

This paper describes a ternary protein complex consisting of junctional adhesion molecule-A (JAM-A), tetraspanin CD9, and αvβ3 integrin in endothelial cells. In this complex, CD9 links JAM-A to αvβ3 integrin to regulate basic fibroblast growth factor–specific mitogen-activated protein kinase activation, endothelial cell migration, and tube formation. Our findings contribute to a better understanding of the signaling events during angiogenesis.
Abstract
Junctional adhesion molecule-A (JAM-A) is a member of the immunoglobulin family with diverse functions in epithelial cells, including cell migration, cell contact maturation, and tight junction formation. In endothelial cells, JAM-A has been implicated in basic fibroblast growth factor (bFGF)-regulated angiogenesis through incompletely understood mechanisms. In this paper, we identify tetraspanin CD9 as novel binding partner for JAM-A in endothelial cells. CD9 acts as scaffold and assembles a ternary JAM-A-CD9-αvβ3 integrin complex from which JAM-A is released upon bFGF stimulation. CD9 interacts predominantly with monomeric JAM-A, which suggests that bFGF induces signaling by triggering JAM-A dimerization. Among the two vitronectin receptors, αvβ3 and αvβ5 integrin, which have been shown to cooperate during angiogenic signaling with bFGF and vascular endothelial growth factor (VEGF), respectively, CD9 links JAM-A specifically to αvβ3 integrin. In line with this, knockdown of CD9 blocks bFGF- but not VEGF-induced ERK1/2 activation. JAM-A or CD9 knockdown impairs endothelial cell migration and tube formation. Our findings indicate that CD9 incorporates monomeric JAM-A into a complex with αvβ3 integrin, which responds to bFGF stimulation by JAM-A release to regulate mitogen-activated protein kinase (MAPK) activation, endothelial cell migration, and angiogenesis. The data also provide new mechanistic insights into the cooperativity between bFGF and αvβ3 integrin during angiogenic signaling.
INTRODUCTION
Junctional adhesion molecule-A (JAM-A) is the founding member of the JAM family of immunoglobulin (Ig)-like proteins (Bazzoni, 2003; Ebnet et al., 2004). Originally identified on the surface of human platelets, JAM-A is expressed by many different cell types, including epithelial and endothelial cells, leukocytes, Sertoli cells and spermatozoa, macroglia cells in the brain, and smooth muscle cells. In epithelial cells, JAM-A regulates various processes, including cell migration, cell proliferation, the maturation of intercellular junctions, and the formation of barrier-forming tight junctions (Liu et al., 2000; Rehder et al., 2006; Severson et al., 2009; Nava et al., 2011; Iden et al., 2012). Some of these functions of JAM-A depend on its ability to associate with cytoplasmic proteins through its C-terminal PDZ domain–binding motif. Among those proteins are the scaffolding proteins ZO-1, MUPP1, PAR-3, and AF-6/afadin (Bazzoni et al., 2000; Ebnet et al., 2000, 2001; Itoh et al., 2001; Hamazaki et al., 2002). As one example, during cell–cell contact formation, JAM-A recruits the cell polarity protein PAR-3 to primordial, spot-like adherens junctions (pAJs) to promote the formation of an active PAR-3–aPKC–PAR-6 complex that regulates the development of pAJs into mature cell junctions with barrier-forming tight junctions (Ebnet et al., 2001; Itoh et al., 2001; Rehder et al., 2006; Iden et al., 2012). The physiological relevance of this function is indicated by the loss of the intestinal barrier in JAM-A–deficient mice (Laukoetter et al., 2007; Vetrano et al., 2008).
In endothelial cells, JAM-A has been intensively analyzed in view of its role in regulating leukocyte–endothelial cell interaction during inflammation (Weber et al., 2007). During inflammation, JAM-A redistributes away from cell–cell junctions and colocalizes with integrin αLβ2 in ring-like structures at the junctional interface between transmigrating neutrophils and endothelial cells (Ostermann et al., 2002; Shaw et al., 2004). In addition, JAM-A has been found to regulate angiogenesis (Naik et al., 2003). Antibody blockade of JAM-A function or inactivation of the F11r gene in mice results in a blunted basic fibroblast growth factor (bFGF) response in sprouting assays (Naik et al., 2003; Cooke et al., 2006). This activity seems to be related to the ability of JAM-A to interact with integrin αvβ3 and to regulate the migration of endothelial cells on vitronectin (Naik et al., 2003; Naik and Naik, 2006).
Tetraspanins are a large family of evolutionarily conserved cell surface membrane proteins expressed in a wide variety of cell types (Hemler, 2005; Charrin et al., 2009). They are characterized by four transmembrane domains and two extracellular loops (Levy and Shoham, 2005b). One of the most distinctive functional features of tetraspanins is probably their ability to interact with a large number of other proteins present in the same membrane, including other tetraspanins, through different tetraspanin domains (Hemler, 2005; Yanez-Mo et al., 2009). As one example, the tetraspanin CD81 interacts with the cytoplasmic domain of the Ig-superfamily (IgSF) protein EWI-2 (Stipp et al., 2001), with the extracellular domain of the IgSF protein CD19 (Bradbury et al., 1993), and with the transmembrane domain of the same or other tetraspanins (Charrin et al., 2009). The ability of tetraspanins to undergo multiple cis-interactions through different domains enables tetraspanins to bring different proteins into close proximity and to form networks of interactions at the cell surface. Not surprisingly, tetraspanins are involved in a variety of biological processes, such as cell fusion and aggregation, cell adhesion and migration, antigen presentation, and viral infection (Hemler, 2005; Charrin et al., 2009; Yanez-Mo et al., 2009).
CD9 (TSPAN29) is among the best-studied tetraspanins and is expressed in a wide variety of cell types in which it interacts with many different proteins, including integrins, IgSF members, proteoglycans, claudins, and others (Yanez-Mo et al., 2009). In endothelial cells, the role of CD9 is incompletely understood. As in other cell types, CD9 most likely has different functions depending on its subcellular localization and on the type of associated proteins. For example, upon tumor necrosis factor-α (TNF-α) stimulation CD9 interacts with the IgSF member ICAM-1 at the apical membrane and, together with tetraspanins CD81 and CD151 and IgSF member VCAM-1, forms adhesive platforms for leukocytes (Barreiro et al., 2005, 2008). Through its association with α2β1 integrin, CD9 regulates proliferation arrest of endothelial cells (Cailleteau et al., 2010). The absence of CD9 impairs the formation of new blood vessels in various model systems, suggesting a role for CD9 in angiogenesis (Cailleteau et al., 2010; Kamisasanuki et al., 2011).
In this study, we identify CD9 as a new binding partner for JAM-A as well as for αvβ3 integrin. Our findings indicate that CD9 physically connects JAM-A to αvβ3 integrin to assemble a ternary complex that mediates bFGF-regulated mitogen-activated protein kinase (MAPK) activation, endothelial cell migration, and tube formation.
RESULTS
JAM-A interacts with tetraspanin CD9 by means of a primary tetraspanin interaction
In a yeast two-hybrid screen using the cytoplasmic domain of JAM-A as bait, we isolated two clones with an open reading frame representing amino acids Glu-82 to Val-203 of murine CD9 (Rubinstein et al., 1993). To confirm this association in cells, we performed coimmunoprecipitation (CoIP) experiments. Tetraspanins can interact with their nontetraspanin partners either through primary interactions (retained in strong detergents, such as Triton X-100 or NP-40) or through tertiary interactions that are mediated through other tetraspanins (retained only in the presence of mild detergents, such as Brij97; Boucheix and Rubinstein, 2001; Hemler, 2005). The interaction between JAM-A and CD9 was detectable both after Triton X-100 and Brij97 lysis (Figure 1A), indicating that the JAM-A–CD9 interaction is a primary tetraspanin interaction. CoIP experiments from transfected HEK293T cells confirmed a robust interaction between JAM-A and CD9 in cells (Figure 1B). Because tetraspanins can interact with their partners within so-called tetraspanin-enriched microdomains (TEMs) through both the extracellular and cytoplasmic regions, we next tested whether the interaction of JAM-A with CD9 is mediated by the cytoplasmic tail of JAM-A. The cytoplasmic tail of JAM-A fused to glutathione S-transferase (GST) was sufficient to pull down CD9 from HEK293T cells (Figure 1C). Deletion of three or nine C-terminal amino acids strongly impaired the association of GST-JAM-A with CD9 (Figure 1C). In agreement with this observation, Flag-JAM-A constructs with C-terminal deletions or triple-alanine mutations within the extreme C-terminus of the cytoplasmic tail failed to interact with CD9 in cells (Figure 1D). Together these observations indicate that JAM-A and CD9 interact in cells by way of a primary tetraspanin interaction that is mediated by the extreme C-terminus of the cytoplasmic domain of JAM-A.
FIGURE 1:

JAM-A interacts with CD9. (A) JAM-A interacts with CD9 by way of a primary tetraspanin interaction. Top and middle panels, HeLa cells were lysed in either Triton X-100– or Brij97-containing lysis buffer as indicated. JAM-A immunoprecipitates were immunoblotted for CD9 (90% of input) and for JAM-A (10% of input). Bottom panel, Immunoprecipitation was performed in the reverse order: CD9 was immunoprecipitated, and immunoprecipitates were immunoblotted for JAM-A (90% of input) or CD9 (10% of input). Note that JAM-A efficiently interacts with CD9 under both lysis conditions. (B) JAM-A strongly interacts with CD9 in HEK293T cells. CD9 immunoprecipitates obtained from Flag-JAM-A–transfected HEK293T cells were immunoblotted with anti-Flag antibodies (top, 90% of input) or anti-CD9 antibodies (middle, 10% of input). Postnuclear supernatants (PNS) were immunoblotted with anti-Flag antibodies (bottom, 2.5% of total lysate). The asterisk denotes signals resulting from IgG heavy chains. (C) CD9 interacts with the cytoplasmic tail of JAM-A. GST precipitates obtained from HEK293T cells with GST fusion proteins containing the full-length cytoplasmic tail of JAM-A (GST-JAM-A/f.l.) or deletion mutants lacking 3 or 9 C-terminal amino acid residues (GST-JAM-A/Δ3, GST-JAM-A/Δ9) were immunoblotted for CD9 (top panel). Equal loading of GST fusion proteins was verified by immunoblotting aliquots with anti-GST antibodies (bottom panel). Arrowheads indicate GST-JAM-A/f.l. constructs resulting from proteolytic cleavage. (D) The interaction between JAM-A and CD9 requires the PDZ domain-binding motif of JAM-A. CD9 immunoprecipitates obtained from HEK293T cells transfected with full-length JAM-A (Flag-JAM-A), C-terminal truncation mutants (Flag-JAM-A/Δ3, -/Δ6, -/Δ9), or triple alanine substitutions (Flag-JAM-A/3A1, Flag-JAM-A/3A2) were immunoblotted with anti-Flag antibodies (top, 90% of input), or with anti-CD9 antibodies (middle, 10% of input). Expression levels of Flag constructs were analyzed by immunoblotting the PNS with anti-Flag antibodies (bottom, 2.5% of total lysate). In addition, all Flag constructs localize to the cell surface as analyzed by flow cytometry (Supplemental Figure S5). Experiments shown in this figure are representative of at least three independent experiments. IP, immunoprecipitation; IB, immunoblotting.
CD9 interacts with JAM-A and αvβ3 integrin in endothelial cells to form a ternary JAM-A–CD9–αvβ3 integrin complex
Integrins and IgSF members are the most common binding partners for tetraspanins (Boucheix and Rubinstein, 2001; Levy and Shoham, 2005a). Because JAM-A had been found in a complex with αvβ3 integrin in endothelial cells (Naik and Naik, 2006), we hypothesized that CD9 might form a link between JAM-A and αvβ3 integrin. Immunoprecipitation experiments from various endothelial cell lines and from primary human umbilical vein endothelial cell (HUVEC) lysates indicated that CD9 and JAM-A exist in the same protein complex (Figure 2A and Supplemental Figure S1). Immunofluorescence analysis indicated that both CD9 and αvβ3 integrin colocalize with JAM-A at cell–cell contacts of cultured endothelial cells (Figure 2B). In addition, whole-mount isolectin B4 labelings of the retinal vasculature revealed that JAM-A and CD9 are coexpressed at the angiogenic front in areas of sprouting angiogenesis of the mouse retina at postnatal day 6 (Figure 2C), suggesting that the association of CD9 and JAM-A is relevant for angiogenic processes in vivo. We next analyzed the presence of αvβ3 integrin in the JAM-A–CD9 complex, and we found that αvβ3 integrin is associated with both JAM-A and CD9 (Figure 2D). The interaction of CD9 with both JAM-A and αvβ3 integrin suggested the existence of a ternary JAM-A–CD9–αvβ3 integrin complex but did not rule out the possibility of independent binary interactions among the three proteins. To address this question, we analyzed the association between JAM-A and αvβ3 integrin in CD9 knockdown cells. In the absence of CD9, JAM-A did not interact with αvβ3 integrin (Figure 2E), indicating that CD9 is required to link JAM-A to αvβ3 integrin. JAM-A did not interact with αvβ5 integrin (Figure 2F), indicating that CD9 links JAM-A specifically to αvβ3 integrin among the two vitronectin receptors expressed by endothelial cells. Together our observations identify JAM-A and αvβ3 as novel binding partners of CD9 in endothelial cells, and they point to the existence of a ternary JAM-A–CD9–αvβ3 integrin complex in which CD9 serves to link JAM-A to αvβ3 integrin.
FIGURE 2:

CD9 assembles a ternary complex by linking JAM-A to αvβ3 integrin in endothelial cells. (A) JAM-A interacts with CD9 in endothelial cells. JAM-A immunoprecipitates obtained from HUVECs were immunoblotted with mAbs against CD9 (top, 90% of input) or against JAM-A (bottom, 10% of input). (B) Both CD9 and αvβ3 integrin colocalize with JAM-A at endothelial cell–cell junctions. HUVECs were costained for JAM-A and CD9 (top panels) and for JAM-A and for αvβ3 integrin (bottom panels). Scale bars: 10 μm. (C) JAM-A and CD9 are coexpressed in the vasculature of the P6 mouse retina at the angiogenic front. Whole-mount preparations of a mouse retina were stained with antibodies against JAM-A (green) and CD9 (red), and with isolectin B4 (IB4, blue) to visualize endothelial cells. Scale bar: 24 μm. (D) JAM-A and CD9 interact with β3 integrin. Immunoprecipitates obtained from HUVECs with antibodies against JAM-A (top panel) or CD9 (bottom panel) were immunoblotted for β3 integrin. Specific IP was verified by immunoblotting 10% of the precipitated material with antibodies against the precipitated protein. The two bands present in the IP performed with control IgG in the bottom panel represent unspecific bands as they do not match the molecular weight of β3 integrin and as they did not appear in other IPs performed with the same IgG (see also Figure 3, A and B). (E) CD9 links JAM-A to αvβ3 integrin. JAM-A immunoprecipitates obtained from CD9 knockdown HUVECs were immunoblotted with anti-β3 integrin antibodies (top panel, 90% of input) or JAM-A antibodies (middle panel, 10% of input). Postnuclear supernatants were also blotted with CD9 antibodies to control for knockdown efficiency of CD9 (bottom panel, 2.5% of total PNS). Note that β3 integrin does not interact with JAM-A in the absence of CD9. (F) JAM-A does not interact with β5 integrins in HUVECs. JAM-A immunoprecipitates obtained from HUVECs were analyzed for the presence of β5 integrin (top, 90% of input) or JAM-A (bottom, 10% of input). All biochemical experiments are representative of three independent experiments.
The JAM-A–CD9–αvβ3 integrin complex is regulated by integrin activation and bFGF signaling
Integrins exist at the cell surface in an inactive, closed conformation, and binding of specific ligands stabilizes the active, open conformation (Shattil et al., 2010). In addition, integrins can laterally interact with tetraspanins through their extracellular domains (Yauch et al., 2000; Stipp et al., 2003; Nishiuchi et al., 2005). Because stimulation with Arg-Gly-Asp-Ser (RGDS) peptides has previously been shown to enhance the association of JAM-A with αvβ3 integrin (Naik and Naik, 2006), we analyzed whether the active conformation of αvβ3 integrin promotes the association of JAM-A with CD9 and/or the association of CD9 with αvβ3 integrin. Preincubation of HUVECs with RGDS peptide for 20 min increased the interaction of JAM-A with CD9 and with αvβ3 integrin, as well as the interaction of CD9 with αvβ3 integrin (Figure 3A), and conversely, knockdown of b3 integrin decreased the association between CD9 and JAM-A (Supplemental Figure S2). These observations indicate that ligand-mediated integrin activation promotes the formation of the ternary JAM-A–CD9–αvβ3 complex.
FIGURE 3:

Ternary complex formation is dependent on integrin activation and negatively regulated by bFGF. (A) Integrin activation promotes ternary complex formation. HUVECs were stimulated with RGDS peptide (100 μg/ml, 20 min). After lysis, JAM-A IPs were analyzed for the presence of CD9 (top, left panel), CD9 IPs were analyzed for the presence of β3 integrin (top, right panel), and β3 integrin IPs were analyzed for JAM-A (bottom, left panel). In all cases, equal and specific IP was verified by immunoblotting 10% of the precipitated material with antibodies against the precipitated protein. The asterisks denote unspecific bands derived from Ig light chains. Bottom, right panel, densitometric analysis of the binary interactions; y-axis: relative signal intensity. Densitometric values obtained from unstimulated cells (no RGDS) were arbitrarily set as 1. Error bars denote the mean ± SE from four separate experiments. Statistical significance was evaluated using one-sample t tests; *, p < 0.05. (B) bFGF dissociates JAM-A from the ternary complex. HUVECs were stimulated with bFGF (10 ng/ml, 10 min). After lysis, JAM-A IPs were analyzed for CD9 (top, left panel) or for β3 integrin (top, right panel), and CD9 IPs were analyzed for β3 integrin (bottom, left panel). In all cases, equal and specific IP was verified by immunoblotting 10% of the precipitated material with antibodies against the precipitated protein. The asterisks denote unspecific bands derived from Ig light chains. Bottom, right panel, densitometric analysis of JAM-A–CD9, JAM-A–β3 integrin and CD9–β3 integrin CoIPs; y-axis: relative signal intensity. Densitometric values obtained from unstimulated cells (no bFGF) were arbitrarily set as 1. Error bars denote the mean ± SE from three separate experiments. Statistical significance was evaluated using one-sample t tests; *, p < 0.05.
During angiogenesis, bFGF selectively cooperates with αvβ3 integrin to activate a specific Ras-Raf-ERK signaling pathway (Friedlander et al., 1995; Hood et al., 2003; Yan et al., 2008). Because JAM-A has been described to be involved in bFGF-mediated MAPK activation (Naik et al., 2003), we analyzed the composition of the ternary JAM-A–CD9–αvβ3 complex in the presence of bFGF. Incubation with bFGF for 10 min reduced the amount of JAM-A associated with CD9, as well as the amount of JAM-A associated with αvβ3 integrin (Figure 3B). The association of CD9 with αvβ3 integrin was not affected by bFGF (Figure 3B). These findings indicate that bFGF dissociates JAM-A from the ternary complex, leaving the binary CD9–αvβ3 integrin complex intact.
CD9 recruits predominantly monomeric JAM-A into the ternary complex
The bFGF-triggered release of JAM-A opened up the possibility that JAM-A linked to αvβ3 integrin serves to inhibit αvβ3 integrin–mediated MAPK activation under steady-state conditions that would be abrogated after bFGF-induced release of JAM-A. Alternatively, JAM-A could be actively involved in MAPK activation after its dissociation from the complex, possibly by assembling a cytoplasmic signaling complex (Severson et al., 2009). Studies in epithelial cells indicate that many functions of JAM-A, including its role in tight junction barrier formation and β1 integrin–mediated cell adhesion and migration, depend on JAM-A dimerization (Mandell et al., 2004; Rehder et al., 2006; Severson et al., 2008). To test whether the ternary JAM-A–CD9–αvβ3 complex contains monomeric or dimeric JAM-A, we analyzed the association of two dimerization-deficient JAM-A mutants (ΔV-JAM-A, which lacks the V-type Ig domain, and JAM-A/E60RK62E, which contains mutations at two residues in the V-type Ig domain that mediate cis-dimerization [ Prota et al., 2003]) with CD9. Both mutants interacted much more strongly with CD9 than with wild-type JAM-A (Figure 4), indicating that the ternary JAM-A–CD9–αvβ3 complex contains predominantly monomeric JAM-A.
FIGURE 4:
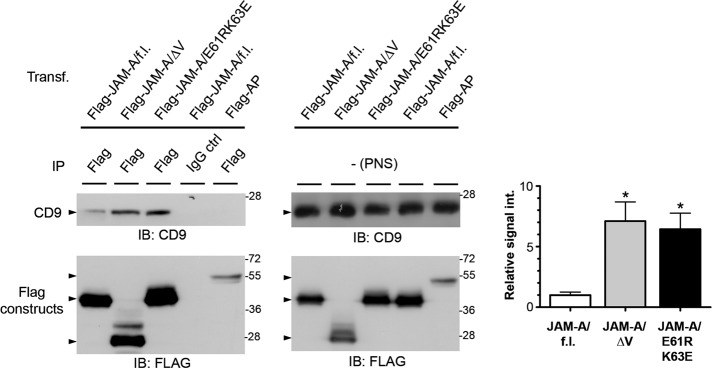
CD9 interacts preferentially with monomeric JAM-A. HEK293T cells were transfected with either full-length Flag-JAM-A (Flag-JAM-A/f.l.) or dimerization-defective JAM-A mutants (Flag-JAM-A/ΔV, Flag-JAM-A/E61RK63E); Flag-alkaline phosphatase (Flag-AP) served as negative control. Left panels, Flag constructs were immunoprecipitated with anti-Flag antibodies or IgG control antibodies as indicated, and the immunoprecipitates were immunoblotted with anti-CD9 antibodies (top, 90% of input) or anti-Flag antibodies (bottom, 10% of input). Middle panels, PNS from the samples used for IP were analyzed for the levels of CD9 (top) and of Flag constructs (bottom). Right panel, densitometric analysis of the amount of CD9 associated with JAM-A and JAM-A mutants; y-axis: relative signal intensity. Densitometric values obtained from the interaction of CD9 with wild-type JAM-A (JAM-A/f.l., left bar) was arbitrarily set as 1. Error bars denote the mean ± SE from four separate experiments. Statistical significance was evaluated using one-sample t tests; *, p < 0.05.
CD9 links JAM-A to αvβ3 integrin to assemble a protein complex that specifically mediates bFGF-induced MAPK activation
To test whether the JAM-A–CD9–αvβ3 integrin complex is required for bFGF to stimulate MAPK signaling, we analyzed bFGF-induced ERK1/2 activation in the absence of CD9. To distinguish between contributions of several integrins from those mediated by the two vitronectin receptors αvβ3 and αvβ5 integrin, we grew cells either on plastic or on vitronectin. In control cells, bFGF induced a strong ERK1/2 phosphorylation irrespective of whether cells were grown on plastic or on vitronectin (Figure 5A). CD9 knockdown cells showed a similarly strong bFGF response when grown on plastic. However, when grown on vitronectin, CD9 knockdown cells failed to respond to bFGF (Figure 5A). These observations indicate that CD9 is required for bFGF-induced ERK1/2 activation specifically when cells are costimulated by the two vitronectin receptors αvβ3 and αvβ5 integrin.
FIGURE 5:

Both CD9 and JAM-A specifically cooperate with bFGF in angiogenic signaling. (A) CD9 is required for ERK1/2 phosphorylation in cells grown on vitronectin. CD9 siRNA-treated HUVECs grown either on plastic or on vitronectin were stimulated with bFGF (10 ng/ml, 20 min) as indicated. Cell lysates were analyzed for total ERK1/2 and phosphorylated ERK1/2. Note that the absence of CD9 blocks bFGF-induced Erk1/2 phosphorylation only when cells are grown on vitronectin. (B) CD9 mediates bFGF- but not VEGF-induced ERK1/2 phosphorylation. CD9 siRNA-treated HUVECs were grown on plastic or on vitronectin and stimulated with bFGF (10 ng/ml, 10 min) or VEGF (20 ng/ml, 10 min) as indicated. Cell lysates were analyzed for total ERK1/2 and phosphorylated ERK1/2. Top, right panel, CD9 and α-tubulin (α-tub) levels in ctrl siRNA- and CD9 siRNA-transfected cells. Bottom, right panel, quantification of ERK1/2 phosphorylation; y-axis: relative signal intensity. Bars represent ERK1/2 phosphorylation in CD9 siRNA-treated cells relative to ERK1/2 phosphorylation in control siRNA-treated cells. Phosphorylation levels were quantified as detailed in the Materials and Methods section. Error bars denote the mean ± SE from three independent experiments. Statistical significance was evaluated using one-sample t tests; **, p < 0.01. (C) JAM-A mediates bFGF- but not VEGF-induced ERK1/2 phosphorylation. JAM-A siRNA-transfected HUVECs were grown on plastic or on vitronectin and stimulated with bFGF (10 ng/ml, 10 min) or VEGF (20 ng/ml, 10 min) as indicated. Cell lysates were analyzed for total ERK1/2 and phosphorylated ERK1/2. Top, right panel, JAM-A and α-tubulin (α-tub) levels in ctrl siRNA- and JAM-A siRNA-transfected cells. Bottom, right panel, quantification of ERK1/2 phosphorylation performed as described in (B). Error bars denote the mean ± SE from three independent experiments. Statistical significance was evaluated using one-sample t tests; *, p < 0.05.
Because previous observations indicated a functional and selective cooperation between bFGF and αvβ3 integrin and between vascular endothelial growth factor (VEGF) and αvβ5 integrin during angiogenesis (Friedlander et al., 1995; Hood et al., 2003; Yan et al., 2008), we next addressed the question of whether CD9 is specific for the bFGF–αvβ3 integrin pathway of angiogenesis. For testing this, CD9 knockdown cells were plated either on plastic or on vitronectin and stimulated with either bFGF or VEGF. Control cells responded to both bFGF and VEGF with increased ERK1/2 phosphorylation (Figure 5B, top panel) under both growth conditions. CD9 knockdown cells showed a strong response to both bFGF and VEGF when grown on plastic. As observed before (Figure 5A), CD9 knockdown cells failed to respond to bFGF when grown on vitronectin. However, their ability to respond to VEGF was unchanged (Figure 5B). These observations indicate that CD9 selectively participates in the bFGF–αvβ3 integrin–regulated pathway of MAPK activation. We then performed the same experiment in JAM-A knockdown cells. Control small interfering RNA (siRNA)- and JAM-A siRNA-treated cells grown on either plastic or vitronectin were stimulated with bFGF or VEGF (Figure 5C). Control siRNA-treated cells responded to both bFGF and VEGF with increased ERK1/2 phosphorylation irrespective of whether they were grown on plastic or on vitronectin (Figure 5C, top panel). JAM-A knockdown cells were able to respond to both growth factors with increased ERK1/2 phosphorylation when grown on plastic (Figure 5C, bottom panel). However, as observed for CD9 knockdown cells, when grown on vitronectin, JAM-A knockdown cells failed to respond to bFGF, whereas they responded normally to VEGF. Finally, knockdown of CD9 or JAM-A did not affect the ability of endothelial cells to respond to bFGF when grown on fibronectin or collagen (Supplemental Figure S4). These observations indicate that both CD9 and JAM-A are selectively involved in the bFGF–αvβ3 integrin pathway of MAPK activation and identify CD9 and JAM-A as upstream regulatory components of the bFGF–αvβ3 pathway of angiogenesis (Friedlander et al., 1995; Hood et al., 2003).
Down-regulation of CD9 or JAM-A impairs endothelial cell migration and tube formation
To address the relevance of the JAM-A–CD9–αvβ3 complex during processes linked to angiogenesis, we analyzed the ability of endothelial cells to migrate and to form tube-like structures in a three-dimensional matrix. Knockdown of either JAM-A or CD9 significantly reduced the ability of endothelial cells to migrate through a Matrigel-coated filter in response to bFGF (Figure 6A). In addition, knockdown of either JAM-A or CD9 significantly impaired the ability of endothelial cells to develop a branched network of tube-like structures when incubated for 24 h in the presence of bFGF in Matrigel (Figure 6B). Finally, knockdown of CD9 resulted in reduced endothelial cell proliferation (Supplemental Figure S3). These observations indicate that both JAM-A and CD9 regulate angiogenesis-related processes such as endothelial cell migration and tube formation. This function of JAM-A and CD9 is most likely due to their ability to associate with αvβ3 integrin in a ternary protein complex that triggers the membrane-proximal signaling events in response to bFGF.
FIGURE 6:

CD9 and JAM-A are required for invasive growth and in vitro tube formation. (A) HUVECs were transfected with siRNAs against JAM-A or CD9. Left panel, knockdown efficiencies were analyzed by immunoblotting. Right panel, siRNA-transfected HUVECs were allowed to invade a Matrigel matrix for 16 h in the presence of bFGF. Invasion was analyzed by counting the number of cells at the bottom surface of the filter. Statistical significance was evaluated using one-way ANOVA with Dunnett's post hoc test. **, p < 0.01. (B) siRNA-transfected HUVECs were seeded on basement membrane extracts and incubated for 24 h in the presence of bFGF. Top panel, knockdown efficiency was analyzed by indirect immunofluorescence. Scale bars: 20 μm. Bottom, left panel, representative phase-contrast micrographs of tube-like structures 24 h after seeding in Matrigel. Original magnification: 10×. Bottom, right panel, total tube length after 24 h. Statistical significance was evaluated using repeated-measures ANOVA with Dunnett's post hoc test. *, p < 0.05; **, p < 0.01.
DISCUSSION
In this study, we identify CD9 as a novel binding partner for JAM-A in endothelial cells. In addition, we identify αvβ3 integrin as novel binding partner for CD9. CD9 acts as a scaffolding protein that assembles a ternary JAM-A–CD9–αvβ3 integrin complex (Figure 7). This complex contains predominantly monomeric JAM-A, which is released upon bFGF stimulation. Our observations identify CD9 as critical upstream component of the bFGF/αvβ3 integrin–regulated angiogenic signaling pathway and suggest that this function of CD9 is due to its ability to link JAM-A to αvβ3 integrin.
FIGURE 7:

Model of angiogenic signaling regulated by JAM-A and CD9. JAM-A, CD9, and αvβ3 integrin form a ternary complex in the membrane of endothelial cells that contains predominantly monomeric JAM-A. JAM-A is linked to CD9 via its cytoplasmic domain, and this interaction is probably indirect. Complex formation is promoted by αvβ3 integrin activation, most likely by lateral association of the extended conformation of the integrin with CD9. This complex is signaling-competent, yet not active. Stimulation with bFGF releases monomeric JAM-A from the ternary complex through an unknown mechanism. We speculate that once monomeric JAM-A is released from the complex it forms homodimers that mediate MAPK activation.
The ternary complex is stable in the presence of Triton X-100. The stability of tetraspanin-containing protein complexes in different detergents can be used to distinguish primary tetraspanin interactions, in which the tetraspanin is linked to its nontetraspanin partner (either directly or indirectly) without involvement of another tetraspanin, from secondary/tertiary tetraspanin interactions, in which the tetraspanin interacts with its nontetraspanin partner through another tetraspanin (Boucheix and Rubinstein, 2001; Hemler, 2005). The latter interactions are unstable in Triton X-100– or NP40-based lysis buffers but stable in lysis buffers containing Brij97, which preserves tetraspanin–tetraspanin interaction (Charrin et al., 2009). Because all Co-IP experiments from endothelial cell lysates were performed in the presence of Triton X-100, we conclude that the associations of CD9 with JAM-A and with αvβ3 integrin are primary tetraspanin interactions. The interaction of JAM-A with CD9 is mediated through the cytoplasmic tail of JAM-A and requires the C-terminal PDZ domain-binding motif of JAM-A. Because CD9 does not contain a PDZ domain, the interaction of JAM-A with CD9 in cells is probably indirect and mediated by an as yet unidentified cytoplasmic protein. The interaction of αvβ3 integrin with CD9 is probably mediated by the extracellular domains, since integrin activation enhances the interaction (Figure 3A). We speculate that the open conformation exposes new binding sites for the lateral association with CD9 and that the concomitant increase in affinity further intensifies the association between CD9 and αvβ3 integrin to stabilize the ternary JAM-A–CD9–αvβ3 integrin complex.
An important finding in our study is the observation that CD9 interacts preferentially with monomeric JAM-A, and that bFGF weakens this interaction. As suggested from previous studies, monomeric JAM-A is most likely signaling-inactive (Mandell et al., 2004, 2005; Rehder et al., 2006; Severson et al., 2008, 2009). As one example, JAM-A dimerization regulates the close apposition of the two Rap1 regulatory proteins AF-6/afadin and PDZ-GEF2, which both interact with JAM-A (Ebnet et al., 2000; Severson et al., 2009). We therefore speculate that the association of monomeric JAM-A with CD9 serves to assemble a signaling-competent JAM-A–CD9–αvβ3 integrin complex that, however, is inactive in the absence of an angiogenic stimulus. On stimulation with bFGF, monomeric JAM-A is released from CD9 and αvβ3 integrin, dimerizes, and adopts its signaling activity.
The selective incorporation of monomeric versus dimeric JAM molecules into specific protein complexes seems to emerge as a common mechanism by which the activity of JAM molecules is regulated. The JAM family–related protein JAM-like (JAM-L) is expressed by different leukocyte subsets and serves to mediate leukocyte endothelial/epithelial cell interactions by interacting with coxsackievirus and adenovirus receptor (CAR) in a trans-heterophilic manner (Moog-Lutz et al., 2003; Zen et al., 2005; Luissint et al., 2008). In resting monocytes and T-cells, JAM-L associates in cis with α4β1 integrin, and interestingly, it is predominantly monomeric JAM-A that is associated with α4β1 integrin (Luissint et al., 2008). Activation of α4β1 integrin by Mn2+ or the chemokine SDF-1α releases JAM-L from α4β1 integrin, allowing for dimerization followed by interaction with CAR (Luissint et al., 2008). In this scenario, the inactive α4β1 integrin keeps JAM-L inactive by inhibiting its dimerization, and integrin activation coactivates the adhesive function of JAM-L by releasing it from the integrin.
The molecular mechanism by which JAM-A is released is not understood. As CD9 knockdown does not induce an increase in ERK1/2 phosphorylation in the absence of bFGF (Figure 5B), the mere release of JAM-A is not sufficient to induce a signal downstream of JAM-A dimerization. It is rather likely that bFGF induces a posttranslational modification of JAM-A that is required for the signaling activity of JAM-A. Interestingly, the bFGF receptor FGFR1 has been identified in a complex with αvβ3 integrin but not αvβ5 integrin in response to stimulation with bFGF and fibrinogen (Sahni and Francis, 2004), which opens up the possibility that JAM-A becomes physically associated with FGFR1 after bFGF stimulation within a JAM-A–CD9–αvβ3 integrin–FGFR1-containing microdomain. As a result of this close proximity, JAM-A could be phosphorylated by FGFR1. Indirect evidence suggests that Tyr-280 phosphorylation of JAM-A is involved in MAPK activation by JAM-A (Naik et al., 2003), which further supports the requirement of a posttranslational modification of JAM-A for its signaling activity.
Another key finding of this study is the specificity of CD9 and JAM-A for the bFGF/αvβ3–mediated angiogenic signaling pathway. Two distinct cytokine-dependent pathways of angiogenesis have been defined based on the involvement of two distinct αv integrins: angiogenesis induced by bFGF or TNF-α is cooperatively regulated by integrin αvβ3, whereas angiogenesis induced by VEGF or TGF-α is cooperatively regulated by integrin αvβ5 (Friedlander et al., 1995). Both pathways result in ERK1/2 activation but differ in the signaling components upstream of Raf activation: the bFGF–αvβ3 pathway activates c-Abl and p21-activated kinase-1, resulting in Raf phosphorylation at Ser-338; the VEGF–αvβ5 pathway depends on PKC and Src kinase and results in Raf phosphorylation at Tyr-340 (Eliceiri et al., 2002; Hood et al., 2003; Yan et al., 2008). Our results identify JAM-A and CD9 as upstream components of the bFGF–αvβ3 pathway. The ability of CD9 to connect JAM-A selectively to αvβ3 integrin but not to αvβ5 integrin may thus be a critical factor contributing to the specificity in the cooperativity of bFGF with αvβ3 integrin.
We propose the following molecular mechanism is involved in the membrane-proximal signaling events regulating the bFGF/αvβ3–dependent activation of the ERK1/2 pathway (Figure 7). JAM-A, CD9, and αvβ3 integrin are associated in a ternary protein complex at cell–cell contacts of endothelial cells, whose formation is enhanced by integrin engagement. In this complex, CD9 serves to link monomeric JAM-A to αvβ3 integrin. A signal mediated by bFGF releases monomeric JAM-A from the complex through an unknown mechanism. Liberated JAM-A monomers probably dimerize to form a signaling-active complex, perhaps by bringing molecules associated with the cytoplasmic tail of JAM-A into close proximity. The next important step to further understand the molecular mechanism downstream of JAM-A dimerization will be the identification of these JAM-A–associated molecules.
MATERIALS AND METHODS
Cell culture and transfections
HEK293T and HeLa cells were maintained in DMEM (Life Technologies, Darmstadt, Germany) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine (Lonza, Basel, Switzerland), and 100 U/ml penicillin/streptomycin (Lonza, Basel). MyEnd cells were cultured in the same medium supplemented with 1 mM Na-pyruvate (Biochrom, Berlin, Germany). Human brain microvascular endothelial cells (kindly provided by the Institute of Infectiology, ZMBE, Münster, Germany) were grown in RPMI supplemented with 10% FCS, 10% NuSerum (BD Biosciences, Heidelberg, Germany), 2 mM glutamine, and 100 U/ml penicillin/streptomycin, 1:100 diluted MEM vitamins (PAA, Cölbe, Germany), 1:100 nonessential amino acids (Biochrom), and 1 mM Na-pyruvate. HUVECs were isolated from umbilical veins by dispase treatment and were maintained in EGM medium (Clonetics, Heidelberg, Germany). Transient transfections of plasmids in HEK293T cells were performed using GeneJammer transfection reagent (Stratagene, Amsterdam, Netherlands). Transfections of siRNA oligonucleotides in HUVECs were performed using the Amaxa HUVEC Nucleofector Kit (Lonza, Cologne, Germany) according to the manufacturer's instructions.
Antibodies and reagents
The following antibodies were used: mouse monoclonal antibody (mAb) anti-CD9 (Millipore, Billerica, MA), rat anti-CD9 mAb KMC8 (eBioscience, Frankfurt, Germany), mouse mAb anti–JAM-A (BD Biosciences), rat anti–JAM-A mAb 106 (Malergue et al., 1998), rabbit polyclonal antibody (pAb) anti-GST (Santa Cruz Biotechnology, Heidelberg, Germany), mouse mAb anti–β3 integrin (BD Biosciences), mouse mAb anti–human integrin αvβ3 (Millipore), mouse mAb anti–α-tubulin (Sigma-Aldrich, Munich, Germany), rabbit mAb anti-ERK1/2 and rabbit mAb anti–Thr-202/Tyr-204–phosphorylated ERK1/2 (Cell Signaling Technology, Frankfurt, Germany), mouse mAb anti–Flag-tag and rabbit pAb anti–Flag-tag (Sigma-Aldrich). A polyclonal antibody against human JAM‑A was generated by immunizing rabbits with a fusion protein consisting of the extracellular part of human JAM-A fused to the Fc-part of human IgG, as described previously (Ebnet et al., 2003). The antibodies were affinity-purified by adsorption on the same fusion protein covalently coupled to cyanogen bromide (CnBr)-activated Sepharose beads (GE Healthcare, München, Germany), and antibodies directed against the Fc-portion were depleted by adsorption on human IgG coupled to CnBr-activated Sepharose beads. The following reagents were used: human vitronectin (Peprotech, Hamburg, Germany), bFGF (Peprotech), VEGF (Sigma-Aldrich), and RGDS peptide (Peprotech).
Yeast two-hybrid screen
A yeast two-hybrid screen using the cytoplasmic domain of murine JAM-A as bait was performed essentially as previously described (Ebnet et al., 2000). Briefly, 250 μg of plasmid DNA derived from a day 9.5/10.5 mouse embryo cDNA library (Hollenberg et al., 1995) was transformed into the Saccharomyces cerevisiae reporter strain L40 expressing the cytoplasmic domain of JAM-A (aa 261–300) fused to LexA. The transformants were plated onto synthetic medium lacking tryptophane, histidine, uracil, leucine, and lysine. After 3 d at 30°C, large colonies were transferred to new plates and grown for an additional 3 d on selective medium. DNA was isolated from clones grown in liquid selective medium using a plasmid isolation kit (USB, Cleveland, OH). The plasmid derived from the library was isolated by transforming Escherichia coli HB101 with the isolated plasmid DNA; this was followed by growing the HB101 transformants on M9 minimal medium lacking leucine. Plasmid DNA was isolated from HB101 transformants and sequenced using standard procedures.
DNA constructs, site-directed mutagenesis, and recombinant protein expression
For transient expression of Flag-tagged JAM-A constructs, the human JAM-A cDNA lacking the leader peptide sequence (Flag-hJAM-A, aa 26–299), C-terminal deletion constructs lacking either three or six or nine C-terminal amino acids (Flag-hJAM-A/Δ3, aa 26–296; Flag-hJAM-A/Δ6, aa 26–293; Flag-hJAM-A/Δ9, aa 26–290), and a human JAM-A construct lacking the membrane-distal, V-type Ig domain (Flag-JAM-A/ΔV) were cloned into the pFlag-CMV-1 vector (Sigma-Aldrich). The two hJAM-A mutants with sets of three amino acids at the C-terminus exchanged with alanines (Flag-JAM-A/3A1, F292Q293K294–A292A293A294; Flag-JAM-A/3A2, T295S296S297–A295A296A297), as well as the dimerization mutant with point mutations within the dimerization interface (Flag-JAM-A/E61RK63E), were generated by a PCR-based approach using mismatch primer pairs with wild-type Flag-hJAM-A as a template. The mouse JAM-A cDNA cloned into pFLAG-CMV-1 has been described before (Ebnet et al., 2001). For recombinant protein expression in E. coli, the pGEX-4T-1 vector containing the entire cytoplasmic tail of murine JAM-A (aa 261–300) or C-terminal deletion mutants (GST-JAM-A/Δ3, aa 261–297; GST-JAM-A/Δ9, aa 261–291) was used as described (Ebnet et al., 2000). Recombinant proteins were purified from E. coli BL21 as has been described before (Ebnet et al., 2000).
RNA interference
For depleting JAM-A and CD9 in HUVECs, the following siRNA heteroduplexes were used: 5′-GGACGUACUCGAAACCUUCTT-3′ (CD9), 5′-GAAGUGAGGGGGAAUUCAATT-3′ (JAM-A). For depleting β3 integrin, a pool of four different siRNA oligonucleotides (On-TARGETplus Smart Pool; Thermo Fisher Scientific, Schwerte, Germany) was used. As control siRNA, a nontargeting siRNA (On-TARGETplus Non-targeting siRNA; Thermo Fisher Scientific) was used. HUVECs (2 × 106) were transfected with 200 pmol of siRNAs by electroporation using the Amaxa HUVEC Nucleofector Kit (Lonza, Cologne, Germany) according to the manufacturer's instructions. After 48 h, the cells were harvested and analyzed.
Immunoprecipitation and Western blot analysis
Immunoprecipitations were performed essentially as has been described before (Ebnet et al., 2001). Cells were either grown under normal culture conditions or, when stimulated with bFGF or RGDS peptide, serum-starved overnight by growth in medium containing 1% bovine serum albumin (BSA) in place of 10% FCS. Cells were lysed in either Triton X-100–containing lysis buffer (50 mM Tris HCl, pH 7.4, 1% [vol/vol] Triton X-100, 150 mM NaCl, protease inhibitors [Protease Inhibitor Cocktail tablets “Complete”; Roche Diagnostics, Mannheim, Germany]) or Brij97-containing lysis buffer (10 mM Tris HCl, pH 7.4, 1% [vol/vol] Brij97, 150 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, protease inhibitors [Protease Inhibitor Cocktail tablets; Roche Diagnostics]) for 30 min on ice and then centrifuged at 4°C. The supernatants were precleared by incubation with 15 μl of protein A or protein G Sepharose beads (GE Healthcare), followed by centrifugation. Postnuclear supernatants were incubated with 3 μg of antibodies coupled to protein A or protein G Sepharose beads overnight at 4˚C. Immune complex–captured beads were washed five times with lysis buffer without inhibitors and boiled in SDS sample buffer containing 2.5% β-mercaptoethanol. The proteins were separated by SDS–PAGE and analyzed by Western blotting. Under these conditions, the anti-CD9 mAbs used in the study showed a specific signal that was abolished after CD9 knockdown. All CoIP experiments from endothelial cells were performed using Triton X-100 lysis buffer. The results of the CoIP experiments are representative for at least three independent experiments. Quantification of signal intensities was performed using the Odyssey imaging system (LI-COR Biosciences, Lincoln, NE). For each band, the integrated intensity (in kilocounts) was calculated with Odyssey application software (version 3.0). Mean values and SEs were calculated from three independent experiments. Statistical significance was evaluated using one-sample t tests. p values below 0.05 were considered significant.
Analysis of ERK1/2 phosphorylation
HUVECs were transfected with JAM-A–specific or CD9-specific siRNAs and incubated for 48 h on regular or vitronectin-coated tissue culture plates. For 14 h prior to stimulation with growth factors, the cells were grown in medium containing 1% BSA instead of FCS (serum starvation). The serum-starved cells were stimulated with either 10 ng/ml bFGF for 10 min or with 20 ng/ml VEGF for 10 min, then lysed with hot SDS sample buffer. Cell lysates were separated by 12% SDS–PAGE, transferred to nitrocellulose membranes, and probed with antibodies against total ERK1/2 or Thr-202/Tyr-204–phosphorylated ERK1/2. The results of the ERK1/2 phosphorylation experiments are representative for at least three independent experiments. Quantification of signal intensities was performed using the Odyssey imaging system, as described above. Phosphorylation signals were corrected for differences in total ERK1/2 levels. Values obtained from unstimulated cells (baseline phosphorylation) were subtracted from the values obtained from bFGF- or VEGF-stimulated cells, resulting in normalized phosphorylation levels. Bars in Figure 5, B and C, show the increase or decrease in ERK1/2 phosphorylation levels in CD9 (Figure 5B) or JAM-A (Figure 5C) knockdown cells relative to the levels in wild-type cells, which were arbitrarily set as 1.
Immunofluorescence microscopy
Immunofluorescence analyses were performed with HUVECs grown on vitronectin-coated Lab-Tek Chamber Slides (ThermoFisher Scientific, Waltham, MA). Cells were fixed in ice-cold EtOH for 30 min and acetone for 3 min at room temperature (RT); this was followed by rehydration in blocking buffer (phosphate-buffered saline [PBS]/10% FCS). After 1 h of blocking, cells were incubated with primary antibodies in 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.01% Tween-20, 0.1% BSA for 1 h at RT or overnight at 4°C. After being washed, cells were incubated with Alexa Fluor 488– or Alexa Fluor 568–conjugated, highly cross-adsorbed secondary antibodies for 1 h at RT. After washing, cells were mounted in fluorescence mounting medium (Dako, Hamburg, Germany) and stored at 4°C. Immunofluorescence microscopy was performed using a confocal microscope (Zeiss LSM 510 Meta, Jena, Germany) equipped with Zeiss Plan-Apochromat lenses (Zeiss Plan-Apochromat DIC, oil, 63× magnification, 1.4 numerical aperture).
Stainings of mouse retina vasculature
Retina strainings were performed essentially as previously described (Wang et al., 2010). Briefly, eyes were dissected from neonatal mice (postnatal day 6) and fixed in 4% PFA for 2 h on ice. Retinas were permeabilized and blocked in 1% BSA (#A4378; Sigma-Aldrich) and 0.3% Triton X-100 for 2 h at room temperature with gentle rocking. Next retinas were washed three times in Pblec buffer (1 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2, and 1% Triton X-100 in PBS) and incubated with biotinylated isolectin B4 (#B-1205, Griffonia simplicifolia lectin 1, 1:50; Vector Labs, Burlingame, CA), and with antibodies against JAM-A (mAb 106) and CD9 (mAb KMC8) overnight at 4°C with gentle rocking. Retinas were washed five times with 0.5% BSA and 0.15% Triton X-100 and incubated with Alexa Fluor–conjugated streptavidin (1:100; Invitrogen) and with the corresponding Alexa Fluor–conjugated secondary antibody (1:500; Invitrogen) in blocking buffer for 2 h at room temperature. Retinas were flat-mounted using Fluoromount-G (#0100-01; SouthernBiotech, Birmingham, AL).
Cell proliferation
Cell proliferation was analyzed using a commercially available cell proliferation kit (Cell Proliferation Kit II (XTT); Roche Diagnostics, Mannheim, Germany). Briefly, cells were seeded on vitronectin-coated 96-well plates at a density of 5 × 103 cells per well. After 24 h, XTT reagent was added to each well. At 4 and 24 h after XTT addition, the absorption at 475 nm was measured using an enzyme-linked immunosorbent assay reader. The quantification is based on three independent experiments with quintuplicate samples in each experiment. Statistical significance was analyzed using one-sample t test.
Flow cytometry
Cell surface expression of transfected JAM-A constructs was analyzed by flow cytometry. After harvest, cells were resuspended in FACS buffer (PBS/3% FCS) at 2 × 106 cells/ml, incubated with primary antibodies (5 μg/ml) for 1 h at 4°C in FACS buffer, washed, and incubated with Cy2-conjugated secondary antibodies (5 μg/ml, 1 h, 4°C). After three washing steps, cells were analyzed by flow cytometry with excitation at 488 nm (BD FACSCalibur; BD Biosciences). For each sample, 10,000 cells were counted.
Cell invasion
Twenty-five thousand endothelial cells harvested 48 h after siRNA transfection were resuspended in 0.5 ml serum-free medium and added to the upper compartment of a BioCoat Matrigel Invasion Chamber (BD Biosciences). After overnight incubation, the medium in the lower compartment was supplemented with 50 ng/ml bFGF, and the cells were allowed to invade the lower compartment. After 16 h, the cells present at the bottom surface of the filters were fixed and stained with Diff-Quik dye (Dade Behring, Duedingen, Switzerland). Filter membranes were excised, mounted, and photographed using a Zeiss Axiovert microscope equipped with AxioVision software (Zeiss) at 100× magnification. For each membrane, cells in five visual fields were counted. For each condition, triplicates were analyzed. Statistical significance was evaluated using one-way analysis of variance (ANOVA) with Dunnett's post hoc test. Mean values and SDs were calculated from three independent experiments.
In vitro tube formation assay
In vitro tube formation assays were performed according to the protocol described by Arnaoutova and Kleinman (2010). Briefly, Cultrex basement membrane extract (BME) with reduced growth factors (Trevigen, Gaithersburg, MD) was thawed overnight at 4°C. After thawing, individual wells of a flat-bottom 96-well microtiter plate were coated with 50 μl of BME at 37°C and 5% CO2 for 1 h. siRNA-treated HUVECs were serum-starved overnight, resuspended at 1.5 × 105 cells/ml in basal medium (EBM 2; Lonza, Cologne, Germany), reconstituted with 40 ng/ml bFGF, and seeded into the BME-coated 96-well plates (100 μl/well). Phase-contrast microscopy images were taken immediately after seeding to control for the seeding density. After 24 h, images were taken again, and the total length of the tube network was analyzed using Image J software (http://rsbweb.nih.gov/ij). Each condition was performed in triplicate. Statistical analysis was performed using repeated-measures ANOVA with Dunnett's post hoc test. Mean values and SDs were calculated from three independent experiments.
Supplementary Material
Acknowledgments
We thank Janina Tomm for initial help in cloning experiments, Boris Günnewig for his help with in vitro binding experiments, Anne-Marie Erpenbeck-Leuer and Frauke Brinkmann for expert technical assistance, and Patrick Seelheim, Institute for Biochemistry, Münster, for helpful discussions. This work was supported by grants from the Medical Faculty of the University of Münster (IMF-EB 1 2 03 23, IZKF Eb2/028/09, and IZKF Ge2/016/10).
Abbreviations used:
- ANOVA
analysis of variance
- bFGF
basic fibroblast growth factor
- BME
basement membrane extract
- BSA
bovine serum albumin
- CAR
coxsackievirus and adenovirus receptor
- CoIP
coimmunoprecipitation
- FCS
fetal calf serum
- GST
glutathione S-transferase
- HUVEC
human umbilical vein endothelial cell
- Ig
immunoglobulin
- IgSF
Ig-superfamily
- IP
immunoprecipitation
- JAM-A
junctional adhesion molecule-A
- JAM-L
JAM-like
- mAb
monoclonal antibody
- MAPK
mitogen-activated protein kinase
- pAb
polyclonal antibody
- PBS
phosphate-buffered saline
- PNS
postnuclear supernatant
- RGDS
Arg-Gly-Asp-Ser
- RT
room temperature
- siRNA
small interfering RNA
- TNF-α
tumor necrosis factor-α
- VEGF
vascular endothelial growth factor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-06-0481) on February 6, 2013.
REFERENCES
- Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nature Protoc. 2010;5:628–635. doi: 10.1038/nprot.2010.6. [DOI] [PubMed] [Google Scholar]
- Barreiro O, Yanez-Mo M, Sala-Valdes M, Gutierrez-Lopez MD, Ovalle S, Higginbottom A, Monk PN, Cabanas C, Sanchez-Madrid F. Endothelial tetraspanin microdomains regulate leukocyte firm adhesion during extravasation. Blood. 2005;105:2852–2861. doi: 10.1182/blood-2004-09-3606. [DOI] [PubMed] [Google Scholar]
- Barreiro O, Zamai M, Yanez-Mo M, Tejera E, Lopez-Romero P, Monk PN, Gratton E, Caiolfa VR, Sanchez-Madrid F. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J Cell Biol. 2008;183:527–542. doi: 10.1083/jcb.200805076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G. The JAM family of junctional adhesion molecules. Curr Opin Cell Biol. 2003;15:525–530. doi: 10.1016/s0955-0674(03)00104-2. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000;275:20520–20526. doi: 10.1074/jbc.M905251199. [DOI] [PubMed] [Google Scholar]
- Boucheix C, Rubinstein E. Tetraspanins. Cell Mol Life Sci. 2001;58:1189–1205. doi: 10.1007/PL00000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury LE, Goldmacher VS, Tedder TF. The CD19 signal transduction complex of B lymphocytes. Deletion of the CD19 cytoplasmic domain alters signal transduction but not complex formation with TAPA-1 and Leu 13. J Immunol. 1993;151:2915–2927. [PubMed] [Google Scholar]
- Cailleteau L, et al. α2β1 integrin controls association of Rac with the membrane and triggers quiescence of endothelial cells. J Cell Sci. 2010;123:2491–2501. doi: 10.1242/jcs.058875. [DOI] [PubMed] [Google Scholar]
- Charrin S, le Naour F, Silvie O, Milhiet PE, Boucheix C, Rubinstein E. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem J. 2009;420:133–154. doi: 10.1042/BJ20082422. [DOI] [PubMed] [Google Scholar]
- Cooke VG, Naik MU, Naik UP. Fibroblast growth factor-2 failed to induce angiogenesis in junctional adhesion molecule-A-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2005–2011. doi: 10.1161/01.ATV.0000234923.79173.99. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Aurrand-Lions M, Kuhn A, Kiefer F, Butz S, Zander K, Meyer Zu Brickwedde MK, Suzuki A, Imhof BA, Vestweber D. The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR-3: a possible role for JAMs in endothelial cell polarity. J Cell Sci. 2003;116:3879–3891. doi: 10.1242/jcs.00704. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Schulz CU, Meyer Zu Brickwedde MK, Pendl GG, Vestweber D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem. 2000;275:27979–27988. doi: 10.1074/jbc.M002363200. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Suzuki A, Horikoshi Y, Hirose T, Meyer Zu Brickwedde MK, Ohno S, Vestweber D. The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM) EMBO J. 2001;20:3738–3748. doi: 10.1093/emboj/20.14.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebnet K, Suzuki A, Ohno S, Vestweber D. Junctional adhesion molecules (JAMs): more molecules with dual functions. J Cell Sci. 2004;117:19–29. doi: 10.1242/jcs.00930. [DOI] [PubMed] [Google Scholar]
- Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, Cheresh DA. Src-mediated coupling of focal adhesion kinase to integrin αvβ5 in vascular endothelial growth factor signaling. J Cell Biol. 2002;157:149–160. doi: 10.1083/jcb.200109079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995;270:1500–1502. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- Hamazaki Y, Itoh M, Sasaki H, Furuse M, Tsukita S. Multi-PDZ domain protein 1 (MUPP1) is concentrated at tight junctions through its possible interaction with claudin-1 and junctional adhesion molecule. J Biol Chem. 2002;277:455–461. doi: 10.1074/jbc.M109005200. [DOI] [PubMed] [Google Scholar]
- Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Sternglanz R, Cheng PF, Weintraub H. Identification of a new family of tissue-specific basic helix-loop-helix proteins with a two-hybrid system. Mol Cell Biol. 1995;15:3813–3822. doi: 10.1128/mcb.15.7.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JD, Frausto R, Kiosses WB, Schwartz MA, Cheresh DA. Differential αv integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J Cell Biol. 2003;162:933–943. doi: 10.1083/jcb.200304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iden S, Misselwitz S, Peddibhotla SS, Tuncay H, Rehder D, Gerke V, Robenek H, Suzuki A, Ebnet K. aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J Cell Biol. 2012;196:623–639. doi: 10.1083/jcb.201104143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S. Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J Cell Biol. 2001;154:491–498. doi: 10.1083/jcb.200103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisasanuki T, Tokushige S, Terasaki H, Khai NC, Wang Y, Sakamoto T, Kosai K. Targeting CD9 produces stimulus-independent antiangiogenic effects predominantly in activated endothelial cells during angiogenesis: a novel antiangiogenic therapy. Biochem Biophys Res Commun. 2011;413:128–135. doi: 10.1016/j.bbrc.2011.08.068. [DOI] [PubMed] [Google Scholar]
- Laukoetter MG, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Shoham T. Protein-protein interactions in the tetraspanin web. Physiology (Bethesda) 2005a;20:218–224. doi: 10.1152/physiol.00015.2005. [DOI] [PubMed] [Google Scholar]
- Levy S, Shoham T. The tetraspanin web modulates immune-signalling complexes. Nat Rev Immunol. 2005b;5:136–148. doi: 10.1038/nri1548. [DOI] [PubMed] [Google Scholar]
- Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113:2363–2374. doi: 10.1242/jcs.113.13.2363. [DOI] [PubMed] [Google Scholar]
- Luissint AC, Lutz PG, Calderwood DA, Couraud PO, Bourdoulous S. JAM-L-mediated leukocyte adhesion to endothelial cells is regulated in cis by α4β1 integrin activation. J Cell Biol. 2008;183:1159–1173. doi: 10.1083/jcb.200805061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malergue F, Galland F, Martin F, Mansuelle P, Aurrand-Lions M, Naquet P. A novel immunoglobulin superfamily junctional molecule expressed by antigen presenting cells, endothelial cells and platelets. Mol Immunol. 1998;35:1111–1119. doi: 10.1016/s0161-5890(98)00102-3. [DOI] [PubMed] [Google Scholar]
- Mandell KJ, Babbin BA, Nusrat A, Parkos CA. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on β1 integrins and Rap1 activity. J Biol Chem. 2005;280:11665–11674. doi: 10.1074/jbc.M412650200. [DOI] [PubMed] [Google Scholar]
- Mandell KJ, McCall IC, Parkos CA. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem. 2004;279:16254–16262. doi: 10.1074/jbc.M309483200. [DOI] [PubMed] [Google Scholar]
- Moog-Lutz C, Cave-Riant F, Guibal FC, Breau MA, Di Gioia Y, Couraud PO, Cayre YE, Bourdoulous S, Lutz PG. JAML, a novel protein with characteristics of a junctional adhesion molecule, is induced during differentiation of myeloid leukemia cells. Blood. 2003;102:3371–3378. doi: 10.1182/blood-2002-11-3462. [DOI] [PubMed] [Google Scholar]
- Naik MU, Mousa SA, Parkos CA, Naik UP. Signaling through JAM-1 and αvβ3 is required for the angiogenic action of bFGF: dissociation of the JAM-1 and αvβ3 complex. Blood. 2003;102:2108–2114. doi: 10.1182/blood-2003-04-1114. [DOI] [PubMed] [Google Scholar]
- Naik MA, Naik UP. Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin avb3 specific. J Cell Sci. 2006;119:490–499. doi: 10.1242/jcs.02771. [DOI] [PubMed] [Google Scholar]
- Nava P, et al. JAM-A regulates epithelial proliferation through Akt/β-catenin signalling. EMBO Rep. 2011;12:314–320. doi: 10.1038/embor.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiuchi R, Sanzen N, Nada S, Sumida Y, Wada Y, Okada M, Takagi J, Hasegawa H, Sekiguchi K. Potentiation of the ligand-binding activity of integrin α3β1 via association with tetraspanin CD151. Proc Natl Acad Sci USA. 2005;102:1939–1944. doi: 10.1073/pnas.0409493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the β2 integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- Prota AE, Campbell JA, Schelling P, Forrest JC, Watson MJ, Peters TR, Aurrand-Lions M, Imhof BA, Dermody TS, Stehle T. Crystal structure of human junctional adhesion molecule 1: implications for reovirus binding. Proc Natl Acad Sci USA. 2003;100:5366–5371. doi: 10.1073/pnas.0937718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehder D, Iden S, Nasdala I, Wegener J, Brickwedde MK, Vestweber D, Ebnet K. Junctional adhesion molecule-A participates in the formation of apico-basal polarity through different domains. Exp Cell Res. 2006;312:3389–3403. doi: 10.1016/j.yexcr.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Rubinstein E, Billard M, Plaisance S, Prenant M, Boucheix C. Molecular cloning of the mouse equivalent of CD9 antigen. Thrombosis Res. 1993;71:377–383. doi: 10.1016/0049-3848(93)90162-h. [DOI] [PubMed] [Google Scholar]
- Sahni A, Francis CW. Stimulation of endothelial cell proliferation by FGF-2 in the presence of fibrinogen requires αvβ3. Blood. 2004;104:3635–3641. doi: 10.1182/blood-2004-04-1358. [DOI] [PubMed] [Google Scholar]
- Severson EA, Jiang L, Ivanov AI, Mandell KJ, Nusrat A, Parkos CA. Cis-dimerization mediates function of junctional adhesion molecule A. Mol Biol Cell. 2008;19:1862–1872. doi: 10.1091/mbc.E07-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA. Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate β1 integrin levels, and enhance cell migration. Mol Biol Cell. 2009;20:1916–1925. doi: 10.1091/mbc.E08-10-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw SK, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200:1571–1580. doi: 10.1084/jem.20040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipp CS, Kolesnikova TV, Hemler ME. EWI-2 is a major CD9 and CD81 partner and member of a novel Ig protein subfamily. J Biol Chem. 2001;276:40545–40554. doi: 10.1074/jbc.M107338200. [DOI] [PubMed] [Google Scholar]
- Stipp CS, Kolesnikova TV, Hemler ME. Functional domains in tetraspanin proteins. Trends Biochem Sci. 2003;28:106–112. doi: 10.1016/S0968-0004(02)00014-2. [DOI] [PubMed] [Google Scholar]
- Vetrano S, et al. Unique role of junctional adhesion molecule-A in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135:173–184. doi: 10.1053/j.gastro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7:467–477. doi: 10.1038/nri2096. [DOI] [PubMed] [Google Scholar]
- Yan W, Bentley B, Shao R. Distinct angiogenic mediators are required for basic fibroblast growth factor- and vascular endothelial growth factor-induced angiogenesis: the role of cytoplasmic tyrosine kinase c-Abl in tumor angiogenesis. Mol Biol Cell. 2008;19:2278–2288. doi: 10.1091/mbc.E07-10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez-Mo M, Barreiro O, Gordon-Alonso M, Sala-Valdes M, Sanchez-Madrid F. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 2009;19:434–446. doi: 10.1016/j.tcb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Kazarov AR, Desai B, Lee RT, Hemler ME. Direct extracellular contact between integrin α3β1 and TM4SF protein CD151. J Biol Chem. 2000;275:9230–9238. doi: 10.1074/jbc.275.13.9230. [DOI] [PubMed] [Google Scholar]
- Zen K, Liu Y, McCall IC, Wu T, Lee W, Babbin BA, Nusrat A, Parkos CA. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol Biol Cell. 2005;16:2694–2703. doi: 10.1091/mbc.E05-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.