

Fluorescent nuclear actin reporters are used to determine the distribution of nuclear actin in live somatic cells and evaluate its potential functions. They reveal distinct monomeric and filamentous actin populations in nuclei of live somatic cells and implicate nuclear actin in mRNA processing and organization of the nucleoplasm.
Abstract
In addition to its long-studied presence in the cytoplasm, actin is also found in the nuclei of eukaryotic cells. The function and form (monomer, filament, or noncanonical oligomer) of nuclear actin are hotly debated, and its localization and dynamics are largely unknown. To determine the distribution of nuclear actin in live somatic cells and evaluate its potential functions, we constructed and validated fluorescent nuclear actin probes. Monomeric actin probes concentrate in nuclear speckles, suggesting an interaction of monomers with RNA-processing factors. Filamentous actin probes recognize discrete structures with submicron lengths that are excluded from chromatin-rich regions. In time-lapse movies, these actin filament structures exhibit one of two types of mobility: 1) diffusive, with an average diffusion coefficient of 0.06–0.08 μm2/s, or (2) subdiffusive, with a mobility coefficient of 0.015 μm2/s. Individual filament trajectories exhibit features of particles moving within a viscoelastic mesh. The small size of nuclear actin filaments is inconsistent with a role in micron-scale intranuclear transport, and their localization suggests that they do not participate directly in chromatin-based processes. Our results instead suggest that actin filaments form part of a large, viscoelastic structure in the nucleoplasm and may act as scaffolds that help organize nuclear contents.
INTRODUCTION
In the cytoplasm, actin filaments form functional networks that enable eukaryotic cells to transport cargo, change shape, and move. These activities organize components of the cytoplasm and help turn a mob of macromolecules into a living cell. Actin is also present in the nucleus (de Lanerolle and Serebryannyy, 2011), but in this compartment its functions are more cryptic. Early studies revealed high concentrations (100 μM) of actin in oocyte germinal vesicles (Clark and Merriam, 1977; Clark and Rosenbaum, 1979; Scheer et al., 1984), but there is disagreement over whether this actin is predominantly monomeric or filamentous (Gall and Wu, 2010; Kiseleva et al., 2004; Bohnsack et al., 2006), and there is no consensus regarding its function.
Somatic cell nuclei contain much less actin than oocyte germinal vesicles, and the forms and functions of this actin are even more hotly debated. Monomeric actin participates as a subunit in several nuclear complexes that regulate chromatin architecture, but its function within these complexes is mysterious (Farrants, 2008). Nuclear actin has also been proposed to form conventional filaments that interact with cytoskeletal regulators, including cofilin (Iida et al., 1992; Dopie et al., 2012), α-actinin (Kumeta et al., 2010), filamin (Loy et al., 2003), coronin 2A (Huang et al., 2011), and nuclear isoforms of myosin (Pestic-Dragovich et al., 2000). There are also reports of unconventional forms of actin in the nucleus, including covalently modified monomers (Hofmann et al., 2009) and nonfilamentous oligomers (Schoenenberger et al., 2005).
Nuclear actin filaments have been proposed to participate in many processes, including control of chromatin architecture, regulation of transcription, and intranuclear cargo transport. Broadly speaking, however, all the functions proposed for nuclear actin filaments fall into one of four categories: 1) providing tracks for micrometer-scale intranuclear movements (Dundr et al., 2007); 2) forming cross-linked networks that limit nucleoplasmic diffusion or increase mechanical strength of the nucleus (Bohnsack et al., 2006); 3) generating short-range forces that regulate DNA-binding proteins or locally alter chromatin architecture (Zhao et al., 1998); or 4) creating small scaffolds for assembly of nuclear regulatory machinery (Percipalle et al., 2003; Huang et al., 2011). Evaluating these proposals is almost impossible without knowing what the nuclear actin “cytoskeleton” actually looks like, but unfortunately there is little information regarding the distribution and dynamics of actin in somatic nuclei.
Progress in understanding the distribution of actin in the nucleus has been limited by the lack of appropriate reporters. No nuclear actin filaments can be detected when cells are stained with fluorescently labeled phalloidin. The data that do exist derive from either 1) immunofluorescence (Krauss et al., 2003; Schoenenberger et al., 2005) or 2) expression of nucleus-targeted, fluorescent actin derivatives (McDonald et al., 2006; Dopie et al., 2012). Neither method discriminates between monomeric and filamentous actin, and, as a result, their use has yielded little functional insight.
The best information on the dynamics of nuclear actin was provided by McDonald et al. (2006), who measured the mobility of nuclear-targeted, green fluorescent protein (GFP)–tagged actin with two different techniques and obtained two different results. Using fluorescence recovery after photobleaching (FRAP), they measured a very low mobility (0.009 μm2/s), whereas using fluorescence correlation spectroscopy (FCS), they obtained significantly higher values (>0.06 μm2/s). Whether these values report on different mobility populations of nuclear actin—for example, the monomer and filament pools—or arise simply from the differences in the techniques used has not been resolved.
To visualize nuclear actin monomers and filaments in live somatic cells and assess their possible functions, we designed, constructed, and validated a set of fluorescent nuclear actin probes. Probes that bind monomeric actin are concentrated in nuclear speckles, globular structures enriched in pre-mRNA splicing factors (Spector and Lamond, 2011). This localization is consistent with proposed interactions between actin and RNA-processing factors. Filamentous actin, however, does not localize to nuclear speckles but forms a set of punctate structures of more or less uniform size. These structures are scattered throughout the interchromatin space and are excluded from chromatin-rich regions, arguing against direct participation of at least the majority canonical actin filaments in gene regulation or chromatin remodeling. In time-lapse movies (Supplemental Movies S1 and S2), most nuclear actin filaments move diffusively (<x2> ∼ t) but very slowly, with mobility ranging from 0.06 to 0.08 μm2/s. A small fraction of filaments moves even more slowly (∼0.015 μm2/s) and subdiffusively (<x2> ∼ t0.67). Analysis of filament trajectories demonstrates that nuclear actin filaments do not undergo directed motion but exhibit anticorrelated motions at short time scales, consistent with entrapment in an elastic mesh.
The small size and lack of directed motions of nuclear actin filaments argue against primary roles in either intranuclear transport or maintenance of nuclear mechanics. We argue that nuclear actin filaments form short scaffolds that interact with—and may help assemble—a viscoelastic structure in the nucleoplasm. To our knowledge, this is the first direct evidence for the presence of cytoskeletal filaments in the nucleoplasm of live somatic cells. This finding is an important milestone in mapping the physical geography of the nucleus and may provide clues into how nuclear contents are organized.
RESULTS
Design of nuclear actin reporters
To visualize actin in nuclei of live cells, we worked against two obstacles: 1) the low concentration of actin in somatic cell nuclei (Stüven et al., 2003), especially compared with the high concentration of actin in the cytoplasm; and 2) the low permeability of the nucleus to fluorescent probes. We dealt with both obstacles by constructing actin reporters localized almost exclusively to the nucleus. We engineered our probes by fusing an actin-binding domain (ABD) to both a fluorescent protein (enhanced GFP [EGFP]) and three tandem repeats of the nuclear localization sequence (NLS) from SV40 (Figure 1a). We tested ABDs from many previously characterized actin-associated proteins (Table 1 and Supplemental Table S1). To distinguish monomeric from polymeric actin, we used ABDs specific for either actin filaments (FABDs) or actin monomers (monomeric [globular] actin–binding domains [GABDs]).
FIGURE 1:
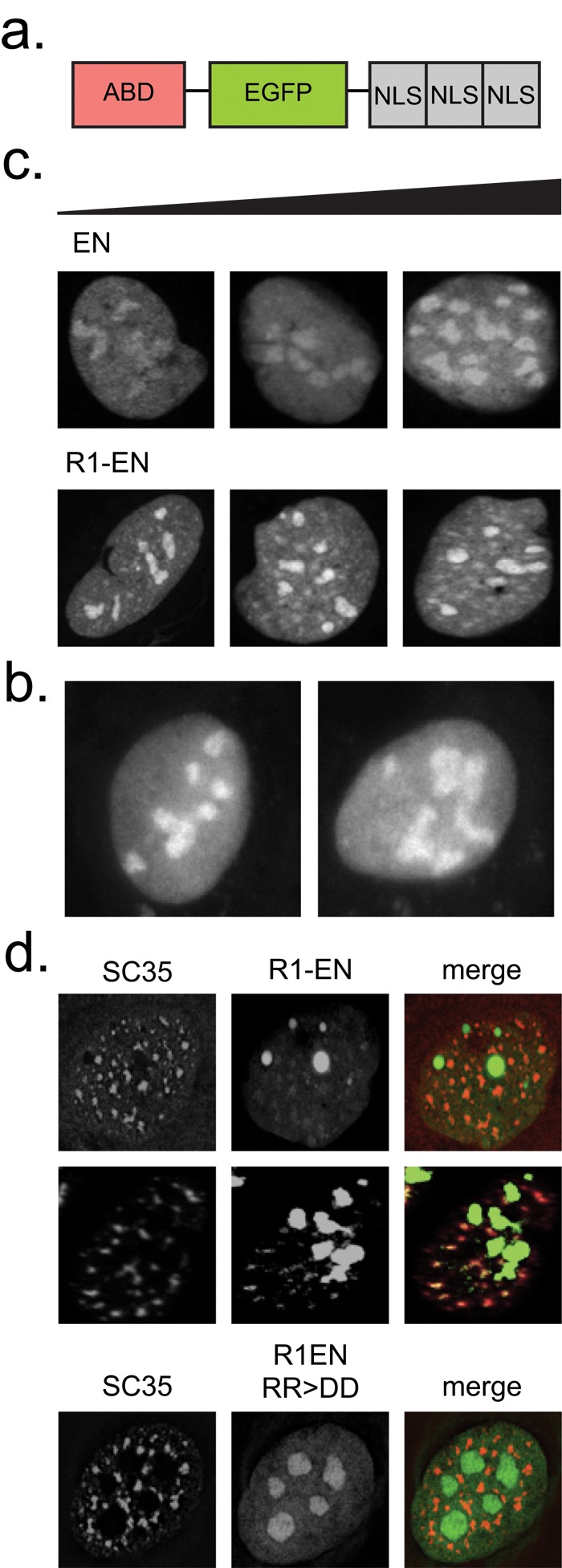
RPEL-based monomeric actin reporters localize to nuclear speckles. (a) Nuclear actin reporter construct design. (b) Localization pattern of EN (right) and NLS-EGFP (left) control constructs in transiently transfected U2OS cells. (c) Localization patterns of EN and R1-EN in transiently transfected U2OS cells. Cellular expression levels for each construct, as determined by the total integrated intensity within the nucleus, increase from left to right. (d) Immunofluorescence assays in U2OS cells transiently transfected with R1-EN or an actin-binding–deficient mutant (RR > DD) and stained with an SC35 antibody. R1-EN images in the second row are enhanced for contrast.
TABLE 1:
Nuclear actin reporter design.
| Domain | Description | Selected member(s) |
|---|---|---|
| WH2 | Actin monomer–binding domain found in nucleators and nucleation-promoting factors | JMY |
| RPEL | Actin monomer–binding domain found in the MAL family of transcriptional coactivators | MAL |
| ERM | Actin filament–binding domain commonly found in focal adhesion components | Ezrin, talin |
| CH | Actin filament–binding domain commonly found in cross-linking proteins | α-Actinin, dystrophin, utrophin |
| HP | Actin filament–binding domain commonly found in focal adhesion components | ABLIM3 |
| Orphan | Unique actin-binding domains | Abl1 FABD, cortactin FABD, vinculin FABD, Lifeact FABD, VASP GABD |
Protein domains tested as candidate nuclear actin reporters. CH, calponin homology; ERM, ezrin/radixin/moesin; HP, headpiece; WH2, WASp-homology 2 domain.
We expressed all our ABD-NLS reporters in human U2OS (osteosarcoma) cells and imaged them by confocal microscopy. We judged the veracity of each construct based on several criteria: 1) whether its localization differed from that of an EGFP-NLS (EN) or NLS-EGFP control; 2) whether it obviously perturbed actin localization or dynamics; and, for FABD-NLS constructs, 3) whether the small amount of reporter remaining in the cytoplasm localized to phalloidin-stainable actin filaments.
Probes that bind monomeric actin localize to nuclear speckles
In U2OS cells, our control construct EN localizes throughout the nucleoplasm and is enriched in nucleoli (Figure 1b). Nucleolar enrichment is commonly observed for proteins targeted strongly to the nucleus by multiple repeats of the SV40 NLS (Shirley et al., 1998). Of our monomer-binding constructs, only the RPEL1 and RPEL2 domains from the transcriptional coactivator, MAL, exhibited a pattern distinct from that of nuclear EGFP controls. RPEL1-EN (R1EN) is distributed through the nucleoplasm and enriched in nucleoli but also localizes to small, nucleoplasmic bodies distinct from nucleoli (Figure 1c). Immunofluorescence reveals that these nucleoplasmic bodies also contain SC35, a marker for nuclear speckles (Figure 1d). To determine whether localization to nuclear speckles reflected interaction with monomeric actin, we expressed an RPEL1 mutant (R81D/R82D) incapable of binding actin (Mouilleron et al., 2008). The point mutations completely abolish colocalization of the probe with SC35 (Figure 1d). These results suggest that monomeric actin is a component of nuclear speckles.
Actin-filament probes identify small particles dispersed through the nucleoplasm
The distributions of most FABD-NLS constructs we tested (Table 1 and Supplemental Table S1) were indistinguishable from nuclear EGFP controls (Supplemental Figure S1) and also failed to detect actin structures in the cytoplasm, suggesting that they are simply not suited for labeling actin filaments in vivo. This failure could be caused by 1) intrinsically low affinity of the ABD for actin filaments or 2) interference from the attached EGFP or NLS. Two of our constructs, however, produced unique nuclear distributions and also recognized actin filaments in the cytoplasm. These constructs contained either the tandem calponin homology (CH) domains from utrophin (Utr261) or the engineered, actin-binding peptide Lifeact, both of which have been used to study in vivo actin filament dynamics (Riedl et al., 2008; Burkel et al., 2007). These two probes recognize filamentous structures in the nucleus that are not observed in nuclear EGFP controls. These structures, however, can also be visualized by phalloidin derivatives and appear to be created by expression of the probes themselves (Figure 2a and Supplemental Figure S1).
FIGURE 2:

Utr230 is a nonperturbing utrophin truncation mutant. (a) Utr261-EN localization in transiently transfected U2OS cells. Cells were fixed and stained with Alexa 564 phalloidin. (b) Structure of Utr261 (Keep et al., 1999; PDB 1QAG). Actin-binding sites are shown in red. Truncation sites for generation of a nonperturbing Utr261 mutant are indicated by yellow arrows. (c) Diagram of Utr261 (human numbering). Truncation positions are indicated by yellow arrows. (d) Localization patterns of EN fusions of utrophin truncation constructs in transiently transfected U2OS cells, consisting of amino acids 1–150 (Utr150-EN), 1–165 (Utr165-EN), 1–205 (Utr205-EN), 1–230 (Utr230-EN), and Utr261-EN.
The mechanism by which Lifeact induces formation of nuclear actin filaments is unclear. Lifeact binds actin monomers with high affinity, even higher than for binding to filaments (Riedl et al., 2008), and so the probe probably promotes import of monomeric actin from the cytoplasm. We could think of no simple way to improve this probe, and so we did not pursue it further.
Utrophin does not bind monomeric actin, so the mechanism by which it perturbs nuclear actin architecture must be different. To understand why the utrophin probe might assemble nuclear actin bundles, we expressed Utr261 in Escherichia coli and purified it to homogeneity. When mixed with purified actin in vitro, Utr261 potently perturbs actin assembly dynamics by stabilizing and bundling actin filaments, even at relatively low concentrations (unpublished observations). We hypothesized that reducing the valency of this interaction might abolish Utr261’s ability to generate ectopic nuclear actin bundles.
Utr261 contains two tandem CH domains, CH type 1 (CH1) and CH type 2 (CH2; Winder et al., 1995). The CH1 domain contains a bipartite binding motif that mediates high-affinity interaction with filaments, whereas CH2 contains a smaller actin-binding motif with weaker affinity (Figure 2, b and c). To create a construct that binds filaments with high affinity but does not bundle them, we made truncation mutants of Utr261. We fused each to EN and expressed them in U2OS cells. The Utr150-EN, Utr165-EN, and Utr205-EN mutants lost the ability to bind cytoplasmic actin filaments and exhibited a nuclear localization similar to the EN control (Figure 2d). However, one truncation mutant, Utr230-EN, bound actin in the cytoplasm and exhibited a remarkably distinct localization pattern in the nucleus: small, distributed puncta. We observed similar localization patterns of Utr230-EN in nuclei of multiple mammalian cell types, including U2OS, UMUC3, and HeLa (Figure 3a).
FIGURE 3:

Utr230-EN binds to native punctate nuclear actin filaments. (a) Confocal sections of HeLa, U2OS, and UMUC3 cells stably expressing Utr230-EN. The second row shows magnifications of the nuclear area. (b) Comparison of average nuclear intensity of U2OS cells stably expressing Utr230-EN with average puncta size (top) and average number of puncta per nucleus (bottom). Data were collected from 97 nuclei. (c) Alignment of the MAL and Myoc RPEL1 domains (adapted from Mouilleron et al., 2008) with actin-binding site 1 of the human α-actinin, utrophin, and dystrophin CH1 domains, revealing a conserved Q R/K R/K T motif. (d) Localization of Utr230-E and Utr230-E QKK > DDD in transiently transfected U2OS cells. (e) Nuclear localization of Utr230-EN and of a transiently transfected predicted actin-binding–deficient mutant (QKK > DDD).
The amount of ectopic actin created by Lifeact- and Utr261-EN probes is proportional to probe expression level. To determine, therefore, whether the punctate nuclear structures identified by Utr230-EN are artifactual, we quantified their number and size as a function of probe expression level. We created a cell line stably expressing Utr230-EN and determined the number and size of actin-containing puncta per nucleus across a range of Utr230-EN expression levels (determined by integrating fluorescence intensity across the entire nucleus). Neither the size nor the number of these nuclear actin structures correlated with the level of Utr230-EN expression (Figure 3b). Although we cannot rule out potential changes in size that occur below the diffraction limit, the absence of large changes in the number or size distribution of the particles argues that they are not created by the probe but represent endogenous actin filament–containing nucleoplasmic structures.
To determine whether Utr230-EN recognizes nucleoplasmic puncta because they contain filamentous actin, we generated a triple point mutant (Utr230-Q33D/K34D/K35D) predicted to be incapable of binding actin. We selected these mutations based on sequence similarity between highly conserved regions of the N-terminal actin-binding site of Utr230 and the RPEL1 domain (Figure 3c). We then tested the mutations’ effect on actin binding in vivo by expressing cytoplasmic versions (lacking nuclear localization signals) of both the mutant and wild-type Utr230-EGFP (Bañuelos et al., 1998; Mouilleron et al., 2008). Wild-type Utr230-EGFP localizes to cytoplasmic actin structures, whereas localization of the triple mutant is completely diffuse (Figure 3d). In the nucleus, the Q33D/K34D/K35D mutant also fails to recognize the punctate structures identified by the wild-type probe (Figure 3e), arguing that they contain filamentous actin.
To further demonstrate that nucleoplasmic puncta identified by Utr230-EN are endogenous structures containing actin filaments, we perturbed nuclear concentrations of actin. Nucleocytoplasmic transport factors importin 9 (IPO9) and exportin 6 (XPO6) shuttle actin into and out of the nucleus, respectively (Stüven et al., 2003; Dopie et al., 2012). We knocked down expression of IPO9 and XPO6 using small interfering RNA (siRNA) and observed the nuclear localization pattern of Utr230-EN puncta. Reduced expression of XPO6 altered the distribution of Utr230-EN from a constellation of small puncta to a handful of much larger, brighter foci (Figure 4, a and b). These large foci appear in the majority (52%) of knockdown cells but only a small fraction of mock-treated (8%) or untreated (9%) cells (Figure 4c). Knocking down expression of the import factor IPO9 produced the opposite effect: the majority of knockdown cells (53%) lack nuclear actin structures, whereas this is true in only a small fraction of mock-treated (12%) or untreated (13%) cells (Figure 4, d–f).
FIGURE 4:

Nuclear actin filament localization is altered by XPO6 and IPO9 levels. (a) Western blot for XPO6 levels 5 d after transient transfection with mock and XPO6 siRNA in lysate prepared from ∼1 million U2OS cells. Hsp70 levels are also indicated as a loading control. (b) Localization of Utr230-EN in mock and XPO6 siRNA cells. (c) Fraction of cells containing aberrant nuclear actin structures 5 d after transient transfection with mock and XPO6 siRNA. Untreated, n = 123; mock, n = 119; XPO6, n = 79; using data pooled from two replicates. (d) Western blot for IPO9 levels 5 d after transient transfection with mock and IPO9 siRNA in lysate prepared from ∼1 million U2OS cells. Hsp70 levels are also indicated as a loading control. (e) Localization of Utr230-EN in mock and IPO9 siRNA cells. (f) Fraction of cells without nuclear actin structures 5 d after transient transfection with mock and IPO9 siRNA. Untreated, n = 123; mock, n = 126; IPO9, n = 95; using data pooled from two replicates.
Phalloidin colocalizes with Utr230-EN after latrunculin B treatment
Previous studies used the actin monomer–sequestering drug latrunculin B (LatB) to probe the function of filamentous actin in the nucleus (Zhao et al., 1998; McDonald et al., 2006; Wu et al., 2006; Ye et al., 2008). To study the effects of LatB on nuclear actin architecture, we treated U2OS cells expressing Utr230-EN with a range of LatB concentrations (0.2–1.0 μM) for 30 min before fixing cells and staining them with 4′,6-diamidino-2-phenylindole (DAPI) and Alexa 568–phalloidin (Figure 5a). After 0.2 and 0.4 μM LatB treatments, significant disassembly of cytoplasmic actin structures occurred while nuclear actin puncta appeared unaffected.
FIGURE 5:

LatB treatment induces intranuclear actin rod formation. (a) U2OS cells stably expressing Utr230-EN treated with LatB concentrations between 0.2 and 1 μM in medium for 30 min at 37°C. Cells were fixed and stained with Alexa Fluor 564–phalloidin and DAPI. (b) Inset from 0.4 μM LatB treatment revealing colocalization between phalloidin and Utr230-EN.
Of interest, even though we detected no change in nuclear actin organization at 0.4 μM LatB treatment, we began to detect phalloidin staining of the nuclear structures recognized by Utr230-EN (Figure 5b). The same nuclear actin puncta were also observed by phalloidin staining in cells treated with 0.4 μM LatB that are not expressing Utr230-EN (Supplemental Figure S2), further arguing that our probe recognizes endogenous actin-containing structures. This concentration of LatB disassembles almost all phalloidin-stainable filaments in the cytoplasm, so the colocalization between Utr230-EN and phalloidin may become visible due to the decreased phalloidin signal from the cytoplasm.
Higher concentrations of LatB perturbed nuclear actin organization. At 0.6 μM the size of nuclear actin puncta actually increased, and at 0.8 μM LatB intranuclear actin rods, detectable with both Utr230-EN and phalloidin, appeared in the majority of cells (as previously reported; Pendleton et al., 2003). Even higher concentrations of LatB eventually resulted in disassembly of nuclear actin structures, and at concentrations ≥1.0 μM we could detect no nuclear actin structures with either phalloidin or Utr230-EN. We observed very similar effects on nuclear actin architecture in cells treated with cytochalasin D (Supplemental Figure S2).
Nuclear actin filaments are enriched in the interchromatin space
We used immunofluorescence to look for colocalization between nuclear actin filaments and a variety of nuclear landmarks and nuclear actin-binding proteins. Curiously, we find that the Utr230-EN actin reporter does not colocalize with RNA polymerase I, II, or III and is not associated with H3K9me3 heterochromatin (Figure 6, a–d). DAPI staining of Utr230-EN–expressing cells throughout the cell cycle reveals that nuclear actin puncta are generally excluded from chromatin (Figure 6e). In fact, nuclear actin filaments do not strongly colocalize with any nuclear landmark we tested, including elements of the nuclear lamina, nuclear matrix proteins, nucleoli, nuclear speckles, PML bodies, and telomeres (Supplemental Figure S3).
FIGURE 6:

Nuclear actin filaments are enriched in the interchromatin space. Immunofluorescence in U2OS cells stably expressing Utr230-EN and stained with antibodies for (a) RNA polymerase I marker PAF49, (b) RNA polymerase II, (c) RNA polymerase II marker POLR3F, (d) H3K9me3 heterochromatin, (f) nuclear myosin 1 (NM1), (g) Baf53a/Arp4, (h) coronin 2A (CORO2A), (i) lamin A/C. (e) DAPI staining in fixed U2OS cells stably expressing Utr230-EN throughout the cell cycle.
We next looked for colocalization between nuclear actin filaments and several actin-associated proteins reported to be in the nucleus: nuclear myosin I (NM1), Baf53a/Arp4 (a marker for all human actin-containing chromatin remodelers), coronin 2A (CORO2A), and lamin A/C (Figure 6, f–i). We observed little or no colocalization of nuclear actin with these proteins or with other chromatin-remodeling markers, including p400 (Swr1 complex), Brg1 (BAF complex), and histone H2AZ (Supplemental Figure S3).
Binding of Utr230-EN to nuclear actin puncta may occlude interactions between actin and chromatin-based complexes. Several reports suggest that the interaction of actin with transcription and remodeling factors is required for normal nucleic acid synthesis (e.g., Ye et al., 2008). To test whether the binding of Utr230-EN with actin blocks the association of actin with chromatin, we performed incorporation assays for 5-bromo-2′-deoxyuridine (BrdU) and 5-ethynyl uridine (EU; a BrUTP analogue) in cells expressing Utr230-EN to determine whether nucleic acid synthesis was inhibited. The distribution of cells in the 4N state as reported by BrdU incorporation, a measure of DNA synthesis, was reduced by Utr230-EN expression (Supplemental Figure S4). However, since a similar reduction was observed for cells expressing EN, we believe this effect is a consequence of nuclear protein overexpression. In our EU incorporation assays, there was no significant decrease in RNA synthesis in cells expressing Utr230-EN (Supplemental Figure S4). The absence of defects in nucleic actin synthesis specific to Utr230-EN expression indicates that Utr230-EN–bound actin is likely distinct from the pools of nuclear actin that participate directly in chromatin-based processes.
Dynamics of nuclear actin filaments
We imaged live U2OS cells expressing Utr230-EN by time-lapse confocal microscopy to determine the dynamics of nuclear actin structures. The trajectories of individual particles (Figure 7a) were calculated using the MATLAB particle tracking package u-Track (Jaqaman et al., 2008). On average, nuclear actin particles move with an apparent diffusion coefficient of ∼0.07 μm2/s and a time-dependent value of α, the scaling exponent for the relationship of the mean-squared displacement versus time (Figure 7b, inset, and Supplemental Figure S5). Theoretically, time dependence in the α value can indicate that the particles are confined within a finite volume, in which the displacement of the particles approaches some limiting value. In our case, however, we find that the time dependence of α is simply an artifact of confocal imaging. The fastest and most highly diffusive particles are the most likely to exit the imaging plane early in the time course, resulting in an enrichment of the remaining, less mobile particles as the observed trajectory length increases. This selection for slower particles at long time scales decreases the apparent average particle speed over time, causing the displacement curve to appear to plateau. Consistent with this interpretation, binning our data for particle trajectories by their length reveals that both the apparent diffusion coefficient and α decrease as trajectory length increases (Figure 7, b and c).
FIGURE 7:

Dynamics of nuclear actin filaments. (a) Trajectories of F-actin puncta in a single confocal slice of a live U2OS nucleus over 30 s at 50-ms resolution. 100 pixels = 9.1 μm. (b) Double-logarithmic plot of mean-squared displacement (MSD) vs. time for nuclear actin puncta with variable trajectory lengths. The fitted line is for the longest trajectories only (>15 s in length) and has a slope of ∼0.67. Inset, linear plot of MSD vs. time, with average values for all nuclear actin trajectories (red) and for a simulated random walk with a diffusion coefficient 0.07 μm2/s (blue). y-axis, MSD in μm2/s; x-axis, time in seconds. N = 25,000 particles for both observed and simulated data. (c) Average apparent diffusions coefficients of all nuclear actin particles as a function of trajectory length. N = 25,000 particles. (d) Distribution of apparent diffusion coefficients for all nuclear actin particles. N = 25,000. (e) SCI values for 10 representative nuclear actin trajectories from a single cell during the first 50 frames (2.5 s) of their trajectories. (f) VCF values averaged from all 0.5-s windows within nuclear actin trajectories (red) and trajectories for a simulated random walk (blue). N = 25,000 particles for both observed and simulated data. (g) Time-lapse image series of U2OS nuclei in cells stably expressing Utr230-mEos2-NLS before and after photoconversion at 405 nm. (h) Average relative mEos2 fluorescence recovery at 488 nm after photoactivation in nuclear and cytoplasmic actin filaments. Cytoplasmic actin, n = 12,000 foci; nuclear actin, n = 16,000 foci.
Particles with shorter recorded trajectories appear to move diffusively, with an apparent diffusion coefficient of ∼0.07 μm2/s and α approaching 1. Particles with the longest recorded trajectories are slower and move more subdiffusively, with an apparent diffusion coefficient of ∼0.015 μm2/s and α of ∼0.67. These diffusion coefficients are unexpectedly low, being at least an order of magnitude below the 0.5–5 μm2/s range reported for transcription factors in the nucleus (Gorski et al., 2008) and the 57 μm2/s value reported for soluble GFP (Houtsmuller et al., 1999). The particles observed over short and long trajectories might represent functionally distinct populations of actin since, in addition to the variation in α, the apparent diffusion coefficients among short- and long-lived particles do not fall into a single uniform distribution (Figure 7d).
We looked for directed motion of nuclear actin filaments by calculating the speed correlation index (SCI) for all of our calculated particle trajectories (Bouzigues and Dahan, 2007). Briefly, the SCI reveals how persistently a particle moves in the same direction; periods of directed motion produce high, positive SCI values across multiple, consecutive time points. Overall we found that nuclear actin particles undergo less persistent directed motion than simulated random walks. In our data, only 0.21% of 3343 nuclear actin trajectories (each with a length of 50–100 frames) scored an SCI of >0.75 for five or more consecutive frames, compared with 1.08% of 5000 trajectories of a simulated random walk. Not only are the positive directional correlations weak, but also our data are generally skewed toward negative correlations. That is, for a particle moving in a given direction at a given time, its movement soon afterward is biased in the opposite direction. This negative bias is clear in plots of SCI values of individual trajectories (Figure 7e). From the absence of directed motion and the small size of the actin-containing structures (Supplemental Figure S5), we conclude that this nuclear actin pool is unlikely to contribute to long-range (micrometer-scale) transport events.
We were intrigued by the negative skew in the speed correlation index of nuclear actin-containing particles. To investigate this phenomenon further, we calculated the average correlation between the direction of particle motion at the beginning of a trajectory (the first two frames) and its direction of motion at all subsequent time points in the trajectory (Weber et al., 2010a,b). The resulting velocity autocorrelation function (VCF) reveals that, over a short time scale, the velocity autocorrelation is indeed negative for actin trajectories (Figure 7f). One physical interpretation of this result is that nuclear filaments are embedded in a viscoelastic medium, such as a cytoskeletal or nucleic acid polymer networks, that provides an elastic force opposing motion (Weber et al., 2010a). Because nuclear actin puncta are excluded from chromatin, we conclude that that this force must arise from a viscoelastic network within the interchromatin domain, perhaps consisting of nuclear matrix proteins.
Finally, to estimate the rate of filament turnover within nuclear actin structures, we performed photoconversion experiments in cells expressing a photoswitchable variant of our nuclear actin reporter, Utr230-mEos2-NLS (McKinney et al., 2009; Figure 7g). Fluorescence recovery curves for nuclear F-actin puncta show an average t1/2 of 195 s, remarkably similar to the 217 s that we calculated for cytoplasmic actin stress fibers analyzed in the same experiments (Figure 7h). From these curves we conclude that the kinetics of nuclear F-actin assembly and disassembly are comparable to those found in cytoplasmic actin structures.
DISCUSSION
Before trusting a nuclear actin probe, one must demonstrate that 1) it actually binds actin in vivo, 2) its localization in the nucleus reflects binding to actin, and 3) its expression does not alter the distribution of nuclear actin. When targeted to the nucleus, the most widely used probes for studying filamentous actin in live cells, Lifeact and Utr261, perturb nuclear actin architecture. We therefore designed and tested several new families of probes for nuclear actin. Our best nuclear actin filament probe, Utr230-EN, passed all three of the tests listed. In the cytoplasm it labels actin filaments in a pattern indistinguishable from that of fluorescent phalloidin. In the nucleus, its localization is disrupted by point mutations that abolish actin binding and by perturbation of the nuclear actin concentration. Changes in its expression level do not alter the number or size of particles recognized by Utr230-EN. Finally, when cytoplasmic actin is depolymerized by latrunculin B, fluorescent phalloidin recognizes similar nuclear structures in untransfected cells.
The size distribution of nuclear actin filaments, as judged by Utr230-EN intensity, is approximately Gaussian (Supplemental Figure S5), which suggests that the filaments have a more or less fixed size. Filaments free to assemble and disassemble, in contrast, would eventually achieve an exponential length distribution (Sept et al., 1999), whereas filaments that break and anneal should follow a power-law distribution (Brangwynne et al., 2011; Foret et al., 2012). The peak in the Utr230-EN distribution corresponds to a short (<100 subunit), fixed-length actin filament, possibly similar to fixed-length filaments found in the membrane skeleton of red blood cells or associated with the Golgi apparatus (Colón-Franco et al., 2011). It is tempting to speculate that nuclear actin filaments share functional and regulatory features with these short, membrane-associated filaments.
The localization and morphology of nuclear actin filaments are inconsistent with several functions previously proposed for them. For example, the spatial restriction of actin filaments predominantly to the interchromatin space argues against direct involvement of conventional actin filaments in transcription and chromatin remodeling (Fomproix and Percipalle, 2004). They are also not long enough (Supplemental Figure S5) to serve as tracks for long-range transport of cargo (e.g., genetic loci) through the nucleus (Dundr et al., 2007). In addition, we observed no directed motion of nuclear actin filaments and little or no colocalization between nuclear actin filaments and known nuclear myosins.
Our results shed new light on the physical nature of somatic cell nucleoplasm. Some nuclear actin filaments move diffusively, whereas the motion of others is restricted. The subdiffusive motions we observe for some filaments likely result from interactions with larger nuclear structures of very low mobility. Of interest, at short time scales, the motion of most actin filaments was anticorrelated, characteristic of particles embedded in a viscoelastic medium (Weber et al., 2010a). What is responsible for the viscoelasticity of the nucleoplasm? The mechanics of the cytoplasm are dominated by the properties of actin filament networks, but the actin filaments we observe in the nucleus are too small and dispersed to themselves account for a significant amount of viscoelasticity. One obvious candidate is chromatin, but this is not consistent with the localization of nuclear actin to the interchromatin space. A better candidate is a nucleoplasmic structure consisting of lamins and other filamentous or filament-associated proteins in the nucleus (Shimi et al., 2008). More work is required to determine whether nuclear actin filaments are simply trapped in such a structure or function as scaffolds that help organize it.
Although our results rule out direct participation of actin in motor-directed intranuclear transport, actin filaments may interact with other nucleoplasm components (e.g., lamins) to form a three-dimensional mesh or matrix. Such a structure would explain previous observations of actin's effect on intranuclear transport. Dundr et al. (2007), for example, tracked the motion of an array of U2 small nuclear RNA minigenes to Cajal bodies after transcriptional activation. They observed two modes of motion: short-range diffusion interspersed with transient periods of high mobility in the direction of the Cajal body. Because these high mobility bursts were absent after expression of the dominant-negative, nonpolymerizing R62D actin mutant, they speculated that the high-mobility bursts result from motor-dependent transport of the U2 array along actin tracks. It is possible, however, that the bursts of higher mobility represent “hopping” of particles between zones of confinement, or corrals (Saxton, 1995).
Our time-lapse imaging results explain some discrepancies with respect to the previous study on nuclear actin dynamics by McDonald et al. (2006). The different mobilities of nuclear actin that they measured using FRAP (0.009 μm2/s) and FCS (>0.06 μm2/s) correspond surprisingly well to the two distinct populations of nuclear actin filaments we observed: the small subset of subdiffusive puncta (<5% of all particles) with an apparent mobility of 0.015 μm2/s, and the larger, diffusive population with an average diffusion coefficient of 0.07 μm2/s. The FCS measurements of McDonald et al. (2006) likely counts more molecules from the fast-moving pool, whereas the FRAP measurement may be more sensitive to the slow-moving filament pool.
The RPEL domain from MAL binds monomeric but not filamentous actin, so we constructed an RPEL-based probe to study monomeric actin in the nucleus. In addition to a diffuse, nucleoplasmic localization, this probe accumulates in globular, nuclear bodies that contain SC35. This colocalization indicates that actin monomers concentrate in nuclear speckles, a compartment previously shown to also contain phosphorylated forms of myosin V (Pranchevicius et al., 2008). This suggests a role for monomeric actin in RNA processing or the staging of RNA-processing factors, which are believed to be the primary function of nuclear speckles. The difference in localization between nuclear actin filaments and monomers argues that these actin pools are functionally distinct.
Our Utr230-EN– and RPEL-based actin probes represent a significant expansion of the molecular toolbox for the in vivo study of actin dynamics. They gave us the first detailed view of the architecture and dynamics of nuclear actin. Further progress in understanding the function of nuclear actin will require not only nucleus-specific probes, but also nucleus-specific tools for perturbing actin assembly and localization.
MATERIALS AND METHODS
Molecular biology
FABD constructs were cloned from full-length human cDNA using standard techniques. JMY WH2 and MAL RPEL domain constructs were cloned from full-length human recombinant proteins. The VASP GABD was cloned from a full length Dictyostelium recombinant protein. p-RSETA-mEos2 (McKinney et al., 2009) was acquired from Addgene (Cambridge, MA; plasmid 20341). Mutagenesis was performed using the QuikChange mutagenesis kit and primer design tool (Stratagene, Santa Clara, CA). We used pEGFP-C1 (Clontech, Mountain View, CA) as the host vector for all EGFP fusions, with N-terminal EGFP fusions inserted into the unique AgeI and NheI sites. Primer sequences are available upon request.
Cell culture
HeLa, U2OS, and UMUC3 cells (American Type Culture Collection, Manassas, VA) were cultured in DMEM supplemented with 10% fetal bovine serum, 2 mM l-glutamine, nonessential amino acids, and penicillin–streptomycin (University of California, San Francisco [UCSF], Cell Culture Facility) at 37°C with 5% CO2. For transient transfection of all ABD constructs other than Utr230-EN, cells were transfected using Lipofectamine LTX (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. For transient transfection of Utr230-EN, cells were transfected with Xtreme-GENE HP (Roche, Indianapolis, IN) according to the manufacturer's protocol. All transient transfections were performed 24–72 h before data collection. Stable cell lines of Utr230-EGFP-NLS and Utr230-mEos2-NLS were generated by lentiviral expression constructs based on the plasmids from the Trono lab (see Li et al., 2004) and containing the selectable marker from pSMPUW-IRES-Blasticidin (Cell Biolabs, San Diego, CA).
siRNA
Mock siRNA and human XPO6 and IPO9 Silencer Select siRNAs were purchased from Invitrogen. Transient reverse transfection using Lipofectamine RNAiMAX was performed on stable Utr230-EN U2OS lines according to the manufacturer's protocol. At 15–20 h after transient transfection, the cell medium was replaced. Cells were split into flasks and/or fibronectin coverslips 3 d after transfection and were either fixed for imaging or lysed for Western blotting at 5 d after transfection.
Western blotting
About 1 million cells were harvested, washed with ice-cold phosphate-buffered saline (PBS), and pelleted in a microfuge by spinning at 1000 rpm for 5 min at 4ºC. Cell pellets were lysed by resuspension in 2 × SDS sample buffer with 1 mM Pefabloc and boiled before SDS–PAGE. For each sample we blotted against HSP70 as a loading control. Standard methods were used for immunoblotting, using 1:500 Rb anti-XPO6 (Abcam, Cambridge, MA), 1:500 Rb anti-IPO9 (Abcam), and 1:1000 mouse anti-HSP70 (Santa Cruz Biotechnology, Santa Cruz, CA) with overnight incubation at 4ºC. Horseradish peroxidase–conjugated secondaries (Jackson ImmunoResearch Laboratories, West Grove, PA) were used at 1:10,000, and ECL reagent (SuperSignal West Pico; Pierce, Rockford, IL) was used according to the manufacturer's instructions.
Immunofluorescence and staining
Cells were passaged onto glass coverslips coated with 10 μg/ml fibronectin (Invitrogen) and cultured overnight to 30–60% confluence. Coverslips were fixed at room temperature for 30 min in 3.7% paraformaldehyde (prepared from Formalin; Sigma-Aldrich, St. Louis, MO) in PBS (UCSF Cell Culture Facility). Cells were permeabilized in 0.1% Triton-X-100 in PBS for 3–5 min and blocked in 5% goat serum/PBS at room temperature for 60 min. Primary antibody incubations were performed for 60 min at room temperature in 5% goat serum/PBS. Cells were incubated in Alexa Fluor 568–labeled secondary antibodies (Invitrogen) in 5% goat serum/PBS for 30 min at room temperature and mounted on slides with fluorescent mounting medium (DakoCytomation, Hamburg, Germany). For phalloidin and DAPI staining, cells were fixed as described and stained for 15 min in 0.1% Triton/PBS with 0.5 μg/ml DAPI (Sigma-Aldrich) or 0.7 U/ml Alexa Fluor 568–phalloidin (Invitrogen). Dilutions and product numbers for antibodies used for immunofluorescence are available upon request.
BrdU and EU incorporation
For BrdU incorporation, cells grown on coverslips were incubated in 1 × BrdU labeling reagent (Invitrogen) in complete medium for 2 h at 37°C with 5% CO2. After incubation, cells were fixed, permeabilized, and blocked as described. Coverslips were then incubated for 60 min at room temperature in 1:500 anti-EGFP antibody (Abcam) in 5% goat serum/PBS. The cells were washed and incubated for an additional 30 min at room temperature in 1:500 Alexa 568 secondary (Invitrogen). Cells were washed in PBS and permeabilized in 1% Triton-X/PBS for 3 × 5 min. The nuclear BrdU epitope was exposed by treatment with 1 N HCl for 10 min at room temperature, followed by 10 min at room temperature and 20 min at 37°C in 2 N HCl. The acid was neutralized in 0.1 M sodium borate for 12 min at room temperature. The cells were permeabilized in 1% Triton-X/PBS for 3 × 5 min at room temperature and incubated overnight at 4°C in 1:50 anti-BrdU (Invitrogen). Anti-BrdU–stained coverslips mounted on slides with fluorescent mounting medium (DakoCytomation). EU incorporation was performed using the Click-It RNA Alexa 594 imaging kit from Invitrogen. Cells were incubated in 1 mM EU in complete medium for 45 min at 37°C with 5% CO2. After EU incubation, the EU detection reaction was performed according to the manufacturer's protocol, with a twofold reduction in Alexa 594 azide concentration from the recommended procedure to reduce background signal. BrdU and EU incorporation levels in fixed cells were acquired using epifluorescence microscopy. The integrated intensity of the nuclear signals for BrdU and EU nucleic acids was measured in ImageJ (National Institutes of Health, Bethesda, MD). The intensities were normalized by nuclear area and plotted using R.
Microscopy
Confocal images for particle tracking were collected using an Eclipse TI-E Motorized Inverted Microscope (Nikon, Melville, NY) equipped with a Yokogawa CSU22 Spinning Disk Confocal and Photometrics Evolve electron-multiplying charge-coupled device camera using a 100×/1.40 numerical aperture (NA) Plan Apo Nikon objective (UCSF Nikon Imaging Center). Confocal data were acquired with MicroManager software (Stuurman et al., 2007). All other images were taken using DeltaVision RT system (Applied Precision, Issaquah, WA) with a CoolSnapHQ camera (Photometrics, Tucson, AZ) using a 100×/1.40 NA UPlanSApo objective (Olympus, Tokyo, Japan). For live-cell imaging, cells were split onto polylysine-coasted Mat-Tek dishes and kept at 37°C with 5% CO2 during image acquisition. Images were processed for contrast enhancement and noise reduction using ImageJ.
Photoconversion
U2OS cells stably expressing the Utr230-mEos2-NLS construct were photoconverted with a 405-nm laser and then imaged at 568 nm in 10-s intervals for 10 min. To reduce bleaching of the highly unstable green state, images were taken at 488 nm only every fifth time point. The time courses for the two channels were then synced and cropped into converted or unconverted regions of interest (ROIs) in FIJI (Fiji Is Just ImageJ; http://fiji.sc), and the particles inside each ROI were analyzed in CellProfiler (Carpenter et al., 2005). Normalization for photobleaching and curve fitting were mathematically calculated in Python, primarily with the SciPy package (www.scipy.org). Plots of recovery curves were generated in R.
Particle tracking and analysis
Particle trajectories were calculated using the u-Track MATLAB software package (Jaqaman et al., 2008) using the default parameters. Calculation of the speed correlation index was performed as described in Bouzigues and Dahan (2007) using a window size of three frames for averaging and implemented in Python. Calculation of the VCF was performed as described in Weber et al. (2010a) and implemented in Python. All additional analysis of particle trajectories was implemented in Python, and all figures were generated using R. To briefly expand on the description of VCF calculation given in Weber et al. (2010a) in the velocity autocorrelation function calculation, the velocity of each particle was determined for every pair of adjacent frames within its trajectory (e.g., particle velocity v0 was calculated between frames 1 and 2, velocity v1 was calculated between frames 2 and 3, and so on). The velocity autocorrelation was then calculated as the sine of the angles between the particle velocity at each time t and the velocity at time t + δ for δ ranging from 50 to 500 ms. Thus a value of 1 indicates a strong positive velocity autocorrelation, in which the particle motion at time t is in the same direction as at time t + δ, and a value of −1 indicates a strong anticorrelation, in which the particle motion at time t is in the opposite direction as at time t + δ. The plotted velocity autocorrelation function is the average of the velocity autocorrelation values for all particle trajectories. Documentation and code for all particle trajectory analyses are available upon request.
Drug treatments
Latrunculin B (Biomol International, Plymouth, PA) and cytochalasin D (Sigma-Aldrich) were diluted in EtOH and used in warm medium. After replacement of old media with drugged medium, cells were incubated for the indicated time at 37°C with 5% CO2.
Supplementary Material
Acknowledgments
This project was conducted in part at the Marine Biological Laboratory (Woods Hole, MA) and relied heavily on the Nikon Imaging Center at the University of California, San Francisco. This bulk of this work was supported by a grant from the National Institutes of Health to R.D.M. (5R01GM061010-12). Additional support was provided by National Institutes of Health Grant R01 CA096840 (E.H.B.), a National Science Foundation Predoctoral Fellowship (B.B.), a National Institutes of Health Ruth L. Kirschstein Predoctoral Fellowship (B.B.), and a Genentech Fellowship (B.C.). We are grateful to Barbara Panning for the use of antibodies for numerous nuclear markers; Richard Cameron for his MYO16B antibodies; and members of the Vale, Blackburn, and Mullins labs, especially J. Brad Zuchero, for reagents and helpful discussion. We also thank Cliff Brangwynne, Thoru Pederson, and Robert Goldman for insightful comments on the manuscript.
Abbreviations used:
- ABD
actin-binding domain
- CH
calponin homology
- EN
enhanced green fluorescent protein–3X nuclear localization sequence
- FABD
filamentous actin–binding domain
- FCS
fluorescence correlation spectroscopy
- GABD
monomeric (globular) actin–binding domain
- IPO9
importin 9
- LatB
latrunculin B
- MSD
mean-squared displacement
- SCI
speed correlation index
- VCF
velocity autocorrelation function
- XPO6
exportin 6
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-09-0685) on February 27, 2013.
REFERENCES
- Bañuelos S, Saraste M, Djinovic’ Carugo K. Structural comparisons of calponin homology domains: implications for actin binding. Structure. 1998;6:1419–1431. doi: 10.1016/s0969-2126(98)00141-5. [DOI] [PubMed] [Google Scholar]
- Bohnsack MT, Stüven T, Kuhn C, Cordes VC, Gorlich D. A selective block of nuclear actin export stabilizes the giant nuclei of Xenopus oocytes. Nat Cell Biol. 2006;8:257–263. doi: 10.1038/ncb1357. [DOI] [PubMed] [Google Scholar]
- Bouzigues C, Dahan M. Transient directed motions of GABAA receptors in growth cones detected by a speed correlation index. Biophys J. 2007;92:654–660. doi: 10.1529/biophysj.106.094524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Mitchison TJ, Hyman AA. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc Natl Acad Sci USA. 2011;108:4334–4339. doi: 10.1073/pnas.1017150108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkel BM, Von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton. 2007;64:822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T, Merriam R. Diffusible and bound actin in nuclei of Xenopus laevis oocytes. Cell. 1977;12:881–891. doi: 10.1016/0092-8674(77)90152-0. [DOI] [PubMed] [Google Scholar]
- Clark T, Rosenbaum J. An actin filament matrix in hand-isolated nuclei of X. laevis oocytes. Cell. 1979;18:1101–1108. doi: 10.1016/0092-8674(79)90223-x. [DOI] [PubMed] [Google Scholar]
- Colón-Franco JM, Gomez TS, Billadeau DD. Dynamic remodeling of the actin cytoskeleton by FMNL1γ is required for structural maintenance of the Golgi complex. J Cell Sci. 2011;124:3118–3126. doi: 10.1242/jcs.083725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle P, Serebryannyy L. Nuclear actin and myosins: life without filaments. Nat Cell Biol. 2011;13:1282–1288. doi: 10.1038/ncb2364. [DOI] [PubMed] [Google Scholar]
- Dopie J, Skarp KP, Kaisa Rajakylä E, Tanhuanpää K, Vartianen MK. Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci USA. 2012;109:544–52. doi: 10.1073/pnas.1118880109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr M, Ospina JK, Sung M, John S, Upender M, Ried T, Hager GL, Matera AG. Actin-dependent intranuclear repositioning of an active gene locus in vivo. J Cell Biol. 2007;176:1095–1103. doi: 10.1083/jcb.200710058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrants AO. Chromatin remodelling and actin reorganization. FEBS Lett. 2008;585:2041–2050. doi: 10.1016/j.febslet.2008.04.032. [DOI] [PubMed] [Google Scholar]
- Fomproix N, Percipalle P. An actin-myosin complex on actively transcribing genes. Exp Cell Res. 2004;294:140–148. doi: 10.1016/j.yexcr.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Foret L, Dawson JE, Villaseñor R, Collinet C, Deustch A, Brusch L, Zerial M, Kalaidzidis Y, Jülicher F. A general theoretical framework to infer endosomal network dynamics from quantitative image analysis. Curr Biol. 2012;22:1381–1390. doi: 10.1016/j.cub.2012.06.021. [DOI] [PubMed] [Google Scholar]
- Gall JG, Wu Z. Examining the contents of isolated Xenopus germinal vesicles. Methods. 2010;51:45–51. doi: 10.1016/j.ymeth.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski S, Snyder S, John S, Grummt I, Misteli T. Modulation of RNA polymerase assembly dynamics in transcriptional regulation. Mol Cell. 2008;30:486–497. doi: 10.1016/j.molcel.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann WA, Arduini A, Nicol SM, Camacho CJ, Lessard JL, Fuller-Pace FV, de Lanerolle P. SUMOylation of nuclear actin. J Cell Biol. 2009;186:193–200. doi: 10.1083/jcb.200905016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtsmuller AB, Rademakers S, Nigg AL, Hoogstraten D, Hoeijmakers JH, Vermeulen W. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science. 1999;284:958–961. doi: 10.1126/science.284.5416.958. [DOI] [PubMed] [Google Scholar]
- Huang W, et al. Coronin 2A mediates actin-dependent de-repression of inflammatory response genes. Nature. 2011;470:414–418. doi: 10.1038/nature09703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida K, Matsumoto S, Yahara I. The KKRKK sequence is involved in head-shock induced nuclear translocation of the 18-kDa actin-binding protein, cofilin. Cell Struct Funct. 1992;17:39–46. doi: 10.1247/csf.17.39. [DOI] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keep NH, Winder SJ, Moores CA, Walke S, Norwood FL, Kendrick-Jones J. Crystal structure of the actin-binding region of utrophin reveals a head-to-tail dimer. Structure. 1999;7:1539–1546. doi: 10.1016/s0969-2126(00)88344-6. [DOI] [PubMed] [Google Scholar]
- Kiseleva E, Drummond SP, Goldberg MW, Rutherford SA, Allen TD, Wilson KL. Actin- and protein-4.1-containing filaments link nuclear pore complexes to subnuclear organelles in Xenopus oocyte nuclei. J Cell Sci. 2004;117:2481–2490. doi: 10.1242/jcs.01098. [DOI] [PubMed] [Google Scholar]
- Krauss S, Chen C, Penman S, Heald R. Nuclear actin and protein 4.1: Essential interactions during nuclear assembly in vitro. Proc Natl Acad Sci USA. 2003;100:10752–10757. doi: 10.1073/pnas.1934680100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumeta M, Yoshimura SH, Harata M, Takeyasu K. Molecular mechanisms underlying nucleoplasmic shuttling of actinin-4. J Cell Sci. 2010;123:1020–1031. doi: 10.1242/jcs.059568. [DOI] [PubMed] [Google Scholar]
- Li S, Rosenberg JE, Donjacour AA, Botchkina IL, Hom YK, Cunha GR, Blackburn EH. Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and Anti-telomerase short-interfering RNA. Cancer Res. 2004;64:4833–40. doi: 10.1158/0008-5472.CAN-04-0953. [DOI] [PubMed] [Google Scholar]
- Loy CJ, Sim KS, Yong EL. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc Natl Acad Sci USA. 2003;100:4562–4567. doi: 10.1073/pnas.0736237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Carrero G, Andrin C, de Vries G, Hendzel MJ. Nucleoplasmic b-actin exists in a dynamic equilibrium between low-mobility polymeric species and rapidly diffusing populations. J Cell Biol. 2006;172:541–552. doi: 10.1083/jcb.200507101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat. Methods. 2009;6:131–3. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouilleron S, Guettler S, Langer CA, Treisman R, McDonald NQ. Molecular basis for G-actin binding to RPEL motifs from the serum response factor coactivator MAL. EMBO J. 2008;27:3198–208. doi: 10.1038/emboj.2008.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton A, Pope B, Weeds A, Koffer A. Latrunculin B or ATP depletion induces cofilin-dependent translocation of actin into nuclei of mast cells. J Biol Chem. 2003;278:14394–14400. doi: 10.1074/jbc.M206393200. [DOI] [PubMed] [Google Scholar]
- Percipalle P, Fomproix N, Kylberg K, Miralles F. An actin-ribonucleoprotein interaction is involved in transcription by RNA polymerase II. Proc Natl Acad Sci USA. 2003;100:6475–6480. doi: 10.1073/pnas.1131933100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestic-Dragovich L, Stojiljkovic L, Philimonenko A, Nowak G, Ke Y, Settiage RE, Shabanowitz J, Hunt DF, Hozek P, de Lanerolle P. A myosin I isoform in the nucleus. Science. 2000;290:337–341. doi: 10.1126/science.290.5490.337. [DOI] [PubMed] [Google Scholar]
- Pranchevicius MC, Baqui MM, Ishikawa-Ankerhold HC, Lourenço EV, Leão RM, Banzi SR, dos Santos CT, Roque-Barreira MC, Espreafico EM, Larson RE. Myosin Va phosphorylated on Ser1650 is found in nuclear speckles and redistributes to nucleoli upon inhibition of transcription. Cell Motil Cytoskeleton. 2008;65:441–456. doi: 10.1002/cm.20269. [DOI] [PubMed] [Google Scholar]
- Riedl J, et al. Lifeact: a versatile marker to visualize F-actin. Nat Meth. 2008;5:606–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton MJ. Single-particle tracking: effects of corrals. Biophys J. 1995;69:389–98. doi: 10.1016/S0006-3495(95)79911-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer U, Hinssen S, Franke WW, Jockusch BM. Microinjection of actin-binding proteins and actin antibodies demonstrates involvement of nuclear actin in transcription of lampbrush chromosomes. Cell. 1984;39:111–112. doi: 10.1016/0092-8674(84)90196-x. [DOI] [PubMed] [Google Scholar]
- Schoenenberger CA, Buchmeier S, Boerries M, Sütterlin R, Aebi U, Jockusch BM. Conformation-specific antibodies reveal distinct actin structures in the nucleus and in the cytoplasm. J Struct Biol. 2005;152:157–168. doi: 10.1016/j.jsb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Sept D, Xu J, Pollard TD, McCammon JA. Annealing accounts for the length of actin filaments formed by spontaneous polymerization. Biophys J. 1999;77:2911–2919. doi: 10.1016/s0006-3495(99)77124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimi T, et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 2008;22:3409–3421. doi: 10.1101/gad.1735208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley RL, Lelivelt MJ, Schenkman LR, Dahlseid JN, Culbertson MR. A factor required for nonsense-mediated mRNA decay in yeast is exported from the nucleus to the cytoplasm by a nuclear export signal sequence. J Cell Sci. 1998;111:3129–3143. doi: 10.1242/jcs.111.21.3129. [DOI] [PubMed] [Google Scholar]
- Spector DL, Lamond AI. Nuclear speckles. Cold Spring Harb Perspect Biol. 2011;3(2):a000646. doi: 10.1101/cshperspect.a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuurman N, Amodaj N, Vale RD. Micro-Manager: open source software for light microscope imaging. Microscopy Today. 2007;15:42–43. [Google Scholar]
- Stüven T, Hartmann E, Görlich D. Exportin 6: a novel nuclear export receptor that is specific for profiling-actin complexes. EMBO J. 2003;22:5928–5940. doi: 10.1093/emboj/cdg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber SC, Spakowitz AJ, Theriot JA. Bacterial chromosomal loci move subdiffusively through a viscoelastic cytoplasm. Phys Rev Lett. 2010a;104:238102. doi: 10.1103/PhysRevLett.104.238102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber SC, Theriot JA, Spakowitz AJ. Subdiffusive motion of a polymer composed of subdiffusive monomers. Phys Rev E Stat Nonlin Soft Matter Phys. 2010b;82:011913. doi: 10.1103/PhysRevE.82.011913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder SJ, Hemmings L, Maciver SK, Bolton SJ, Tinsley JM, Davies KE, Critchley DR, Kendrick-Jones J. Utrophin actin binding domain: analysis of actin binding and cellular targeting. J Cell Sci. 1995;108:63–71. doi: 10.1242/jcs.108.1.63. [DOI] [PubMed] [Google Scholar]
- Wu X, Yoo Y, Okuhama N, Tucker PW, Liu G, Guan L. Regulation of RNA-polymerase-II-dependent transcription by N-WASP and its nuclear binding partners. Nat Cell Biol. 2006;8:756–763. doi: 10.1038/ncb1433. [DOI] [PubMed] [Google Scholar]
- Ye J, Zhao J, Hoffmann-Rohrer U, Grummt I. Nuclear myosin I acts in concert with polymeric actin to drive RNA polymerase I transcription. Genes Dev. 2008;22:322–330. doi: 10.1101/gad.455908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Wang W, Rando OJ, Xue Y, Swiderek K, Kuo A, Crabtree GR. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T-lymphocyte receptor signaling. Cell. 1998;95:625–638. doi: 10.1016/s0092-8674(00)81633-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.