

To further understand the functional role of lasp-2, a focal adhesion protein, a survey identifies two novel binding partners: vinculin and paxillin. The data suggest that lasp-2 has an important role in regulating the composition and dynamics of focal adhesions, as well as significant roles in metastasis, with dual functions in migration and invasion.
Abstract
Focal adhesions are intricate protein complexes that facilitate cell attachment, migration, and cellular communication. Lasp-2 (LIM-nebulette), a member of the nebulin family of actin-binding proteins, is a newly identified component of these complexes. To gain further insights into the functional role of lasp-2, we identified two additional binding partners of lasp-2: the integral focal adhesion proteins vinculin and paxillin. Of interest, the interaction of lasp-2 with its binding partners vinculin and paxillin is significantly reduced in the presence of lasp-1, another nebulin family member. The presence of lasp-2 appears to enhance the interaction of vinculin and paxillin with each other; however, as with the interaction of lasp-2 with vinculin or paxillin, this effect is greatly diminished in the presence of excess lasp-1. This suggests that the interplay between lasp-2 and lasp-1 could be an adhesion regulatory mechanism. Lasp-2’s potential role in metastasis is revealed, as overexpression of lasp-2 in either SW620 or PC-3B1 cells—metastatic cancer cell lines—increases cell migration but impedes cell invasion, suggesting that the enhanced interaction of vinculin and paxillin may functionally destabilize focal adhesion composition. Taken together, these data suggest that lasp-2 has an important role in coordinating and regulating the composition and dynamics of focal adhesions.
INTRODUCTION
Focal adhesions are protein-dense regions that occupy extracellular, transmembrane, and cytoplasmic compartments of the cell. These complex protein assemblies make contact with the extracellular matrix and facilitate cell attachment, migration, and cellular communication. The number of focal adhesion proteins identified is growing and comprises a mixture of cytoskeletal and signaling proteins (for reviews see Wozniak et al., 2004; Lo, 2006). Focal adhesions display an extremely well-organized molecular composition with layers of distinct protein–protein interactions (Kanchanawong et al., 2010). Although focal adhesions might appear to be relatively static structures, many components cycle in and out at different rates depending on the activation and posttranslational modifications of several key proteins (e.g., Humphries et al., 2007; Deramaudt et al., 2011). In fact, it is the dynamics and turnover of new focal adhesions forming at the leading edge of the cell and older focal adhesions disassembling that are important contributors for cell spreading and migration (for reviews see Le Clainche and Carlier, 2008; Gardel et al., 2010). Although cell migration and spreading is required for normal biological processes, aberrant regulation of the adhesion and cytoskeletal machinery is the fundamental mechanism of cancer cell metastasis and invasion.
Adding to the list of focal adhesion components is lasp-2 (LIM-nebulette), a member of the nebulin family of actin-binding proteins (Katoh, 2003; Li et al., 2004; Terasaki et al., 2004). Proteins in the nebulin family contain differing numbers of the characteristic “nebulin repeat,” which is an ∼35-residue repeat containing an actin-binding SDxxYK motif (Labeit et al., 1991). Lasp-2 is a splice variant of the cardiac-specific nebulin family member nebulette (Moncman and Wang, 1995; Katoh, 2003). Although it is an isoform of nebulette, lasp-2 differs significantly in its modular domain organization since it has four unique exons and is also likely transcribed from a promoter that is not specific to striated muscle (Li et al., 2004). The tissue expression profile of lasp-2 includes abundant protein levels in the brain but also expression in the lung, kidney, and striated muscle (Li et al., 2004; Terasaki et al., 2004; Zieseniss et al., 2008). Lasp-2 exhibits a high degree of homology to another nebulin family member, lasp-1 (Tomasetto et al., 1995), although these proteins are transcribed from separate genes (Katoh, 2003). Both lasp-2 and lasp-1 contain an N-terminal LIM domain, either three nebulin repeats (lasp-2) or two nebulin repeats (lasp-1), and a C-terminal SH3 domain (Schreiber et al., 1998; Terasaki et al., 2006). Lasp-1 is ubiquitously expressed and, like lasp-2, is a component of focal adhesions and binds zyxin and F-actin (Chew et al., 2002; Li et al., 2004). Changes in protein levels of lasp-1 have been shown to have effects on cell migration, focal adhesion dynamics, and proliferation (Lin et al., 2004; Grunewald et al., 2006, 2007; Zhang et al., 2009). Increased lasp-1 protein expression is also found in tumors associated with metastatic ovarian, breast, and colorectal cancer (Tomasetto et al., 1995; Grunewald et al., 2006, 2007; Zhao et al., 2010). Although lasp-1’s link to metastasis is significant, it is notable that lasp-2’s potential role in this phenomenon has not yet been examined.
Lasp-2 is reported to localize in focal adhesions and actin filament bundles, based on the assembly patterns of GFP–lasp-2 in several cell lines (HeLa, NIH3T3, PtK2, C2C12, and NG108-15; Li et al., 2004; Terasaki et al., 2004; Panaviene and Moncman, 2007; Deng et al., 2008; Nakagawa et al., 2009). In striated muscle, green fluorescent protein (GFP)–lasp-2 also localizes to focal adhesions, as well as to Z-disks and intercalated disks of cardiomyocytes (Panaviene and Moncman, 2007; Zieseniss et al., 2008). Lasp-2 can bind and bundle actin filaments, and the addition of α-actinin, a lasp-2 binding partner, results in thicker, more robust actin filament bundles (Zieseniss et al., 2008).
Recently attention has focused on the role of lasp-2 in focal adhesion function and organization. Not only does lasp-2 bind F-actin and α-actinin (both found at focal adhesions), but it also binds the focal adhesion protein zyxin (Li et al., 2004). Zyxin is an integral focal adhesion molecule with roles in actin organization, stress fiber repair, assembly, and cell motility (e.g., Crawford and Beckerle, 1991; Hirata et al., 2008; Smith et al., 2010; for review see Beckerle, 1997). Zyxin also has the ability to shuttle to the nucleus, where it can interact with transcription factors (Wang and Gilmore, 2003; Hervy et al., 2006). Lasp-2, through its localization to focal adhesions, interacts with the actin cytoskeleton and, presumably with the cooperation of some of its binding partners, appears to play a role in cell spreading (Deng et al., 2008). Although the localization of lasp-2 to focal adhesions is established, little is known about the functional role of lasp-2 in these structures, its relationship with other focal adhesion proteins, and its function in cell motility.
To provide further insights into the functional role(s) of lasp-2 in actin dynamics and at focal adhesions, we first sought to identify novel lasp-2 binding partners. Two focal adhesion/actin-associated proteins, vinculin and paxillin, were identified as lasp-2 binding partners. The SH3 domain of lasp-2 was mapped as the binding site for both vinculin and paxillin. Of interest, it is the LIM domain and nebulin repeats (and not the SH3 domain) of lasp-2 that are required for the localization of lasp-2 to both focal adhesions and the cortical actin cytoskeleton. Functionally altering the levels of lasp-2 in metastatic cancer cells (SW620 or PC-3B1) results in enhanced cell migration, indicating that lasp-2 has a critical role in the regulation of focal adhesion dynamics. Taken together, our data support the hypothesis that lasp-2 is an important scaffold for several key focal adhesion proteins and has a pivotal role in regulating the composition and dynamics of these cytoskeletal assemblies.
RESULTS
Lasp-2 interacts with the focal adhesion proteins vinculin and paxillin
To probe for the functional role of lasp-2 in mammalian cells, we used human embryonic kidney cells (HEK 293) because reverse transcription (RT)–PCR with lasp-2–specific primers showed that HEK 293 cells express lasp-2 transcripts (Figure 1A). To specifically detect lasp-2, we generated monoclonal antibodies against the C-terminus of lasp-2 and chose a clone that does not recognize lasp-1 in our assays. The reactivity of the antibody to lasp-2 and not to lasp-1 or other cellular proteins was determined by probing Western blots of cell lysates expressing recombinant lasp-1. This is important because all anti–lasp-2 antibodies that are available appear to have the potential to bind lasp-1 (e.g., Supplemental Figure S1). Western blot analysis with our monoclonal anti–lasp-2 antibody shows that lasp-2 is detected in HEK 293 cells (Figure 1B). Because our monoclonal anti–lasp-2-specific antibodies do not work by immunofluorescence microscopy, in order to visualize lasp-2, we transfected HEK 293 cells with GFP–lasp-2 and evaluated them by immunofluorescence microscopy. Lasp-2 localizes to focal adhesion plaques, as indicated by colocalization with the known focal adhesion protein vinculin (Figure 2A). This localization to focal adhesions is consistent with other studies (Li et al., 2004; Deng et al., 2008; Nakagawa et al., 2009).
FIGURE 1:
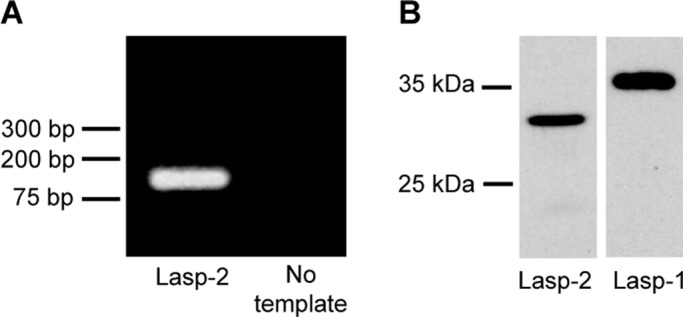
Lasp-2 is expressed in HEK 293 cells. RT-PCR analysis detects lasp-2 transcript in HEK 293 cells. (A) Lasp-2–specific primers amplify a single band of the expected size. (B) HEK 293 cell protein lysates were analyzed by Western blot analysis. Our anti–Lasp-2-specific antibody recognizes a single protein band migrating slightly below 35 kDa, whereas an anti–lasp-1 antibody detects a protein band above 35 kDa.
FIGURE 2:

Lasp-2 interacts with vinculin. (A) Lasp-2 (green) colocalizes with vinculin (red) at focal adhesions. HEK 293 cells were transfected with GFP–lasp-2 and stained for vinculin. Areas of colocalization are highlighted with arrows. Scale bar, 10 μm. (B) For analysis with the yeast two-hybrid (Y2H) system, yeast strain AH109 was cotransfected with a bait coding for lasp-2 and a prey coding for vinculin. Various vinculin prey fragments were then cotransformed with lasp-2, and the binding site for lasp-2 on vinculin was determined to be within the vinculin-tail. (C) Also using the Y2H system, the binding site for vinculin on lasp-2 was found to be the SH3 domain of lasp-2 using different lasp-2 bait constructs. (D) Lasp-2 binds to vinculin-tail in a saturable manner in ELISA. A 10-pmol amount of vinculin-tail or His-peptide (negative control) was immobilized on microtiter plates and incubated with increasing amounts of lasp-2. Bound lasp-2 was detected with an anti–lasp-2 antibody. Lasp-2 bound to vinculin-tail but much less to His-peptide. (E) Lasp-2 coimmunoprecipitates with vinculin. Endogenous vinculin was immunoprecipitated from HEK 293 cell lysates expressing either GFP or GFP–lasp-2. GFP–lasp-2 coimmunoprecipitated with vinculin.
Because we hypothesize that lasp-2 has a role as a molecular scaffold, we next sought to identify other components that interact with Lasp-2 in these structures. We used a candidate yeast two-hybrid approach. Vinculin, an important structural focal adhesion component, was chosen as a candidate because it colocalizes with lasp-2 (Figure 2A) and contains a proline-rich region that might be a target for the SH3 domain of lasp-2. Vinculin is composed of a globular head domain and a tail domain (which includes a short, 40–amino acid, proline-rich neck region). Prey constructs containing full-length as well as the head and tail domains were generated. Initially, full-length lasp-2 was expressed as bait and cotransformed into yeast strain AH109 with full-length vinculin as prey. Lasp-2 was found to interact with vinculin. The tail domain of vinculin but not the head domain interacted with lasp-2 in this assay (Figure 2B). In addition, by using lasp-2 truncations in this assay, we found the binding site for vinculin to be the SH3 domain of lasp-2 (Figure 2C). Proline-rich regions are well-described binding sites for SH3 domains (Yu et al., 1994). Indeed, the proline-rich region of vinculin is contained within the tail/neck domain construct, and so it is likely the target for the SH3 domain of lasp-2.
To confirm the interaction of lasp-2 with vinculin, solid-phase binding assays (enzyme-linked immunosorbent assays [ELISAs]) were performed. Purified recombinant vinculin-tail was absorbed onto microtiter plates and incubated with increasing amounts of recombinant lasp-2. The amount of bound lasp-2 was detected using an anti–lasp-2 antibody. Results from these experiments demonstrate that lasp-2 binds to vinculin-tail directly and the binding was saturable, with an approximate Kd of 140 nM (Figure 2D).
To investigate whether lasp-2 and vinculin can form a molecular complex in cells, we performed coimmunoprecipitation experiments. GFP–lasp-2 was expressed in HEK 293 cells. Endogenous vinculin was immunoprecipitated using an anti-vinculin antibody. GFP–lasp-2 was coimmunoprecipitated with vinculin (Figure 2E). The identity of vinculin and GFP–lasp-2 in the complex was verified by Western blot analysis (Figure 2E). In addition, the SH3 domain of lasp-2 is sufficient to interact with vinculin in coimmunoprecipitation experiments (Supplemental Figure S3).
Given our hypothesis that lasp-2 is a scaffolding protein, we also tested whether it interacted with paxillin, another dynamic, focal adhesion component. Paxillin was a likely candidate because it has a very similar domain organization to zyxin (a lasp-2 binding partner). Lasp-2 also colocalizes with endogenous paxillin in HEK 293 cells (Figure 3A). Via a yeast two-hybrid assay with full-length Lasp-2 as bait and full-length paxillin as prey, we detected an interaction. Further analysis showed that lasp-2 lacking the SH3 domain did not interact with paxillin, indicating that the SH3 domain of lasp-2 is likely to be the domain responsible for paxillin binding (Figure 3B). Note that the SH3 domain alone cannot be used in this yeast two-hybrid assay because it autoactivates; thus the direct interaction involving the SH3 domain of lasp-2 could not be tested using this approach.
FIGURE 3:

Lasp-2 interacts with paxillin. (A) Lasp-2 (green) colocalizes with paxillin (red) at focal adhesions. HEK 293 cells were transfected with GFP–lasp-2 and stained for paxillin. Areas of colocalization are highlighted with arrows. Scale bar, 10 μm. (B) For analysis with the Y2H, yeast strain AH109 was cotransfected with a bait vector coding for lasp-2 and a prey vector coding for paxillin. The binding site for paxillin on lasp-2 was found to be the SH3 domain of lasp-2 using different lasp-2 bait constructs. (C) Paxillin binds to lasp-2 in a saturable manner in ELISA. A 10-pmol amount of GST–lasp-2 or GST alone was immobilized on microtiter plates and incubated with increasing amounts of paxillin. Bound paxillin was detected with an anti-paxillin antibody. (D) Lasp-2 coimmunoprecipitates with paxillin. Endogenous paxillin was immunoprecipitated from HEK 293 cell lysates expressing either GFP or GFP–lasp-2. GFP–lasp–2 coimmunoprecipitated with paxillin.
The direct interaction of lasp-2 with paxillin was confirmed using ELISAs. Purified recombinant glutathione S-transferase (GST)–lasp-2 was absorbed onto microtiter plates and incubated with increasing amounts of recombinant paxillin. Paxillin bound to GST–lasp-2 in a saturable manner, whereas paxillin did not bind to GST alone (Figure 3C). The Kd value for this interaction was ∼20 nM.
To evaluate whether lasp-2 can form a complex with paxillin from cell lysates, we again performed coimmunoprecipitation experiments. GFP–lasp-2 or GFP alone was expressed in HEK 293 cells, and endogenous paxillin was immunoprecipitated using an anti-paxillin antibody. GFP–lasp-2 coimmunoprecipitated with paxillin, whereas GFP alone is nearly undetectable in the precipitate (Figure 3D). Moreover, the SH3 domain of lasp-2, which interacts with vinculin (see earlier discussion), is also sufficient to interact with paxillin in coimmunoprecipitation assays (Supplemental Figure S3). These results indicate not only that lasp-2 is localized to focal adhesions but that it also interacts directly with several important components of focal adhesions—vinculin, paxillin (this study), and zyxin (Li et al., 2004)—and it can do so both directly (yeast two-hybrid; ELISA) and in the context of a cellular environment (coimmunoprecipitation).
Lasp-2 interaction with paxillin and vinculin is reduced in the presence of Lasp-1
As was shown, Lasp-2 will robustly coimmunoprecipitate with vinculin and paxillin. Because lasp-1 is a closely related nebulin family member to lasp-2, we tested whether lasp-1 would also coimmunoprecipitate with vinculin and paxillin. Cherry–lasp-1 was expressed in HEK 293 cells, and endogenous vinculin (Figure 4A) or paxillin (Figure 4B) was immunoprecipitated from these cells. Compared to lasp-2, the amount of lasp-1 that was coimmunoprecipated with paxillin or vinculin was low and/or undetectable (Figure 4). When lasp-1 is coexpressed along with lasp-2, the amount of lasp-2 that coimmunoprecipitates with endogenous vinculin or paxillin is significantly reduced (Figure 4, A and B). The lack of a detectable interaction between lasp-1 with vinculin and paxillin or the loss of the lasp-2 interaction with vinculin and paxillin in the presence of lasp-1 is not due to a shift of vinculin or paxillin into the insoluble lysate fraction (Supplemental Figure S2).
FIGURE 4:

The lasp-2 interaction with paxillin and vinculin is greatly reduced in the presence of lasp-1. (A) GFP–lasp-2 coimmunoprecipitates with endogenous vinculin from HEK 293 cell lysates. Very little, if any, Cherry–lasp-1 is found to coimmunoprecipitate. However, when GFP–lasp-2 and Cherry–lasp-1 are coexpressed in HEK 293 cells, the interaction of GFP–lasp-2 with vinculin is diminished. (B) Similarly, GFP–lasp-2 coimmunoprecipitates with endogenous paxillin from HEK 293 cell lysates, whereas Cherry–lasp-1 is hardly detected. When GFP–lasp-2 and Cherry–lasp-1 are coexpressed, the interaction of lasp-2 with paxillin is nearly undetectable. (C) The potential interaction of lasp-2 with lasp-1 was evaluated via yeast two-hybrid analysis. Lasp-2 as a bait construct interacted with lasp-1 as a prey construct, as indicated by yeast growth on growth selection plates. (D) The interaction of lasp-2 with lasp-1 was confirmed using ELISA. GST–lasp-1 bound to His–lasp-2 in a saturable manner.
These results suggest that the binding affinity of lasp-2 for vinculin or paxillin protein complexes is higher than the affinity of lasp-1 for these same complexes. In addition, lasp-1 appears to interfere with the ability of lasp-2 to bind vinculin or paxillin. One possible explanation for how lasp-1 disrupts the binding of lasp-2 with vinculin or paxillin is that lasp-1 itself binds lasp-2. Indeed, yeast two-hybrid assays using lasp-2 as bait and lasp-1 as prey showed a positive interaction (Figure 4C), indicating that the two proteins can interact. The interaction of lasp-1 and lasp-2 was confirmed using a solid-phase binding assay. Histidine (His)–lasp-2 or His peptide alone was absorbed onto microtiter plates and incubated with increasing amounts of GST–lasp-1. GST–lasp-1 interacted with His–lasp-2 in a saturable manner (Figure 4D). The Kd value for this interaction was ∼15 nM. These data suggest that lasp-1 directly impedes the binding of lasp-2 to vinculin or paxillin.
The interaction of vinculin and paxillin is enhanced in the presence of Lasp-2
Vinculin and paxillin interact with each other (Turner et al., 1990) as well as with lasp-2 (this study). To investigate the effect of lasp-2 on the interaction of vinculin and paxillin, endogenous vinculin was immunoprecipitated in cells expressing GFP-paxillin, GFP–lasp-2, or GFP alone (control). In the presence of GFP–lasp-2, the amount of paxillin pulled down with endogenous vinculin complex was significantly increased (Figure 5). Thus lasp-2 appears to enhance the interaction of vinculin and paxillin, possibly by stabilizing their binding. This enhancement does not appear to be maintained in the presence of lasp-1 (Figure 5).
FIGURE 5:
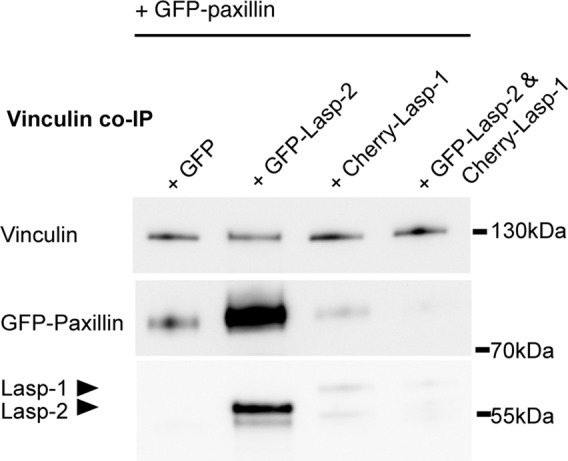
The interaction of vinculin and paxillin is enhanced in the presence of lasp-2 and diminished in the presence of lasp-1. Vinculin and paxillin are known binding partners, and the presence of GFP–lasp-2 enhances the amount of paxillin protein that is coimmunoprecipitated with vinculin in HEK 293 lysates when compared with controls. Endogenous vinculin was immunoprecipitated from cell lysates expressing GFP-paxillin and GFP, GFP–lasp-2, Cherry-lasp-1, or GFP–lasp-2 and Cherry–lasp-1 together. GFP-paxillin is more robustly coimmunoprecipitated in lysates with GFP–lasp-2. This enhancement is not detectable when lasp-2 is lost from the complex as a result of Cherry–lasp-1 expression.
The LIM-nebulin-linker domain is important for localizing Lasp-2 in HEK 293 cells
To further determine how lasp-2 plays a role in focal adhesion composition and to better understand its functional domains, we examined the propensity of lasp-2 fragments to assemble. HEK 293 cells were transfected with GFP, GFP–lasp-2, GFP–lasp-2 1–214 (containing the LIM domain, nebulin repeats, and linker) or GFP–lasp-2 161–273 (containing the linker and SH3 domain), fixed, and stained for vinculin to mark focal adhesions. As expected, full-length GFP–lasp-2 localizes to focal adhesions (Figure 6). Lasp-2 was observed to colocalize with vinculin in some areas, whereas in other areas lasp-2 assembled without detectable vinculin. Unexpectedly, however, GFP–lasp-2 1–214 localized similarly to full-length GFP–lasp-2. This was surprising because studies of lasp-2 in fibroblast cell lines demonstrated that the linker–SH3 domain is necessary for targeting lasp-2 to focal adhesions (Panaviene and Moncman, 2007; Nakagawa et al., 2009). In contrast, GFP–lasp-2 161–273 was not observed to assemble, and in fact its distribution was indistinguishable from the diffuse cytoplasmic and nuclear distribution of GFP alone (Figure 6). The assembly patterns of different lasp-2 fragments suggest that it is the LIM domain and nebulin repeats, and thus potentially the actin-binding activity of lasp-2, that allow for its proper assembly in HEK 293 cells. These data are consistent with a study that showed that it was the LIM domain and the first nebulin repeat that confer F-actin binding and that this F-actin–binding activity was essential for targeting of lasp-2 to filopodial actin bundles (Nakagawa et al., 2009).
FIGURE 6:
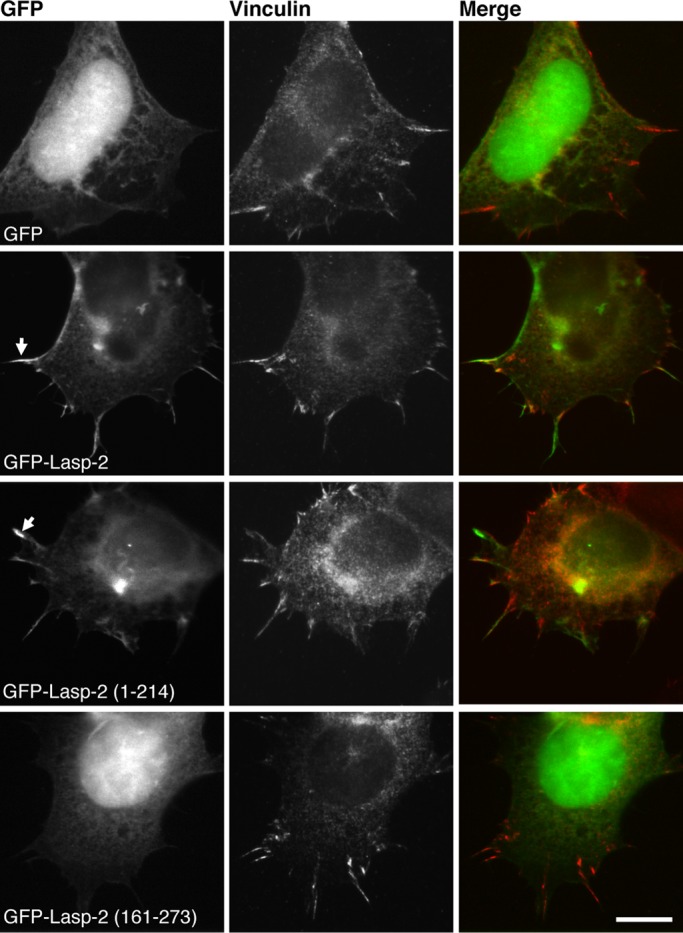
The LIM domain and nebulin repeats are necessary for localizing lasp-2 in HEK 293 cells. GFP–Lasp-2 and GFP–Lasp-2 1–214 assemble in similar patterns at focal adhesions (arrows) in HEK 293 cells, whereas GFP–Lasp-2 161–273 and GFP are predominantly nuclear, with some diffuse cytoplasmic distribution. Staining for endogenous vinculin marks sites of adhesion. These results indicate that the linker–SH3 domain (161–273) of lasp-2 is not necessary for its proper localization. Scale bar, 10 μm.
In addition, even though GFP–lasp-2 161–273 would be predicted to have the ability to bind to vinculin (because it contains the SH3 domain of lasp-2), endogenous vinculin protein localization was not detectably altered in focal adhesions when these fragments were expressed (Figure 6).
Knockdown of Lasp-2 increases cell spreading rates
Lasp-2 is localized to focal adhesions and binds to several key focal adhesion components. To investigate whether the loss of lasp-2 has functional consequences in cell adhesion and dynamics, such as the ability of cells to spread, we performed lasp-2–knockdown experiments using small interfering RNA (siRNA). Cell spreading was observed in HEK 293 cells with depleted levels of lasp-2. Briefly, cells were transfected with lasp-2 siRNA or control scrambled siRNA; protein knockdown of lasp-2 was verified using anti–lasp-2 antibodies via Western blots (Figure 7A). At 72 h later, the cells were trypsinized and replated onto coverslips and allowed to spread for 30 min. The spread area was measured using Cell Profiler (Carpenter et al., 2006). Reproducibly, the spread area in the lasp-2–knockdown cells was increased by ∼15% when compared with control cells. Three different sequences of siRNA to human lasp-2 were used, each yielding similar increases in cell spreading. One representative experiment out of three replicates for one of the siRNA sequences is shown (Figure 7B). Although the measured spread area was enhanced in the cells with reduced lasp-2, the cells had similar morphology and adhesion when compared with the control siRNA cells. All cells exhibited abundant filopodial outgrowths and similar patterns of actin filament organization. These data suggest that the loss of lasp-2 leads to a functional change in the ability of the cell to spread without altering cell morphology or actin filament organization.
FIGURE 7:

The knockdown of lasp-2 increases cell spreading rates. (A) siRNA to human lasp-2 was used to reduce lasp-2 RNA and protein levels in HEK 293 cells. RT-PCR shows that lasp-2 transcript is greatly reduced with lasp-2 siRNA. Lasp-2 protein levels are also significantly reduced with lasp-2 siRNA treatment. (B) In cell-spreading assays, the spread area of lasp-2 siRNA–treated cells was ∼15% increased in comparison to controls.
Overexpression of Lasp-2 increases the migration rate but decreases the invasion of cancer cells
Cancer cell metastasis involves increased cell migration and dysregulation of normal adhesion components and signaling. Given that lasp-2 is a component of focal adhesions with several key binding partners that are involved in cell migration, the role of lasp-2 in cell migration was examined. Two different metastatic cell lines were used, SW620 and PC-3B1. Derived from lymph node cells from a colorectal cancer patient, SW620 cells are metastatic (Leibovitz et al., 1976) and contain low levels of endogenous lasp-2 (unpublished data). PC-3B1 cells are a highly metastatic version of PC-3 cells (Sroka et al., 2009) and were derived from the bone metastasis of a prostate cancer patient (Kaighn et al., 1979). To study the role of lasp-2 in cell migration, we performed a wound-healing assay. GFP–lasp-2 or GFP alone was expressed in SW620 or PC-3B1 cells. The cells were scraped and monitored for wound closure. Both SW620 cells and PC-3B1 cells expressing GFP–lasp-2 closed significantly faster (∼3- and 1.4-fold, respectively) than cells expressing GFP alone (Figure 8A). Our in vitro binding data indicate that the presence of lasp-2 enhances the interaction of vinculin and paxillin. Functionally, the excess lasp-2 could be acting in a dominant-negative capacity by effectively sequestering the complex of vinculin and paxillin. This could reduce the stability of the focal adhesions, leading to increased migration.
FIGURE 8:

The overexpression of lasp-2 in SW620 and PC-3B1 cells increases cell migration rates but reduces cell invasion. (A) SW620 cells, derived from metastatic colorectal cancer, and PC-3B1 cells, derived from metastatic prostate cancer, migrated at a faster rate when GFP–lasp-2 is expressed. There was a threefold increase in the wound-healing rate in cells expressing GFP–lasp-2 in the SW620 cells. There was a 1.4-fold increase in the wound-healing rate in cells expressing GFP–lasp-2 in the PC-3B1 cells. *p < 0.05. (B) Cell invasion is reduced in cells expressing GFP–lasp-2. GFP–lasp-2–expressing cells invaded the chamber an average of 11-fold less than control cells in SW620 cells and invaded the chamber an average of fourfold less than control cells in PC-3B1 cells. *p < 0.005. (C) Loss of lasp-2 protein leads to an increase in cell invasion. Two different siRNA sequences to human lasp-2 were used to reduce lasp-2 protein levels in PC-3 cells. Cells with lasp-2 protein knocked down invaded the chamber approximately twofold more than controls. Data from one of the siRNA sequences are shown. *p < 0.05.
In addition to the ability to migrate, metastatic cells must also be able to invade tissue barriers. To examine whether lasp-2 also had an effect on cell invasion, we performed invasion chamber assays. SW620 or PC-3B1 cells expressing either GFP or GFP–lasp-2 were plated onto Matrigel-coated invasion chambers and allowed to invade. Surprisingly, cells expressing GFP–lasp-2 invaded the chamber an average of 11-fold less in SW620 cells and 4-fold less in PC-3B1 cells than in control cells expressing GFP alone (Figure 8B).
To determine whether the loss of lasp-2 had an opposite effect on invasion compared with lasp-2 overexpression, we assessed cells with lasp-2 knockdown via siRNA (using two different siRNA sequences) for their ability to invade. PC-3 cells (Kaighn et al., 1979)—prostate cancer cells (from which PC-B1 cells are derived)—were used because they express higher levels of endogenous lasp-2 than the related PC-3B1 cells. PC-3 cells with lasp-2 protein knocked down invaded the Matrigel-coated invasion chambers an average of approximately twofold more than control cells treated with a scrambled siRNA (Figure 8C). Similar results were found using both siRNA sequences. Collectively, these experiments reveal that lasp-2 enhances cancer cell migration but reduces cell invasion.
DISCUSSION
To discover insights into the functional role of lasp-2, we identified additional binding partners. Our data show that lasp-2 directly interacts with two well-described focal adhesion components—vinculin and paxillin—through its SH3 domain. Lasp-2 enhances the binding of vinculin and paxillin with each other, whereas the presence of lasp-1 reduces this effect. Although the SH3 domain contains the binding sites for Lasp-2’s focal adhesion–binding partners, the LIM and nebulin repeats containing its actin-binding domains are clearly required for targeting lasp-2 to focal adhesions. Altering the levels of lasp-2 revealed a role for this protein in cell spreading, migration, and invasion. Taken together, these results indicate that lasp-2 is an important component of cell adhesion complexes and has functional roles in cell motility.
The binding site on lasp-2 for vinculin and paxillin is the SH3 domain, which is also the mapped binding site for zyxin on lasp-2 (Li et al., 2004). Because SH3 domains often interact with multiple proteins, it is likely that there are additional mechanisms used by the cell to dictate when and how frequently lasp-2 interacts with each of its binding partners. In fact, because the SH3 domain is a highly conserved protein interaction motif found in hundreds of mammalian proteins and is responsible for governing the assembly of protein complexes and intracellular signaling, there is much discussion on how specificity is achieved in pairwise SH3–ligand interactions (e.g., Ladbury and Arold, 2000; Li, 2005). It has been proposed that compartmentalization of potential interaction partners is one way to confer specificity (Mayer, 2001). To this end, an elegant study by Kanchanawong et al. (2010) using superresolution microscopy showed that focal adhesions have partially overlapping strata of protein interacting zones (i.e., integrin signaling layer, force transduction layer, and an actin regulatory layer). Although these strata are not strict compartments, the location of lasp-2 either in or between one or two of these layers could be one of the mechanisms dictating the duration and frequency of the interactions with each of its binding partners.
Whereas lasp-2 interacts with vinculin and paxillin in cells, the presence of overexpressed lasp-1 disrupts this interaction without itself appearing to be a major component of the complex. A potential explanation for this is that lasp-1 may physically interact with lasp-2, preventing lasp-2 from binding to paxillin or vinculin. In fact, our data show that lasp-2 does interact with lasp-1 with an in vitro Kd of 15 nM. The antagonistic roles of lasp-1 and lasp-2 would be predicted to provide yet another layer of regulation for adhesion structures. The differences in binding partners between lasp-2 and lasp-1 would also be predicted to represent another level of regulation of the two proteins.
Vinculin and paxillin are known binding partners (Turner et al., 1990). It has been suggested that the direct association of vinculin-tail and paxillin in cells is weak and may require an indirect association through another protein (Humphries et al., 2007). Of interest, the interaction of vinculin and paxillin was significantly enhanced in the presence of lasp-2. As such, our data suggest that lasp-2 may, in fact, represent the predicted missing protein that enhances the association of vinculin with paxillin. Consistent with this idea is that the binding site for paxillin on vinculin is located within the vinculin-tail (amino acids 979–1028; Wood et al., 1994) but is separate from the proline-rich region, the likely binding site for lasp-2 (through its SH3 domain). Therefore it would be possible for vinculin to be bound to both paxillin and lasp-2 at the same time. Perhaps lasp-2 helps to facilitate the interaction of vinculin and paxillin by physically keeping the proteins near one another and/or recruiting yet another, unidentified binding protein. Of note, a complication to this possibility is that it would be unlikely for the SH3 domain of lasp-2 to be bound to vinculin and paxillin simultaneously. However, because lasp-2 and vinculin share actin as a binding partner, actin filaments could be involved in bringing different molecules of lasp-2 bound to vinculin and paxillin together in a complex. It is also conceivable that lasp-2 forms dimers (likely through its LIM domain, since this domain is commonly used for dimerization; Feuerstein et al., 1994): one member of the dimer could bind vinculin, and the other could bind paxillin.
In HEK 293 cells lasp-2 does not require its SH3 or linker–SH3 domain for proper localization but instead requires the LIM domain and nebulin repeats, suggesting that it is the actin-binding ability of lasp-2 that first localizes the protein to focal adhesions and the cortical actin cytoskeleton. This idea is consistent with work by Nakagawa et al. (2009), which reported that the LIM and first nebulin repeat allow for proper localization of lasp-2 in neuroblastoma cells (NG-108), and also by (Li et al., 2004), which showed that the lasp-2 SH3 domain alone was primarily localized to the nucleus of HeLa cells and not focal adhesions. In contrast, several studies in fibroblast cell lines concluded that it is the linker and SH3 domain of lasp-2 that are necessary for the assembly of lasp-2 to focal adhesions (Panaviene and Moncman, 2007; Nakagawa et al., 2009). Of interest, it is also the linker–SH3 domain that is mainly responsible for targeting it to the Z-disks of mature cardiomyocytes, although it should be noted that a fragment of lasp-2 that contained the LIM domain, nebulin repeats, and linker was also able to weakly target to the Z-disk (Zieseniss et al., 2008). The present study suggests that HEK 293 cells display a cytoskeletal and cell adhesion organization that differ from common fibroblast lines. Thus it appears that lasp-2 is a multifunctional protein whose domains play different roles depending on the constituents of the cytoskeletal assemblies with which it associates.
Knockdown of lasp-2 in HEK 293 cells resulted in a significant increase (∼15%) in cell spreading rate. The enhanced cell spreading with loss of lasp-2 may be a function of a change in the dynamics of key focal adhesion components. Focal adhesions are in a constant state of assembly, stabilization, and turnover, and cell spreading represents a state in which focal adhesion formation exceeds turnover (for review see Nagano et al., 2012). Because a number of focal adhesion components are lasp-2 binding partners (vinculin, paxillin, and zyxin), perhaps the loss of lasp-2 alters the dynamics of one or more of these proteins and leads to a functional change in cell spreading rates.
A process that is intimately involved in cell adhesion dynamics, cell migration, and invasion is metastasis. The exact mechanisms that lead to metastasis are not fully known, and proteins related to migration and the actin cytoskeleton are often targets of study. Indeed, overexpression of lasp-2 enhanced cell migration but reduced invasion in SW620 colorectal cancer cells and PC-3B1 prostate cancer cells. A recent study that knocked down the lasp-2 related protein lasp-1 in SW620 cells found that both cell migration and invasion were reduced (Zhao et al., 2010); this result suggests that alterations of lasp-1 protein levels affect cell migration in a similar manner to lasp-2, but the invasion potential differs between the two proteins, as lasp-2 did not facilitate cell invasion. Cell invasion involves a complex of different factors, including migration, but also adhesion and proteolysis of extracellular matrix components (for reviews see Friedl and Wolf, 2003; Yamaguchi and Condeelis, 2007). Thus, although the role of lasp-2 and lasp-1 in cell migration is perhaps a shared function, the process of cell invasion highlights key differences between lasp-1 and lasp-2, and interplay in the amount of lasp-1 and lasp-2 in cells could be a regulatory mechanism for cell migration and invasion. For instance, for a cancer cell to become metastatic, enhanced protein levels of lasp-2 would be beneficial for migration but lasp-2 protein levels would need to be down-regulated to penetrate tissue barriers.
Of interest, lasp-2 overexpression in SW620 cells and PC-3B1 cells reveals invasion and migration dynamics more similar to that in cells with decreased vinculin expression. Vinculin-null cells migrate more rapidly but have reduced invasion (Goldmann et al., 1995; Mierke et al., 2010), similar to lasp-2–overexpressing cells. The mechanisms explaining this include that the lack of vinculin destabilizes focal adhesion structures, leading to increased migration, as well as reducing contractile force generation (since it is a mechanoregulating protein; Mierke et al., 2008), and this interferes with invasion. It is possible that the overexpression of lasp-2 in metastatic cells could effectively bind up a significant amount of endogenous vinculin, thus inhibiting vinculin from interacting with its many other binding partners and performing some of its cellular functions, mimicking a vinculin knockdown.
We conclude that lasp-2 is an important member of focal adhesions. Through interactions with its binding partners, lasp-2 clearly has roles in focal adhesion composition, maintenance, and signaling. Lasp-2 also can facilitate the interaction of several key focal adhesion components making it a potentially important scaffolding protein in cell adhesion. Our data are also consistent with Lasp-2 having potentially significant roles in cell motility, with dual functions for migration and invasion.
MATERIALS AND METHODS
Cell culture and transfection
HEK 293 and SW620 cells were maintained in high-glucose DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 4 mM l-glutamine, 1 mM sodium pyruvate, 0.1 mM MEM nonessential amino acids, and 1% penicillin–streptomycin. PC-3B1 and PC-3 prostate cancer cells were a generous gift from Anne Cress (University of Arizona, Tucson, AZ). These cells were maintained in Iscove's modified Dulbecco's medium (Invitrogen) supplemented with 10% FBS and 1% penicillin–streptomycin. Cells were plated into 35-mm tissue culture dishes at a density of 1 × 105 cells/dish. For imaging experiments, the cells were plated on 12-mm-diameter glass coverslips. Transfections of GFP-tagged constructs were performed with Effectene transfection reagent (Qiagen, Valencia, CA) according to the manufacturer's specifications. The culture medium was changed 24 h after transfection.
Construct preparation
For cloning primer design of full-length lasp-2 and lasp-2 truncations see Zieseniss et al. (2008). Briefly, constructs were cloned into pEGFP-C2 (Clontech, Mountain View, CA) using 5′ EcoRI and 3′ XhoI restriction sites. Full-length lasp-1 was amplified from mouse cDNA and cloned into mCherry-C2 (Clontech) using 5′ EcoRI and 3′ XhoI restriction sites (forward primer 5′-TAGAATTCATGAACCCTAACTGTGCC-3′ and reverse primer 5′-ATGTCGACTCAGATGGCCTCCACGTA-3′).
Immunofluorescence microscopy
HEK 293 cells were fixed in 3% formaldehyde for 15 min, permeabilized with 0.2% Triton X-100/phosphate-buffered saline (PBS), and blocked with 2% bovine serum albumin (BSA)/1% normal donkey serum/PBS. Cells were incubated with monoclonal anti-vinculin antibodies (1:2000; Sigma-Aldrich, St. Louis, MO) or monoclonal anti-paxillin antibodies (1:100; BD Biosciences, San Jose, CA), followed by Texas red–conjugated donkey anti-mouse immunoglobulin G (IgG; 1:600; Jackson ImmunoResearch Laboratories, West Grove, PA). For spreading assays, cells were stained with Texas red–phalloidin (1:100; Invitrogen) to mark F-actin. Coverslips were mounted on slides using Aqua PolyMount (Polysciences, Warrington, PA) and analyzed with an inverted fluorescence microscope (Carl Zeiss, Oberkochen, Germany) using a 100×/numerical aperture 1.25 objective, and micrographs were collected as digital images (OrcaER; Hamamatsu, Hamamatsu City, Japan) using OpenLab software (Improvision, PerkinElmer, Waltham, MA). Images were processed using Photoshop (Adobe, San Jose, CA).
Yeast two-hybrid assays
Full-length lasp-2 was cloned into the yeast bait vector pGBKT7 (Clontech) downstream of the DNA-binding domain of GAL4. Vinculin (full-length and head and tail regions), paxillin, and lasp-1 were cloned into the yeast prey vector pGADT7 (Clontech) downstream of the transactivation domain of GAL4. Lasp-2 bait was cotransformed into yeast strain AH109 with the vinculin, paxillin, or lasp-1 prey constructs using the Yeastmaker Yeast Transformation Kit (Clontech) according to the manufacturer's specifications. Selection for positive interactions, and therefore activation of the HIS3 and ADE2, was carried out on agar plates lacking tryptophan, leucine, histidine, and adenine. All constructs were determined not to be toxic to the yeast or to activate the reporter genes independently of a positive interaction (autoactivation).
Protein expression and purification
Full-length lasp-2 and full-length lasp-1 were prepared as GST-fusion proteins (in pGEX-4T; Amersham Biosciences/GE Healthcare, Waukesha, WI) in Escherichia coli cells (BL21DE) and purified using glutathione–Sepharose 4B (GE Healthcare) according to the manufacturer's specifications. Recombinant GST–lasp-2 and GST–lasp-1 were dialyzed against 20 mM NaPO4 and 100 mM KCl, pH 7.2, flash frozen, and stored at −80°C until use. Lasp-2 (full-length), vinculin-tail (amino acids 840–1066), and paxillin (full-length) were prepared as His-fusion proteins (in pET28a; Novagen/EMD Millipore, Billerica, MA) in BL21DE cells using nickel– nitriloacetic acid agarose (Qiagen, Valencia, CA) according to the manufacturer's specifications. Recombinant His–vinculin-tail was dialyzed against 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 80 mM KCl, and 2 mM MgCl2, pH 7.4. Recombinant His-paxillin was dialyzed against PBS, pH 7.4. Both proteins were flash frozen and stored at –80°C until use. His peptide used as a negative control was purchased from Abcam (Cambridge, United Kingdom).
Solid-phase binding assays
ELISAs were used to confirm the interaction of lasp-2 with paxillin, lasp-2 with vinculin, and lasp-2 with lasp-1. For the interaction with vinculin, microtiter plates were coated with 10 pmol of His–vinculin-tail or His-peptide alone. Wells were washed with 0.1% Tween 20 in binding buffer (20 mM HEPES, pH 7.4, 120 mM NaCl, 80 mM KCl, 2 mM MgCl2) and blocked with 2% BSA in binding buffer for 1 h at room temperature. Increasing amounts of His-tagged lasp-2 in 1% BSA/binding buffer (0.1–25 pmol) were added to the wells and incubated for 1.5 h at room temperature. Bound lasp-2 was detected with anti–lasp-2 antibodies (1 μg/ml), followed by a goat anti-mouse alkaline phosphatase–conjugated IgG (Jackson ImmunoResearch Laboratories). For the interaction with paxillin, microtiter plates were coated with 10 pmol of GST–lasp-2 (or GST alone). Increasing amounts of His-tagged paxillin (0.1–25 pmol) were added to the wells, which were incubated for 1.5 h at room temperature. Bound paxillin was detected with anti-paxillin antibodies (0.1 μg/ml; BD BioSciences), followed by a goat anti-mouse alkaline phosphatase–conjugated IgG (Jackson ImmunoResearch Laboratories). For the interaction with lasp-1, microtiter plates were coated with 10 pmol of His–lasp-2 or his peptide alone. Increasing amounts of GST–lasp-1 were added to the wells, which were incubated for 1.5 h at room temperature. Bound GST–lasp-1 was detected using an anti-GST antibody (0.2 μg/ml; Sigma-Aldrich), followed by a goat anti-mouse alkaline phosphatase–conjugated IgG (Jackson ImmunoResearch Laboratories). For all ELISAs, enzyme activity was measured using 4-nitrophenyl phosphate disodium salt hexahydrate (Sigma-Aldrich) as a substrate at 405 nm using a microplate reader (Tecan Group, Mannedorf, Switzerland). Prism (GraphPad, San Diego, CA) was used for analysis and presentation of the data.
Coimmunoprecipitation experiments
HEK 293 cells were plated on 10-cm tissue culture dishes transfected with plasmids encoding GFP, GFP–lasp-2, Cherry–lasp-1, or Cherry–lasp-1 and GFP–lasp-2. At 48 h after transfection, lysate was harvested in ice-cold immunoprecipitation buffer (137 mM NaCl, 1% NP-40, 20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 10% glycerol) with protease inhibitors. After sonication and centrifugation for 15 min at 16,000 × g to remove insoluble debris, total protein levels were measured using a bicinchoninic acid assay (ThermoFisher Scientific, Waltham, MA). We used 1.5 mg of lysate per immunoprecipitation. The lysate was precleared with 50 μl of protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h and then combined with 2 μg of either anti-vinculin (Sigma-Aldrich) or anti-paxillin antibodies (BD Biosciences) overnight. We then added 100 μl of protein A/G beads for an additional 4 h. The beads/antibody/lysate was washed four times with immunoprecipitation buffer and the sample prepared for Western blot analysis. Each coimmunoprecipitation was repeated at least three times, and a representative experiment is shown.
Generation of an anti–Lasp-2 monoclonal antibody
To raise an antibody that is specific to lasp-2 and does not recognize lasp-1 and nebulette, we generated antibodies using a recombinant mouse/chicken (identical amino acid sequence) lasp-2 fragment containing amino acids 1–119, conjugated to KLH (Invitrogen) as an antigen. Amino acids 1–119 represent the four unique exons in lasp-2 that are not found in nebulette. Monoclonal antibodies were generated by BSBS Antibody Facility (Braunschweig, Germany). Clones were screened for reactivity to the antigen by ELISA. Positive clones were screened by Western blot analysis for specificity to lasp-2 protein but not to lasp-1.
RT-PCR
Transcript levels of lasp-2 were evaluated by PCR. Total RNA was extracted from cells with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA concentration was determined using a NanoDrop Spectrophotometer (ThermoFisher Scientific). cDNA was synthesized from 1 μg of total RNA using M-MLV reverse transcriptase (Invitrogen). This cDNA was used to assess transcript levels of genes with endpoint PCR. Primers used included human lasp-2 (forward, 5′-CATTCCCAAGGCTATGGCTA-3′; reverse, 5′-ATCGTACATGGCTCGGTAGG-3′) and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH; forward, 5′-GAAGGTGAAGGTCGGAGTC-3′; reverse, 5′-GAAGATGGTGATGGGATTTC-3′). All primer sets were intron spanning. GoTaq (Promega, Madison, WI) was used for endpoint PCR to amplify lasp-2 cDNA using 28 cycles.
Adenovirus preparation
A replication-defective adenovirus (Adv) expressing GFP or GFP–Lasp-2 was constructed using the AdEasy Adenoviral Vector System from Stratagene (La Jolla, CA). Briefly, GFP or GFP–Lasp-2 cDNA was subcloned into pShuttle-CMV plasmid and linearized according to the manufacturer's instructions before transformation of BJ5183 cells containing the pAdEasy-1 vector. After homologous recombination, the purified pAdEasy-1 vector containing GFP or GFP–Lasp-2 was then transfected into HEK 293 cells for Adv propagation. In each experiment, a replication-defective Adv expressing GFP was used to control for nonspecific effects of Adv infection. All Adv were propagated in HEK 293 cells and purified by CsCl gradient centrifugation. The multiplicity of viral infection (MOI) was determined by viral dilution assay in HEK 293 cells grown in 96-well clusters. At a MOI of 5–10, >95% of the cells were infected, as determined by GFP-positive cells, and there were no cytotoxic effects of Adv infection during the 24 h after Adv infection.
Western blot analysis
Whole-cell lysates from HEK 293 cells were prepared in SDS sample buffer, run on 8 or 10% SDS–PAGE, and transferred onto nitrocellulose membranes (Whatman, Kent, United Kingdom). Membranes were blocked in 2% BSA/PBS for 1 h at room temperature and incubated with primary antibodies to vinculin (1:10,000; Invitrogen), paxillin (1:2000; BD Biosciences), zyxin (1:1000; Invitrogen), lasp-1 (1:2000; Chemicon/EMD Millipore), lasp-2 (0.5 μg/ml; generated in this study; see earlier description), and GAPDH (1:40,000; Ambion/Life Technologies, Carlsbad, CA) for 1–2 h at room temperature. Blots were then incubated with horseradish peroxidase–conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch) for 1 h, followed by chemiluminescence detection using West Pico substrate or West Femto (ThermoFisher Scientific) and visualized with either film or using a G:Box Chemi system (Syngene, Frederick, MD). Protein loading was assessed by GAPDH (loading control) and Coomassie blue staining of proteins in the gel. Protein level changes between experimental and control groups were quantified using ImageJ (National Institutes of Health, Bethesda, MA).
siRNA transfection
Three predesigned siRNA targeted specifically to lasp-2 were obtained from Invitrogen: 1) sense, 5′-CAGCGAUGCUGCCUAUAAAtt-3′, and antisense, 5′-UUUAUAGGCAGCAUCGCUGac-3′; 2) sense, 5′-CAAUGCAGCAUUCACCAAAtt-3′, and antisense, 5′-UUUGGUGAAUGCUGCAUUGac-3′; and 3) sense, 5′-CCCGGAGCCUAUCAGCAAAtt-3′, and antisense, 5′-UUUGCUGAUAGGCUCCGGGac-3′. HEK 293 cells were maintained in complete media without antibiotics at ∼60% confluence and then transfected with lasp-2 siRNA using Lipofectamine RNAiMAX (Invitrogen) according to manufacturer's instructions and transfected a second time after 4 8h. Cells were analyzed 72 h after transfection. Lasp-2 protein knockdown was monitored by Western blot analysis and RT-PCR.
Cell-spreading assay
HEK 293 cells were transfected with siRNA or control scrambled siRNA and cultured for 72 h. Cells were trypsinized and replated onto coverslips coated with collagen (Invitrogen) and allowed to spread for 30 min. The cells were then fixed with 3% formaldehyde and stained with Texas red–phalloidin (1:100; Invitrogen) to mark F-actin. The cells were imaged, and the spread area of each cell was calculated using Cell Profiler (Carpenter et al., 2006). Three different siRNA sequences were used to knock down lasp-2, and similar results were obtained.
Wound-healing assays
SW620 or PC-B1 cells were transfected or infected with either GFP or GFP–lasp-2 plasmids. At 48 h after transfection, a confluent monolayer of cells formed. Cells were serum starved overnight, and wounds were made with a pipette tip. Cells were imaged at wounding, and then the identical cells was imaged again at 24 and 48 h for SW620 cells and 6 and 12 h for PC-B1 cells (the wound closure rates varied for each cell type). The percentage of the wound closure area was evaluated using ImageJ.
Invasion chamber assays
In some experiments, SW620 or PC-3B1 cells were infected with either GFP or GFP–lasp-2 plasmids. In other experiments, PC-3 cells were transfected twice (with an interval of 48 h) with human lasp-2–specific siRNA or control scrambled siRNA. At 48 h after infection with plasmids or 72 h after transfection with siRNA, cells were trypsinized and replated into the top chamber of BD Biocoat Matrigel–coated invasion chambers with 8.0-μm pore sizes (BD Biosciences) at a density of 2.5 × 104 cells/0.5 ml in serum-free media. Complete medium was added to the bottom of the chamber. Cells were allowed to invade through the membrane for 22 h. Cells that did not invade through the membrane were scraped off according to manufacturer's instructions. For overexpression studies, the total number of infected cells that had migrated through the membrane was counted using a fluorescence microscope using GFP fluorescence to mark the cell, and each experiment was done in technical triplicate. For knockdown studies, the cells that had invaded through the chamber were fixed and stained with crystal violet solution to mark the cells. The total number of invaded cells was quantified, and each experiment was done in technical triplicate. Two different siRNA sequences were used, with similar results obtained.
Statistics
A paired Student's t test was used to test significance in the cell-spreading and migration/invasion experiments.
Supplementary Material
Acknowledgments
We thank Anke Zieseniss for initiating these studies and preparing the GFP–lasp-2 constructs, Cathleen Cover for purification of lasp-2 protein, Yasuko Ono and Christine Henderson for consultation and assistance on yeast two-hybrid assays, Chinedu Nworu for assistance with Cell Profiler, and Anne Cress for providing the PC-3B cell line. This work was supported by an American Heart Association Predoctoral Fellowship (10PRE3780013) and a National Heart, Lung, and Blood Institute Training Grant (T32 HL07249) to K.B., the Undergraduate Biology Research Program (HHMI52005889) to C.M.J.W., and National Institutes of Health Grants HL083146 and HL108625 to C.C.G.
Abbreviations used:
- Co-IP
coimmunoprecipitiation
- ELISA
enzyme-linked immunosorbent assay
- F-actin
filamentous actin
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HEK 293
human embryonic kidney 293 cells
- His
histidine
- PBS
phosphate-buffered saline
- SH3
SRC homology 3 domain
- siRNA
small interfering RNA
- Y2H
yeast two-hybrid
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-10-0723) on February 6, 2013.
REFERENCES
- Beckerle MC. Zyxin: zinc fingers at sites of cell adhesion. Bioessays. 1997;19:949–957. doi: 10.1002/bies.950191104. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew CS, Chen X, Parente JA, Jr, Tarrer S, Okamoto C, Qin HY. Lasp-1 binds to non-muscle F-actin in vitro and is localized within multiple sites of dynamic actin assembly in vivo. J Cell Sci. 2002;115:4787–4799. doi: 10.1242/jcs.00174. [DOI] [PubMed] [Google Scholar]
- Crawford AW, Beckerle MC. Purification and characterization of zyxin, an 82,000-dalton component of adherens junctions. J Biol Chem. 1991;266:5847–5853. [PubMed] [Google Scholar]
- Deng XA, Norris A, Panaviene Z, Moncman CL. Ectopic expression of LIM-nebulette (LASP2) reveals roles in cell migration and spreading. Cell Motil Cytoskeleton. 2008;65:827–840. doi: 10.1002/cm.20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deramaudt TB, Dujardin D, Hamadi A, Noulet F, Kolli K, De Mey J, Takeda K, Ronde P. FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol Biol Cell. 2011;22:964–975. doi: 10.1091/mbc.E10-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein R, Wang X, Song D, Cooke NE, Liebhaber SA. The LIM/double zinc-finger motif functions as a protein dimerization domain. Proc Natl Acad Sci USA. 1994;91:10655–10659. doi: 10.1073/pnas.91.22.10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Mechanical integration of actin and adhesion dynamics in cell migration. Annu Rev Cell Dev Biol. 2010;26:315–333. doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann WH, Schindl M, Cardozo TJ, Ezzell RM. Motility of vinculin-deficient F9 embryonic carcinoma cells analyzed by video, laser confocal, and reflection interference contrast microscopy. Exp Cell Res. 1995;221:311–319. doi: 10.1006/excr.1995.1380. [DOI] [PubMed] [Google Scholar]
- Grunewald TG, Kammerer U, Schulze E, Schindler D, Honig A, Zimmer M, Butt E. Silencing of LASP-1 influences zyxin localization, inhibits proliferation and reduces migration in breast cancer cells. Exp Cell Res. 2006;312:974–982. doi: 10.1016/j.yexcr.2005.12.016. [DOI] [PubMed] [Google Scholar]
- Grunewald TG, Kammerer U, Winkler C, Schindler D, Sickmann A, Honig A, Butt E. Overexpression of LASP-1 mediates migration and proliferation of human ovarian cancer cells and influences zyxin localisation. Br J Cancer. 2007;96:296–305. doi: 10.1038/sj.bjc.6603545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervy M, Hoffman L, Beckerle MC. From the membrane to the nucleus and back again: bifunctional focal adhesion proteins. Curr Opin Cell Biol. 2006;18:524–532. doi: 10.1016/j.ceb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Hirata H, Tatsumi H, Sokabe M. Mechanical forces facilitate actin polymerization at focal adhesions in a zyxin-dependent manner. J Cell Sci. 2008;121:2795–2804. doi: 10.1242/jcs.030320. [DOI] [PubMed] [Google Scholar]
- Humphries JD, Wang P, Streuli C, Geiger B, Humphries MJ, Ballestrem C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J Cell Biol. 2007;179:1043–1057. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW. Establishment and characterization of a human prostatic carcinoma cell line (PC-3) Invest Urol. 1979;17:16–23. [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M. Identification and characterization of LASP2 gene in silico. Int J Mol Med. 2003;12:405–410. [PubMed] [Google Scholar]
- Labeit S, Gibson T, Lakey A, Leonard K, Zeviani M, Knight P, Wardale J, Trinick J. Evidence that nebulin is a protein-ruler in muscle thin filaments. FEBS Lett. 1991;282:313–316. doi: 10.1016/0014-5793(91)80503-u. [DOI] [PubMed] [Google Scholar]
- Ladbury JE, Arold S. Searching for specificity in SH domains. Chem Biol. 2000;7:R3–8. doi: 10.1016/s1074-5521(00)00067-3. [DOI] [PubMed] [Google Scholar]
- Le Clainche C, Carlier MF. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 2008;88:489–513. doi: 10.1152/physrev.00021.2007. [DOI] [PubMed] [Google Scholar]
- Leibovitz A, Stinson JC, McCombs WB, 3rd, McCoy CE, Mazur KC, Mabry ND. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 1976;36:4562–4569. [PubMed] [Google Scholar]
- Li B, Zhuang L, Trueb B. Zyxin interacts with the SH3 domains of the cytoskeletal proteins LIM-nebulette and Lasp-1. J Biol Chem. 2004;279:20401–20410. doi: 10.1074/jbc.M310304200. [DOI] [PubMed] [Google Scholar]
- Li SS. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem J. 2005;390:641–653. doi: 10.1042/BJ20050411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Park ZY, Lin D, Brahmbhatt AA, Rio MC, Yates JR, 3rd, Klemke RL. Regulation of cell migration and survival by focal adhesion targeting of Lasp-1. J Cell Biol. 2004;165:421–432. doi: 10.1083/jcb.200311045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SH. Focal adhesions: what's new inside. Dev Biol. 2006;294:280–291. doi: 10.1016/j.ydbio.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Mayer BJ. SH3 domains: complexity in moderation. J Cell Sci. 2001;114:1253–1263. doi: 10.1242/jcs.114.7.1253. [DOI] [PubMed] [Google Scholar]
- Mierke CT, Kollmannsberger P, Zitterbart DP, Diez G, Koch TM, Marg S, Ziegler WH, Goldmann WH, Fabry B. Vinculin facilitates cell invasion into three-dimensional collagen matrices. J Biol Chem. 2010;285:13121–13130. doi: 10.1074/jbc.M109.087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierke CT, Kollmannsberger P, Zitterbart DP, Smith J, Fabry B, Goldmann WH. Mechano-coupling and regulation of contractility by the vinculin tail domain. Biophys J. 2008;94:661–670. doi: 10.1529/biophysj.107.108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncman CL, Wang K. Nebulette: a 107 kD nebulin-like protein in cardiac muscle. Cell Motil Cytoskeleton. 1995;32:205–225. doi: 10.1002/cm.970320305. [DOI] [PubMed] [Google Scholar]
- Nagano M, Hoshino D, Koshikawa N, Akizawa T, Seiki M. Turnover of focal adhesions and cancer cell migration. Int J Cell Biol. 2012;2012:310616. doi: 10.1155/2012/310616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Suzuki H, Machida S, Suzuki J, Ohashi K, Jin M, Miyamoto S, Terasaki AG. Contribution of the LIM domain and nebulin-repeats to the interaction of Lasp-2 with actin filaments and focal adhesions. PLoS One. 2009;4:e7530. doi: 10.1371/journal.pone.0007530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaviene Z, Moncman CL. Linker region of nebulin family members plays an important role in targeting these molecules to cellular structures. Cell Tissue Res. 2007;327:353–369. doi: 10.1007/s00441-006-0305-2. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Moog-Lutz C, Regnier CH, Chenard MP, Boeuf H, Vonesch JL, Tomasetto C, Rio MC. Lasp-1, a novel type of actin-binding protein accumulating in cell membrane extensions. Mol Med. 1998;4:675–687. [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Blankman E, Gardel ML, Luettjohann L, Waterman CM, Beckerle MC. A zyxin-mediated mechanism for actin stress fiber maintenance and repair. Dev Cell. 2010;19:365–376. doi: 10.1016/j.devcel.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroka IC, Pond GD, Nagle RB, Porreca F, King T, Pestano G, Futscher BW, Gard JM, Riley J, Cress AE. Human cell surface receptors as molecular imaging candidates for metastatic prostate cancer. Open Prostate Cancer J. 2009;2:59–66. doi: 10.2174/1876822900902010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki AG, Suzuki H, Ando J, Matsuda Y, Ohashi K. Chromosomal assignment of LASP1 and LASP2 genes and organization of the LASP2 gene in chicken. Cytogenet Genome Res. 2006;112:141–147. doi: 10.1159/000087526. [DOI] [PubMed] [Google Scholar]
- Terasaki AG, Suzuki H, Nishioka T, Matsuzawa E, Katsuki M, Nakagawa H, Miyamoto S, Ohashi K. A novel LIM and SH3 protein (lasp-2) highly expressing in chicken brain. Biochem Biophys Res Commun. 2004;313:48–54. doi: 10.1016/j.bbrc.2003.11.085. [DOI] [PubMed] [Google Scholar]
- Tomasetto C, Moog-Lutz C, Regnier CH, Schreiber V, Basset P, Rio MC. Lasp-1 (MLN 50) defines a new LIM protein subfamily characterized by the association of LIM and SH3 domains. FEBS Lett. 1995;373:245–249. doi: 10.1016/0014-5793(95)01040-l. [DOI] [PubMed] [Google Scholar]
- Turner CE, Glenney JR, Jr, Burridge K. Paxillin: a new vinculin-binding protein present in focal adhesions. J Cell Biol. 1990;111:1059–1068. doi: 10.1083/jcb.111.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gilmore TD. Zyxin and paxillin proteins: focal adhesion plaque LIM domain proteins go nuclear. Biochim Biophys Acta. 2003;1593:115–120. doi: 10.1016/s0167-4889(02)00349-x. [DOI] [PubMed] [Google Scholar]
- Wood CK, Turner CE, Jackson P, Critchley DR. Characterisation of the paxillin-binding site and the C-terminal focal adhesion targeting sequence in vinculin. J Cell Sci. 1994;107(Pt 2):709–717. [PubMed] [Google Scholar]
- Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–119. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 1994;76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chen X, Bollag WB, Bollag RJ, Sheehan DJ, Chew CS. Lasp1 gene disruption is linked to enhanced cell migration and tumor formation. Physiol Genomics. 2009;38:372–385. doi: 10.1152/physiolgenomics.00048.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, et al. Promotion of colorectal cancer growth and metastasis by the LIM and SH3 domain protein 1. Gut. 2010;59:1226–1235. doi: 10.1136/gut.2009.202739. [DOI] [PubMed] [Google Scholar]
- Zieseniss A, Terasaki AG, Gregorio CC. Lasp-2 expression, localization, and ligand interactions: a new Z-disc scaffolding protein. Cell Motil Cytoskeleton. 2008;65:59–72. doi: 10.1002/cm.20244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.