2007b), CENP-A nucleosome deposition represents the major form of chromatin assembly in G1.
FIGURE 2:
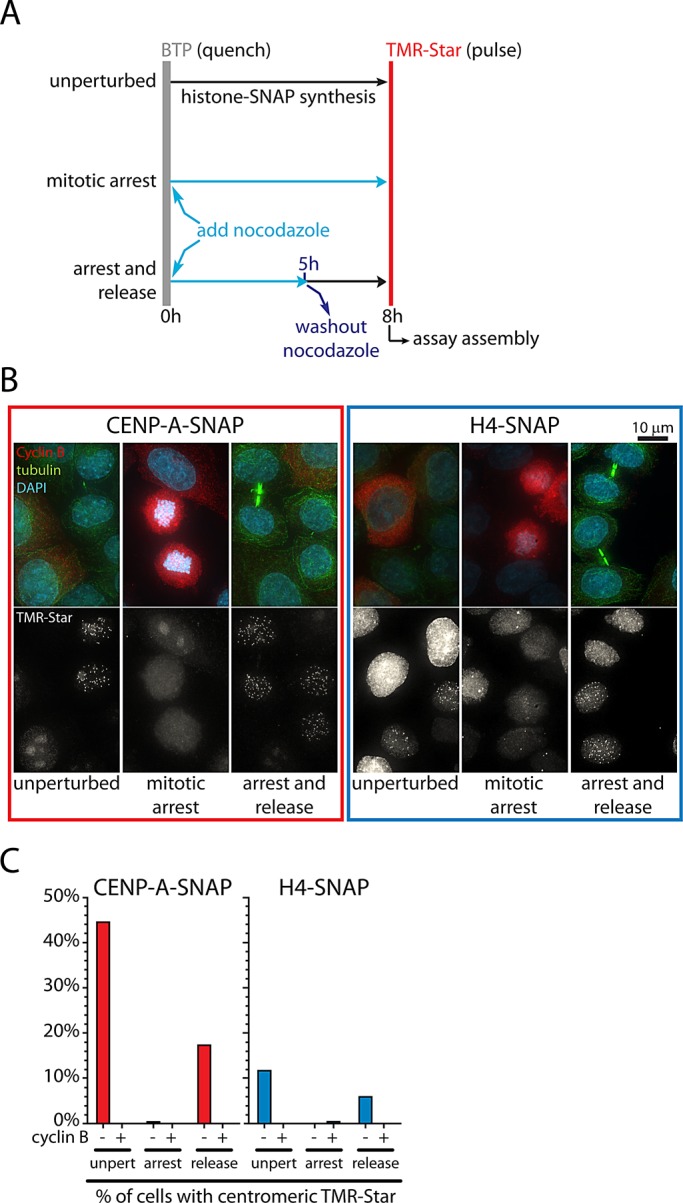
Assembly of CENP-A and H4 depends on passage through mitosis. (A) Outline of quench-chase-pulse experiment in unperturbed cells, combined with nocodazole treatment, or with nocodazole treatment and washout. (B) Results of A for CENP-A–SNAP and H4-SNAP. Cyclin B and tubulin staining indicate G2 and G1 (midbodies) status, respectively. (C) Quantification of D. Approximately 200–300 cells were analyzed for each condition. Note that during the 8-h chase, cells transit through ∼40% of an ∼22 h cell cycle, indicating the maximum expected percentage of cells entering G1 phase.
Taken together, these results strongly suggest that CENP-A and H4 represent the centromeric nucleosome core, which is assembled as a preformed complex during early G1 phase by the CENP-A–loading machinery. The absence of foci of nascent H3.1, H3.3, and H2B indicates that these proteins are not preferentially assembled at centromeres, arguing against general chromatin reorganization during G1 phase.
Quantitative retention of the centromeric nucleosome core
Once incorporated into centromeric chromatin, CENP-A is stably transmitted as cells divide (Jansen et al., 2007) and diluted among nascent sister chromatids during S phase (Dunleavy et al., 2011). To test whether this is also true for other histones at the centromere, we performed pulse-chase experiments of SNAP-tagged proteins (Figure 3A). SNAP-based fluorescent pulse labeling followed by a chase period determines the turnover rate of the labeled protein pool in vivo (Jansen et al., 2007; Bodor et al., 2012). It is remarkable that both CENP-A and H4 retain centromeric enrichment for the duration of the experiment (72 h; Figure 3B) and can still be observed at even longer time scales (up to 120 h for CENP-A and 96 h for H4; Supplemental Figure S3A). To determine the relative stability of centromere enriched histones, we quantified centromeric and noncentromeric TMR-Star fluorescence intensity as a measure of the amount of protein remaining at different time points (Figure 3C; see also Materials and Methods). Strikingly, we find that centromeric pools of CENP-A and H4 are considerably more stable than H3.1 (Figure 3D). Moreover, whereas H3.1 turnover is indifferent to centromere localization, the centromeric pool of H4 has an increased stability compared with H4 outside of the centromere (Figure 3D), indicating that CENP-A/H4–containing nucleosomes are preferentially stabilized compared with general chromatin.
FIGURE 3:

CENP-A and H4 are preferentially maintained at centromeres. (A) Outline of pulse-chase experiment allowing for analysis of a preincorporated pool of SNAP for up to 72 h. At each time point, cells were counted to allow accurate quantification of SNAP turnover per cell division. (B) Results of A for CENP-A–SNAP, H4-SNAP, and H3.1-SNAP. Enlargements show rescaled images of remaining protein pool after 72 h (see also Supplemental Figure S3A). (C) Schematic outline for calculation of histone half-life in D and E (see Materials and Methods). (D) Half-life measurements of centromeric and noncentromeric histone pools as a function of time from experiment in B. Noncentromeric CENP-A is below detection and therefore not measured. Data are obtained from between 570 and 1464 (centromeric) foci for each time point.
Similar to H3.1, no specific stability of H2B or H3.3 was observed at centromeres (Supplemental Figure S3B). This indicates that H2A/H2B dimers exchange on centromeric nucleosomes at similarly high rates as on conventional nucleosomes in bulk chromatin. Moreover, considering that intervening H3.1 and H3.3 nucleosomes are present at centromeres (Blower et al., 2002; Dunleavy et al., 2011), we find that long-term retention of chromatin is restricted to the CENP-A/H4 core of CENP-A nucleosomes, with H3.1/H3.3 nucleosomes turning over at higher rates.
Timing of assembly and stable retention of the centromeric nucleosome core are controlled by the CENP-A–targeting domain
Although centromeres are maintained epigenetically, the unusual properties of CENP-A nucleosomes we uncovered may be dependent on local sequence features at centromeres. Alternatively, timing of centromere assembly and stable retention of CENP-A nucleosomes could be directed in-cis by CENP-A itself.
The CENP-A–targeting domain (CATD), encompassing the L1 loop and α2 helix of the CENP-A histone fold domain, plays a pivotal role in the definition of centromeric chromatin. Replacement of the corresponding domain of canonical H3 with the CATD of CENP-A is sufficient to target the chimeric H3CATD to both canonical centromeres (Black et al., 2004, 2007b) and neocentromeres (Bassett et al., 2010). Furthermore, binding of prenucleosomal CENP-A to its histone chaperone HJURP is mediated through the CATD (Foltz et al., 2009; Shuaib et al., 2010; Bassett et al., 2012). HJURP is itself recruited to centromeric chromatin in early G1 (Dunleavy et al., 2009; Foltz et al., 2009).
We decided to test directly whether, in addition to regulating centromeric targeting, the CATD is sufficient to dictate the timing of histone assembly. When labeling a nascent pool of stably expressed H3CATD-SNAP we detected centromeric H3CATD only in cyclin B–negative cells (Figure 4, A and B; unperturbed), suggesting that cells only load H3CATD into centromeres during G1 phase. In addition, as for CENP-A and H4 (Figure 2A), prevention of mitotic exit by nocodazole treatment abolished centromeric assembly of H3CATD-SNAP, whereas release from this induced arrest resulted in mitotic exit and concomitant centromeric assembly (Figure 4, A and B). We conclude that, apart from centromere localization, the CATD also mediates cell cycle control of CENP-A assembly.
FIGURE 4:
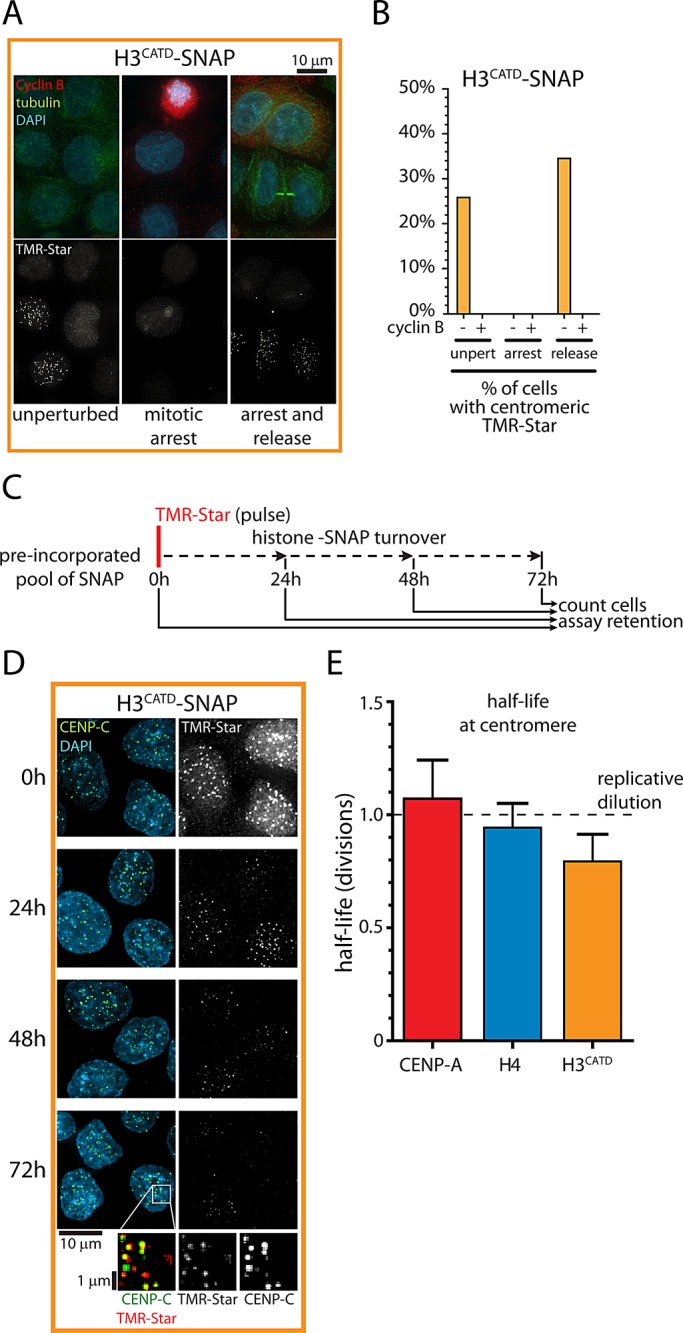
CATD determines G1 phase assembly and stable transmission of CENP-A nucleosomes. (A) Results of experiment as outlined in Figure 2A for H3CATD-SNAP. Cyclin B and tubulin staining indicate G2 and G1 (midbodies) status, respectively. (B) Quantification of A. Approximately 200–300 cells were analyzed for each condition. Note that during the 8-h chase, cells transit through ∼40% of an ∼22 h cell cycle, indicating the maximum expected percentage of cells entering G1 phase. (C) Outline of pulse-chase experiment analogous to experiment in Figure 3. At each time point, cells were counted to allow accurate quantification of SNAP turnover per cell division. (D) Results of C. Enlargements show rescaled images of remaining protein pool after 72 h (see also Supplemental Figure S3A). (E) Determination of centromeric histone half-life as a function of population doublings from experiments shown in Figures 3B and 4D. Dashed line indicates expected values for proteins that are never lost but merely redistributed as cells divide. Average and SEM of three independent experiments.
Next we determined whether long-term retention of CENP-A nucleosomes at centromeres is also an intrinsic property of CENP-A. We carried out pulse-chase experiments on H3CATD-SNAP–expressing cells (Figure 4C) and analyzed retention of H3CATD over time. As for CENP-A and H4, pulse-labeled H3CATD-SNAP remains detectable for multiple cell divisions up to 120 h after labeling (Figure 4D and Supplemental Figure S3A).To compare the stability of centromeric histones, we determined their rate of turnover as a function of the number of cell divisions expressed as the half-life (Figure 4E and see Materials and Methods). In an extreme case in which histones do not turn over at all, loss of histone proteins would be expected to occur only by redistribution among newly replicated sister chromatids during S phase (replicative dilution). In this situation, we would find a 50% reduction of fluorescence after each cell division (i.e., a histone half-life of exactly 1 division; Figure 4E, dashed line). For CENP-A–SNAP (experiment in Figure 3), we observed a half-life of 1.07 ± 0.17 divisions (mean ± SEM; Figure 4E), consistent with turnover by replicative dilution only. Of importance, we observed very similar behavior for both H4-SNAP and H3CATD-SNAP at centromeres, with half-lives of 0.94 ± 0.11 and 0.79 ± 0.12 divisions, respectively (Figure 4E). None of these values is significantly different from a theoretical replicative dilution rate of 1 cell division (one-tailed, one-sample t test; n = 3, α = 0.05 in all cases).
Although the CATD has been implicated in rigidifying the CENP-A/H4 interface within the nucleosome particle (Black et al., 2004, 2007a; Sekulic et al., 2010; Bassett et al., 2012), how this contributes to CENP-A stability in vivo remained untested. Our results now show that CENP-A confers long-term stability to the centromeric (CENP-A/H4)2 subnucleosome core and that this in vivo stability is encoded within the residues that constitute the CENP-A–targeting domain. This feature of CENP-A ensures stable chromatin marking of centromeres across multiple divisions.
Quantitative retention of CENP-A does not require HJURP
We showed that quantitative retention of CENP-A is directed, at least in part, by the CATD. However, the mechanism by which the CATD contributes to CENP-A stability remains unclear. The most clearly defined function of the CATD is to provide the binding interface for the CENP-A chaperone HJURP (Foltz et al., 2009; Shuaib et al., 2010; Hu et al., 2011; Bassett et al., 2012). Of interest, a proportion of endogenous HJURP is stably chromatin bound (Foltz et al., 2006). This raises the possibility that HJURP binding to CENP-A protects it from turning over, that is, by binding to chromatin-incorporated CENP-A or by transiently chaperoning this histone during the transition from parental chromosomes to daughter chromatids during DNA replication. To test this hypothesis directly, we combined SNAP labeling experiments with RNA interference against HJURP, as detailed in Figure 5A.
FIGURE 5:

HJURP is dispensable for stable retention of CENP-A. (A) Outline of quench-chase-pulse and pulse-chase experiment combined with siRNA-mediated protein depletion. Note that quench-chase-pulse and pulse-chase experiments were done in parallel to minimize variation of RNAi efficiency. (B) Results of A after depletion of GAPDH (control), HJURP, or CENP-A. Images are displayed for nascent CENP-A-SNAP and the preincorporated pool 24 h after target protein depletion. (C) Quantification of centromeric TMR-Star fluorescence after depletion of GAPDH (white), HJURP (light gray), or CENP-A (dark gray) for indicated protein pools of CENP-A–SNAP. Results were normalized against control RNAi. Average and SEM for at least three independent experiments. Asterisks and NS, respectively, indicate statistically significant (p < 0.01) and nonsignificant (p > 0.05) differences from control samples in paired t tests.
As expected, nascent centromeric CENP-A–SNAP was readily observed in all cells after siRNA-mediated depletion of a control protein (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]; Figure 5B). However, a large proportion of cells were unable to assemble nascent CENP-A–SNAP after depletion of HJURP (Figure 5B), as was observed previously (Foltz et al., 2009). This result is consistent with the known role of HJURP in the assembly of CENP-A during G1 (Barnhart et al., 2011). Quantification of centromeric signals after HJURP depletion shows that nascent CENP-A–SNAP levels are reduced by ∼50% (Figure 5C). Similar results were obtained when RNA interference (RNAi) was performed against CENP-A itself (Figure 5, B and C; Bodor et al., 2012).
To test whether HJURP is also involved in stabilizing previously incorporated CENP-A nucleosomes, we combined pulse-chase experiments with RNAi. Retention of CENP-A–SNAP at centromeres was analyzed after target protein depletion for 24, 48, and 72 h to allow for assessment of both short- and long-term effects on CENP-A stability. In this assay, centromeric CENP-A–SNAP could be observed in all cells analyzed (Figure 5B), and no quantitative differences were observed between control RNAi or depletion of HJURP or CENP-A (Figure 5C) at any time point. To ensure that we used conditions that effectively reduce protein levels, we performed these pulse-chase experiments in parallel with the quench-chase-pulse experiments described earlier (Figure 5A). Our results strongly suggest that HJURP is dispensable for stabilizing centromeric CENP-A nucleosomes. We thus conclude that the long-term stability of the CENP-A/H4 nucleosome core is due to an HJURP-independent role of the CATD.
Timing of centromeric nucleosome assembly is independent of alphoid DNA
We have shown that the CATD of CENP-A is sufficient to direct G1 phase–restricted assembly of CENP-A chromatin, suggesting that temporal loading is dictated by the CENP-A protein itself. However, this does not exclude a role for involvement of local sequence context in regulating cell cycle timing. Mammalian centromeres are assembled on arrays of α satellite DNA. Although overall centromere function is not strictly dependent on this DNA sequence, it may play a role in regulating centromere assembly and maintenance. This is clear from efforts to produce centromeres de novo on artificial chromosomes. Although in some systems de novo centromeres can be formed on any DNA (Yuen et al., 2011), success in mammalian cells has only been reported with constructs containing large fragments of alphoid DNA (Ohzeki et al., 2002). In addition, the inner centromere component Aurora B was found to be mislocalized at a stably maintained human non–alphoid-containing neocentromere, resulting in an impaired mitotic error correction mechanism (Bassett et al., 2010). Thus, although neocentromeres can exist on non–alphoid DNA, the role of DNA sequences in maintenance of existing centromeres remains elusive.
To determine the contribution of cis DNA elements in alphoid sequences on the timing of CENP-A assembly, we stably expressed CENP-A-SNAP in pseudodicentric-neocentric chromosome 4 (PD-NC4) cells. In these cells, the centromere on the paternally inherited chromosome 4 (but not the maternal one) is repositioned to chromosomal position 4q21.3, which does not contain alphoid DNA sequences (Figure 6A; Amor et al., 2004). By combining quench-chase-pulse experiments with fluorescent in situ hybridization (FISH) against 4q21.3 (NeoCEN4), we were able to determine that CENP-A assembly at neocentromeres occurred contemporaneously with canonical centromeres of the same cell (Figure 6, B and C). Of importance, although a subset of cells displayed diffuse nucleolar staining, indicative of the prenucleosomal pool of CENP-A in G2 phase (Jansen et al., 2007; Silva et al., 2012), CENP-A assembly was never observed at the NeoCEN4 alone, that is, when no assembly occurred on other centromeres (Figure 6D). To corroborate these results, we stably expressed a green fluorescent protein (GFP)–tagged version of Mis18α, an essential component of the Mis18 complex, in PD-NC4 cells. Of interest, one member of this complex, M18BP1, contains a Myb domain (Fujita et al., 2007; Maddox et al., 2007), a protein domain that is often involved in site-specific DNA binding (Lipsick, 1996). Nevertheless, GFP-Mis18α is consistently recruited to NeoCEN4 and alphoid DNA–bearing centromeres simultaneously (Figure 6E). Taken together, these results show that CENP-A assembly at the NeoCEN4 occurs concurrently with canonical centromeres, indicating that temporal control of the CENP-A assembly machinery is maintained independently of alphoid DNA. This is consistent with a dominant role for the CENP-A–encoded CATD in directly controlling temporal assembly of CENP-A chromatin.
FIGURE 6:

Timing of CENP-A assembly is maintained at neocentromeres. (A) Cartoon of maternal (canonical centromere) and paternal (neocentric) chromosome 4 in PD-NC4 cells. Indicated is chromosomal position 4q21.3, the site of neocentromere formation and the hybridization site of the FISH probe used. (B) Outline of quench-chase-pulse experiment in CENP-A–SNAP–expressing PD-NC4 cells. (C, D) Results of B for cells in G1 phase (C) or G2 phase (D), as indicated by nucleolar TMR staining, shown in rescaled inset. CENP-T indicates centromere positions. Enlargements display images of the hybridization sites of the FISH probe. Green arrows indicate the neocentromere, and red arrows show the homologous region on the maternal chromosome. (E) GFP-Mis18α–expressing PD-NC4 cells were stained for GFP and for 4q21.3 by FISH to detect Mis18α and the NeoCEN4, respectively. Enlargements as described above. Paternal (neocentric) and maternal 4q21.3 positions are indicated by p and m, respectively, in C–E.
DISCUSSION
Maintenance of epigenetic identity requires the inheritance of structural information from one cell generation to the next. Chromatin proteins and their modifications have been implicated in such cellular memory (Talbert and Henikoff, 2010; Gardner et al., 2011). However, transmission of chromatin-based information faces many challenges throughout the cell cycle that may disturb epigenetic inheritance, including nucleosome disruption during DNA replication and chromatin (de)condensation during mitosis. Previous work identified an atypical timing of assembly of CENP-A, as well as centromere retention of the existing pool of CENP-A throughout the cell cycle (Jansen et al., 2007). We now extend these findings and determine that a distinct phase of centromeric loading in G1, as well as quantitative centromeric retention, is restricted to CENP-A and H4, rather than being a general property of centromeric chromatin. Metabolic labeling experiments and photobleaching studies of GFP-tagged histones previously established that histones H3 and H4 are stable components, whereas H2A and H2B are more dynamic (Kimura and Cook, 2001; Xu et al., 2010). However, apart from CENP-A itself (Jansen et al., 2007), locus-specific assembly and turnover was not previously determined for these or other histones. Our results now show that at the centromere, CENP-A/H4 forms a stable subnucleosomal core that is quantitatively retained throughout multiple cell divisions to maintain centromere identity (Figure 7). Retention of H4 specifically at the centromere, but not elsewhere, indicates that the centromeric CENP-A/H4 species is more stable than general chromatin, outlasting most, if not all, other nucleosome types.
FIGURE 7:

Model depicting unique features of centromeric nucleosomes. Cell cycle dynamics of different types of nucleosomes are indicated. CENP-A nucleosomes are assembled at centromeres in G1 phase, whereas H3.1 and H3.3 nucleosomes are assembled into general chromatin in S phase and throughout the cell cycle, respectively (Ray-Gallet et al., 2011). Neither H3.1 nor H3.3 nucleosomes are preferentially loaded into centromeric chromatin during G1 phase or any other cell cycle stage. Whereas H2A and H2B are dynamic in all types of nucleosomes, the centromeric CENP-A/H4 core is stable at time scales far surpassing the cell division rate. However, H3.1, H3.3, and noncentromeric H4 turn over more rapidly than CENP-A, and no preferential centromeric maintenance of H3.1 or H3.3 is observed. Key to both temporal assembly and stable transmission is the CATD domain of CENP-A, which forms a stable interface with H4 in both CENP-A and H3CATD nucleosomes (Sekulic et al., 2010; Bassett et al., 2012).
Of interest, many of the unique features of the CENP-A/H4 centromeric core are directed through the CATD region of CENP-A. It has been shown that this region is responsible for 1) targeting of CENP-A to centromeres (Black et al., 2004, 2007b) in a sequence-independent manner (Bassett et al., 2010); 2) binding to the CENP-A–specific histone chaperone HJURP (Foltz et al., 2009; Shuaib et al., 2010; Bassett et al., 2012); 3) a unique, highly rigid CENP-A/H4 dimerization interface (Black et al., 2004; Sekulic et al., 2010; Bassett et al., 2012); and 4) binding of CENP-N, which in turn is required for efficient centromeric recruitment of nascent CENP-A (Carroll et al., 2009). In addition, we now show that 5) CATD is the element in CENP-A that mediates correct timing of CENP-A assembly, independent of underlying DNA sequence, and 6) critically, this region confers in vivo hyperstability to centromeric nucleosomes in a manner independent of HJURP binding. Of importance, parts of CENP-A outside of the CATD region have been shown to be required for kinetochore assembly, for example through binding of the centromere protein CENP-C to the six most carboxy-terminal residues of CENP-A (Carroll et al., 2010; Guse et al., 2011). Thus, although different domains of CENP-A are likely to be involved in full centromere function, all of the key properties of CENP-A for epigenetically maintaining centromere position are mediated through the CATD.
Our results identify the CENP-A/H4 complex as the primary component of the centromere that is selectively assembled each cell division in a manner that leads to its long-term maintenance. A key future challenge is to determine whether this unusual stability is an intrinsic property of CENP-A nucleosomes or depends on external factors that ensure stable transmission of CENP-A and centromere identity.
MATERIALS AND METHODS
Constructs and cell lines
Human H2B-SNAP, H4-SNAP, and H3CATD-SNAP constructs were created by PCR cloning of histone open reading frames (ORFs) into pSS26m (Covalys/New England Biolabs, Ipswich, MA) to create C-terminal SNAP fusion proteins. A triple hemagglutinin (3XHA) tag was placed at the C-terminus of SNAP. Histone H4–yellow fluorescent protein (YFP) was generated by PCR cloning of the human H4 ORF into pEYFP-N1 (Clontech, Mountain View, CA) carrying Q69M (citrin) and A206K (monomerization) mutations. The histone-SNAP-3XHA and H4-YFP ORFs were subcloned into pBABE-Blast to generate retroviral expression constructs. These constructs were delivered into HeLa cells via Moloney murine leukemia retroviral delivery, as described previously (Morgenstern and Land, 1990; Burns et al., 1993). Cells stably expressing the SNAP fusion proteins were selected with 5 μg/ml blasticidin S (Calbiochem, La Jolla, CA) and were isolated and individually sorted by flow cytometry (except H4-YFP, which was analyzed as a polyclonal cell population). The resulting monoclonal lines were selected for proper levels of the SNAP fusion proteins by fluorescence microscopy after TMR-Star labeling. The following clones were selected and used throughout this study: H2B-SNAP clone #5; H4-SNAP clone #3; and H3CATD-SNAP clone #37. We previously described HeLa monoclonal cell lines stably expressing H3.1-SNAP or H3.3-SNAP (clone #7 or #2, respectively; Ray-Gallet et al., 2011) or CENP‑A-SNAP (clone #23; Jansen et al., 2007). All HeLa cell lines were grown at 37°C and 5% CO2 in DMEM containing 10% newborn calf serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (henceforth referred to as complete medium). In addition, SNAP-expressing cells were maintained by addition of 1 μg/ml blasticidin S. PD-NC4 stable transgenic cell lines were created by Moloney murine leukemia retroviral delivery of constructs expressing CENP–A-SNAP (Jansen et al., 2007) or GFP-Mis18α (gift from Iain Cheeseman [Whitehead Institute]; Silva et al., 2012). PD-NC4 cells were grown at 37°C and 5% CO2 in DMEM supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 μg/ml neomycin,100 U/ml penicillin, and 100 μg/ml streptomycin. Stable transgenic PD-NC4 lines were selected with 2.5 μg/ml blasticidin (CENP–A-SNAP) or 500 ng/ml puromycin (GFP-Mis18α).
SNAP labeling
SNAP labeling was performed essentially as described (Jansen et al., 2007; Bodor et al., 2012). Briefly, cells were labeled for 30 min with 2 μM BTP (SNAP-Cell Block; New England Biolabs) or 15 min with 2μM TMR-Star (New England Biolabs) in complete medium for quench or pulse labeling, respectively, after which cells were washed twice with phosphate-buffered saline (PBS) and reincubated with complete medium. After an additional 30 min, cells were washed once more with PBS and either reincubated with complete medium or fixed and further treated for analysis, as indicated.
Cell synchronization and RNAi
Cells were synchronized in early S phase by double-thymidine block as described previously (Jansen et al., 2007; Bodor et al., 2012). Nocodazole was used at a concentration of 500 ng/ml except for the experiment in Supplemental Figure S2F, for which 200 ng/ml was used.
RNAi was performed in a 24-well format with 60 pmol of small interfering RNAs (siRNAs) using Oligofectamine (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. All siRNAs were obtained from Dharmacon (Lafayette, CO): SMARTpools were used to deplete HJURP and GAPDH; for CENP-A depletion, siRNA target 5′-ACAGUCGGCGGAGACAAGG-3′ was used.
Immunofluorescence
Fixation, immunofluorescence, and 4′,6-diamidino-2-phenylindole (DAPI) staining of HeLa cells was performed as described (Bodor et al., 2012). Preextraction was performed for 5 min using 0.3% Triton X-100 (Sigma-Aldrich, St. Louis, MO) in PBS before fixation. Antibodies against CENP-C (mouse monoclonal), cyclin B (sc-245; Santa Cruz Biotechnology, Santa Cruz, CA), and α-tubulin (YL1/2; AbD Serotec, Raleigh, NC) were used at dilutions of 1:10,000, 1:50, and 1:2500, respectively. Fluorescent secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA) and used at a dilution of 1:200.
Immuno-FISH
FISH was performed as previously described (Black et al., 2007a), with the following alterations: On cell fixation and the freeze/thaw cycles, cells were prepared for immunofluorescence as described. GFP-Mis18α was detected by immunofluorescence, as GFP signal is lost during the FISH fixation procedure. GFP-Booster (ChromoTek, Martinsried, Germany), CENP-T (Barnhart et al., 2011) and anti-rabbit Dy680 (Rockland Immunochemicals, Gilbertsville, PA) were used at dilutions of 1:100, 1:1000, and 1:50, respectively. Subsequently, cells were fixed with 2% formaldehyde for 10 min at room temperature and washed with PBS. FISH protocol was then continued as described (Black et al., 2007a). A chromosome 4q21.3-specific probe was generated by labeling a mixture of BAC clones (RP11-113G13, RP11-204I22, RP11-209G6, RP11-458J15; BACPAC Resources Center, Oakland, CA) with either tetramethyl-rhodamine-5-dUTP or fluorescein-12-dUTP (Roche, Indianapolis, IN) to detect colocalization with GFP-Mis18α or with CENP-A–SNAP, respectively. Coverslips were washed in 2× SSC (0.3 M NaCl, 30 mM sodium citrate, pH 7.0), containing 60% formamide before DAPI staining and mounting.
Microscopy
Cells were imaged on a DeltaVision Core system (Applied Precision, Issaquah, WA) controlling an inverted microscope (IX-71; Olympus, Tokyo, Japan), which is coupled to a Cascade II electron-multiplying charge-coupled device camera (Photometrics, Tucson, AZ). Images were collected at 1× binning using a 100× oil objective (numerical aperture 1.40, UPlanSApo) with 0.2-mm Z-sections scanning the entire nucleus. Images were subsequently deconvolved using softWoRx (Applied Precision). Unless otherwise indicated, maximum-intensity projections of deconvolved images are shown. Centromere quantification was performed using a custom-made macro for ImageJ (National Institutes of Health, Bethesda, MD), called CRaQ (Bodor et al., 2012). For quantitative purposes, images were collected on a 512 × 512 pixel chip and flat field and camera noise corrected during acquisition using softWoRx. Fluorescence quantification was performed on nondeconvolved images. For centromere quantification, CRaQ was set to measure peak intensity values within a 7 × 7 pixel box around the centroid position of the centromere. For noncentromeric values (Figure 3D), a 2 × 2 pixel box was placed at a position shifted away from the centromere centroid by 5 pixels in both x and y. In Figure 3D, to enable the measurement of diffuse nuclear signals, fluorescence immediately outside nuclei was used for background correction. For Figure 4E, centromeric fluorescence was corrected for local background for each centromere. To quantify the rate of division of SNAP-tagged cells, we seeded one additional coverslip of CENP-A–SNAP cells for each time point and treated it identically to the other cells throughout the duration of the experiment (TMR-Star and BTP were omitted, and cells were mock treated with dimethyl sulfoxide instead). At the time of fixation, the extra coverslip was trypsinized, and cells were counted in a hemocytometer. To calculate histone half-life, we measured fluorescence intensities as a function of number of cell divisions at 24, 48, and 72 h. From this, we calculated the best-fit one–phase decay regression line (F = e−kt, where F is fluorescence and t is time or number of divisions) using Prism software (GraphPad Software, La Jolla, CA) with a constrained plateau at 0 and F0 = 1. Half-life equals ln(2)/k (Figure 3C).
Supplementary Material
Acknowledgments
We are indebted to Don W. Cleveland, who hosted preliminary experiments in his laboratory. We thank Mariluz Gómez Rodríguez for help with the immunofluorescence-FISH procedure and Nuno Moreno for help with image quantification. D.L.B. and L.P.V. are supported by Fundação para a Ciência e a Tecnologia Fellowships SFRH/BD/74284/2010 and SFRH/BPD/69115/2010, respectively. This work is supported by National Institutes of Health Grant GM082989, a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund, and a Rita Allen Foundation Scholar Award to B.E.B. and by Fundação Calouste Gulbenkian and Fundação para a Ciência e a Tecnologia Grants BIA-BCM/100557/2008 BIAPRO/100537/2008, the European Commission FP7 Program, and an EMBO Installation Grant to L.E.T.J.
Abbreviations used:
- CATD
CENP-A targeting domain
- CENP-A
centromere protein A
- CRaQ
centromere recognition and quantification macro
- DAPI
4′,6-diamidino-2-phenylindole
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- NeoCEN4
neocentromere of chromosome 4 in PD-NC4 cells
- PD-NC4
pseudodicentric-neocentric chromosome 4
- RNAi
RNA interference
- siRNA
small interfering RNA
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-01-0034) on January 30, 2013.
REFERENCES
- Ahmad K, Henikoff S. Histone H3 variants specify modes of chromatin assembly. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16477–16484. doi: 10.1073/pnas.172403699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor DJ, Bentley K, Ryan J, Perry J, Wong L, Slater H, Choo KHA. Human centromere repositioning “in progress.”. Proc Natl Acad Sci USA. 2004;101:6542–6547. doi: 10.1073/pnas.0308637101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart MC, Kuich PHJL, Stellfox ME, Ward JA, Bassett EA, Black BE, Foltz DR. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J Cell Biol. 2011;194:229–243. doi: 10.1083/jcb.201012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett EA, DeNizio J, Barnhart-Dailey MC, Panchenko T, Sekulic N, Rogers DJ, Foltz DR, Black BE. HJURP uses distinct CENP-A surfaces to recognize and to stabilize CENP-A/histone H4 for centromere assembly. Dev Cell. 2012;22:749–762. doi: 10.1016/j.devcel.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett EA, Wood S, Salimian KJ, Ajith S, Foltz DR, Black BE. Epigenetic centromere specification directs Aurora B accumulation but is insufficient to efficiently correct mitotic errors. J Cell Biol. 2010;190:177–185. doi: 10.1083/jcb.201001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernad R, et al. Xenopus HJURP and condensin II are required for CENP-A assembly. J Cell Biol. 2011;192:569–582. doi: 10.1083/jcb.201005136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Brock MA, Bédard S, Woods VL, Cleveland DW. An epigenetic mark generated by the incorporation of CENP-A into centromeric nucleosomes. Proc Natl Acad Sci USA. 2007a;104:5008–5013. doi: 10.1073/pnas.0700390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Foltz DR, Chakravarthy S, Luger K, Woods VL, Cleveland DW. Structural determinants for generating centromeric chromatin. Nature. 2004;430:578–582. doi: 10.1038/nature02766. [DOI] [PubMed] [Google Scholar]
- Black BE, Jansen LET, Maddox PS, Foltz DR, Desai AB, Shah JV, Cleveland DW. Centromere identity maintained by nucleosomes assembled with histone H3 containing the CENP-A targeting domain. Mol Cell. 2007b;25:309–322. doi: 10.1016/j.molcel.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Blower MD, Sullivan BA, Karpen GH. Conserved organization of centromeric chromatin in flies and humans. Dev Cell. 2002;2:319–330. doi: 10.1016/s1534-5807(02)00135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor DL, Rodríguez MG, Moreno N, Jansen LET. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr Protoc Cell Biol. 2012 doi: 10.1002/0471143030.cb0808s55. Chapter 8: Unit 8.8. [DOI] [PubMed] [Google Scholar]
- Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci USA. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF. Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol. 2010;189:1143–1155. doi: 10.1083/jcb.201001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Silva MCC, Godek KM, Jansen LET, Straight AF. Centromere assembly requires the direct recognition of CENP-A nucleosomes by CENP-N. Nat Cell Biol. 2009;11:896–902. doi: 10.1038/ncb1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol. 2008;9:33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- Dunleavy EM, Almouzni G, Karpen GH. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus. 2011;2:146–157. doi: 10.4161/nucl.2.2.15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell. 2009;137:485–497. doi: 10.1016/j.cell.2009.02.040. [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LET, Bailey AO, Yates JR, Bassett EA, Wood S, Black BE, Cleveland DW. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell. 2009;137:472–484. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LET, Black BE, Bailey AO, Yates JR, Cleveland DW. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol. 2006;8:458–469. doi: 10.1038/ncb1397. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Hayashi T, Kiyomitsu T, Toyoda Y, Kokubu A, Obuse C, Yanagida M. Priming of centromere for CENP-A recruitment by human hMis18α, hMis18β, and M18BP1. Dev Cell. 2007;12:17–30. doi: 10.1016/j.devcel.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Gardner KE, Allis CD, Strahl BD. OPERating ON chromatin, a colorful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature. 2011;477:354–358. doi: 10.1038/nature10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerich P, Weidtkamp-Peters S, Hoischen C, Schmiedeberg L, Erliandri I, Diekmann S. Dynamics of inner kinetochore assembly and maintenance in living cells. J Cell Biol. 2008;180:1101–1114. doi: 10.1083/jcb.200710052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, et al. Structure of a CENP-A-histone H4 heterodimer in complex with chaperone HJURP. Genes Dev. 2011;25:901–906. doi: 10.1101/gad.2045111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen LET, Black BE, Foltz DR, Cleveland DW. Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol. 2007;176:795–805. doi: 10.1083/jcb.200701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc Natl Acad Sci USA. 2004;101:9955–9959. doi: 10.1073/pnas.0401923101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Cook PR. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2b. J Cell Biol. 2001;153:1341–1354. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsick JS. One billion years of Myb. Oncogene. 1996;13:223–235. [PubMed] [Google Scholar]
- Liu S-T, Rattner JB, Jablonski SA, Yen TJ. Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J Cell Biol. 2006;175:41–53. doi: 10.1083/jcb.200606020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox PS, Hyndman F, Monen J, Oegema K, Desai A. Functional genomics identifies a Myb domain-containing protein family required for assembly of CENP-A chromatin. J Cell Biol. 2007;176:757–763. doi: 10.1083/jcb.200701065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall OJ, Chueh AC, Wong LH, Choo KHA. Neocentromeres: new insights into centromere structure, disease development, and karyotype evolution. Am J Hum Genet. 2008;82:261–282. doi: 10.1016/j.ajhg.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, Duronio RJ. Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol. 2002;14:692–699. doi: 10.1016/s0955-0674(02)00387-3. [DOI] [PubMed] [Google Scholar]
- Mellone BG, Grive KJ, Shteyn V, Bowers SR, Oderberg I, Karpen GH. Assembly of Drosophila centromeric chromatin proteins during mitosis. PLoS Genet. 2011;7:e1002068. doi: 10.1371/journal.pgen.1002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiburo MJ, Padeken J, Fülöp S, Schepers A, Heun P. Drosophila CENH3 is sufficient for centromere formation. Science. 2011;334:686–690. doi: 10.1126/science.1206880. [DOI] [PubMed] [Google Scholar]
- Moree B, Meyer CB, Fuller CJ, Straight AF. CENP-C recruits M18BP1 to centromeres to promote CENP-A chromatin assembly. J Cell Biol. 2011;194:855–871. doi: 10.1083/jcb.201106079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki J, Nakano M, Okada T, Masumoto H. CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. J Cell Biol. 2002;159:765–775. doi: 10.1083/jcb.200207112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson CG, Yeh E, Gardner M, Odde D, Salmon ED, Bloom K. Stable kinetochore-microtubule attachment constrains centromere positioning in metaphase. Curr Biol. 2004;14:1962–1967. doi: 10.1016/j.cub.2004.09.086. [DOI] [PubMed] [Google Scholar]
- Ray-Gallet D, Quivy J-P, Scamps C, Martini EM-D, Lipinski M, Almouzni G. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol Cell. 2002;9:1091–1100. doi: 10.1016/s1097-2765(02)00526-9. [DOI] [PubMed] [Google Scholar]
- Ray-Gallet D, et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell. 2011;44:928–941. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Schuh M, Lehner CF, Heidmann S. Incorporation of Drosophila CID/CENP-A and CENP-C into centromeres during early embryonic anaphase. Curr Biol. 2007;17:237–243. doi: 10.1016/j.cub.2006.11.051. [DOI] [PubMed] [Google Scholar]
- Sekulic N, Bassett EA, Rogers DJ, Black BE. The structure of (CENP-A-H4)(2) reveals physical features that mark centromeres. Nature. 2010;467:347–351. doi: 10.1038/nature09323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuaib M, Ouararhni K, Dimitrov S, Hamiche A. HJURP binds CENP-A via a highly conserved N-terminal domain and mediates its deposition at centromeres. Proc Natl Acad Sci USA. 2010;107:1349–1354. doi: 10.1073/pnas.0913709107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MCC, Bodor DL, Stellfox ME, Martins NMC, Hochegger H, Foltz DR, Jansen LET. Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev Cell. 2012;22:52–63. doi: 10.1016/j.devcel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. Histone variants—ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- Voullaire LE, Slater HR, Petrovic V, Choo KH. A functional marker centromere with no detectable alpha-satellite, satellite III, or CENP-B protein: activation of a latent centromere. Am J Hum Genet. 1993;52:1153–1163. [PMC free article] [PubMed] [Google Scholar]
- Xu M, Long C, Chen X, Huang C, Chen S, Zhu B. Partitioning of histone H3-H4 tetramers during DNA replication–dependent chromatin assembly. Science. 2010;328:94–98. doi: 10.1126/science.1178994. [DOI] [PubMed] [Google Scholar]
- Yoda K, Ando S, Morishita S, Houmura K, Hashimoto K, Takeyasu K, Okazaki T. Human centromere protein A (CENP-A) can replace histone H3 in nucleosome reconstitution in vitro. Proc Natl Acad Sci USA. 2000;97:7266–7271. doi: 10.1073/pnas.130189697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen KWY, Nabeshima K, Oegema K, Desai A. Rapid de novo centromere formation occurs independently of heterochromatin protein 1 in C. elegans embryos. Curr Biol. 2011;21:1800–1807. doi: 10.1016/j.cub.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.