

Abstract
Breast cancer is a complex disease, with heterogeneous clinical evolution. Several analyses have been performed to identify the risk factors for breast cancer progression and the patients who respond best to a specific treatment. We aimed to evaluate whether the hormone receptor expression, HER2 and MYC genes and their protein status, and KRAS codon 12 mutations may be prognostic or predictive biomarkers of breast cancer. Protein, gene and mutation status were concomitantly evaluated in 116 breast tumors from women who underwent neoadjuvant chemotherapy with doxorubicin plus cyclophosphamide. We observed that MYC expression was associated with luminal B and HER2 overexpression phenotypes compared to luminal A (p<0.05). The presence of MYC duplication or polysomy 8, as well as KRAS mutation, were also associated with the HER2 overexpression subtype (p<0.05). MYC expression and MYC gain were more frequently observed in early-onset compared to late-onset tumors (p<0.05). KRAS mutation was a risk factor of grade 3 tumors (p<0.05). A multivariate logistic regression demonstrated that MYC amplification defined as MYC/nucleus ratio of ≥2.5 was a protective factor for chemotherapy resistance. On the other hand, age and grade 2 tumors were a risk factor. Additionally, luminal B, HER2 overexpression, and triple-negative tumors presented increased odds of being resistant to chemotherapy relative to luminal A tumors. Thus, breast tumors with KRAS codon 12 mutations seem to present a worse prognosis. Additionally, MYC amplification may help in the identification of tumors that are sensitive to doxorubicin plus cyclophosphamide treatment. If confirmed in a large set of samples, these markers may be useful for clinical stratification and prognosis.
Introduction
Several analyses have been performed to identify the risk factors for breast cancer (BC) progression. The histological response to preoperative chemotherapy is one of the most reliable predictors for prognosis of BC patients. Many different chemotherapy regimens have been applied in the preoperative setting. However, the identification of patients who respond best to a specific treatment is still critical to the appropriate management.
Some markers have been described as useful factors for prognostic evaluation or predicting therapeutic response. Several studies demonstrated that lack of estrogen (ER) and/or progesterone (PR) receptors predicts for chemosensitivity [1]. On the other hand, it has been proposed that HER2 (c-erbB2) is a predictor factor for either resistance or sensitivity to different types of chemotherapeutic agents. However, the literature results are still controversial, especially concerning the response to anthracyclines [2].
ER, PR and HER2 had been used to classify tumors according to luminal A (ER+/HER2−), luminal B (ER+/HER2+), HER2 overexpression (ER−/HER2+), and triple-negative (ER−/PR−/HER2−) molecular subtypes. HER2 overexpression and triple-negative are more aggressive and present poor prognosis than the luminal subtypes [3], [4]. Moreover, molecular phenotypes have become increasingly valuable in guiding treatment decisions. However, these markers remain imperfect tools and, therefore, new prognostic and predictive factors are still required to optimize treatments among BC patients [5].
MYC acts as a downstream target of HER2-driven proliferative signals in BC cells in vitro [6] and may be regulated by ER or PR contributing to different cell phenotypes [7]. MYC plays a role in the regulation of cell growth and proliferation, metabolism, differentiation, apoptosis, and angiogenesis [8]. MYC amplification and its protein overexpression have been found in about 15% and 40% of BC, respectively [9]. Due to the elevated frequency of alteration, it has been advocated that MYC is involved in BC development and progression [10].
Activation of HER2 induces activation of RAS, which enhances the accumulation of MYC activity by stabilizing the MYC protein [1], [11]. In a transgenic mouse model, the synergistic effects of Myc and the mutant Kras leads to breast tumor formation, maintenance, and recurrence [12]. These data suggest that the KRAS mutation may have a role in breast carcinogenesis.
In the present study, we evaluated the hormone receptor (HR) expression, HER2 and MYC genes and their protein status, and KRAS codon 12 mutations in BC from women who underwent neoadjuvant chemotherapy, as well as their associations with clinicopathological features and chemotherapy response.
Methods
Patients and tumor samples
During the period from 2005 to 2011, 116 females were selected from a cohort of patients with locally advanced invasive ductal carcinoma who underwent therapeutic surgery for a first incidence of BC. All tumors were at stage III according to TNM staging [13]. Cardiac problems, presence of distant metastasis, pregnancy or lactation were exclusion criteria. The mean age of patients was 52±12 years (range of 31–83).
All patients were treated at Ophir Loyola Hospital (Pará, Brazil) and received Adriamycin (doxorubicin; 60 mg/m2) plus Cytoxan (cyclophosphamide; 600 mg/m2) by intravenous every 21 days for four cycles. The response to chemotherapy was based on the change in the primary tumor size on pre- and post-therapy. The tumor size was assessed by clinical palpation using a caliper. Tumors were classified as sensitive to chemotherapy if complete (macroscopic disappearance) or partial (at least a 50% reduction) response was achieved. Tumors were defined as treatment-resistant if no response (less than 50% reduction or less than 25% increase) or progression (at least a 25% increase or presence of new lesions) was observed.
Tumors were obtained by incisional biopsy before neoadjuvant chemotherapy. The tumor invasion and the nodal status were determined according to TNM staging [13]. The histological grade was assessed using the modified Scarff-Bloom-Richardson system [14].
Tumors were classified as luminal A, luminal B, HER2 overexpression, and triple-negative subtypes based on the ER, PR, and HER2 status [15]. We also classified data into tumors of early-onset (patients with ≤40 years of age) and late-onset (>40 years) [16], [17].
Tumors samples were formalin-fixed paraffin-embedded (FFPE). Sections of FFPE tissue were stained with hematoxylin-eosin for histological evaluation or used for immunohistochemical, FISH and PCR analyses.
The study was approved by the ethics committee of the Federal University of Pará, Brazil. Written informed consent with approval of the ethics committee was obtained from all patients prior to specimen collection.
Immunohistochemistry
Immunohistochemistry was performed with primary monoclonal antibodies against ER (SAB4500810, Sigma, USA), PR (HPA004751, Sigma, USA), HER2 (Clone CB11, Life Technologies, USA) or MYC (clone 289–19510, Life Technologies, USA). Universal peroxidase-conjugated secondary antibody kit (DakoCytomation, USA) was used for the detection system and 3,30-diamino-benzidine/H2O2 (Dakocytomation, Denmark) was used as the chromogen. Positive ER, PR or MYC expression was defined as clear nuclear immunostaining in more than 10% of tumor cells [18], [19], [20], [21]. HER2 protein staining was scored as 0 (negative), 1+(weakly positive), 2+ (moderately positive) and 3+ (strongly positive) [2]. Double-blind analysis was performed on all samples.
A breast tissue sample from a male with gynecomastia was used as negative control. In addition, negative controls with primary antibody replaced with Tris-buffered saline were run with the patient slides.
Dual-color FISH
HER2 and MYC amplification was evaluated by dual-color FISH assay using Dako ERBB2 FISH PharmDX™ Kit and MYC/CEN-8 FISH Probe Mix (Dako A/S, Denmark), respectively. FISH scoring was performed by counting fluorescence signals in at least 60 tumor cells. Double-blind analysis was performed on all samples.
For the detection of HER2 amplification, the ratio of HER2 signals to chromosome 17 (CEP17) signals was calculated according to the established guidelines. Patients were stratified depending on their HER2 gene status as: amplified if HER2/CEP17 ratio >2.2; not amplified if HER2/CEP17<1.8; equivocal if 1.8<HER2/CEP17<2.2 [2].
Since no established guideline was published, MYC amplification was defined using different cutoffs as per previously-criteria: 1) the ratio of MYC signals to chromosome 8 (CEP8) signals >2.2 [22], [23] as applied for the detection of HER2 amplification; 2) MYC/CEP8 ratio≥1.3 (at least gene duplication) or MYC/CEP8 ratio<1.3 with polysomy 8 (3 or more copied of CEP8) [22], [23]; 3)>5 MYC copies/nucleus (high MYC gain) [23]; 4)≥2,5 MYC copies/nucleus, which included low MYC gain [23].
Mutation analysis
DNA was purified using MagMAX™ FFPE DNA Isolation Kit (Life Technologies, USA). KRAS codon 12 point mutation was evaluated by PCR-RFLP as previously described [24]. PCR products were digested with endonuclease BstOI. The digestion products were electrophoresed on polyacrylamide gels with SYBR® Safe DNA Gel Stain (Life Technologies, USA) and visualized using blue light. The mutant-type (non-restricted PCR products) were 189 bp, whereas the wild-type products were 160 bp (Figure 1). The PCR products of muted KRAS were sequenced for confirmation of mutation using an ABI Prism® 377 DNA Sequencer (Life Technologies, USA).
Figure 1. Mutation analysis by PCR-RFLP of KRAS codon 12.
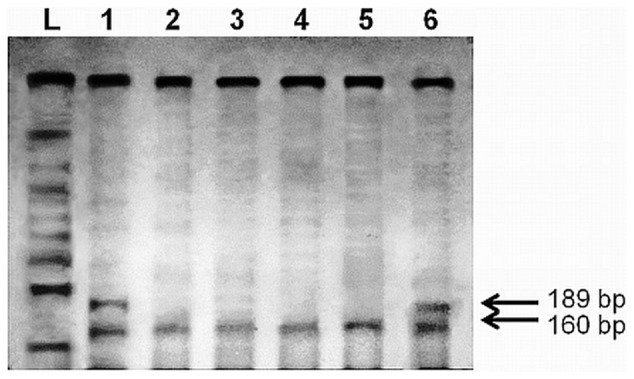
1 and 6: tumors with mutation. 2–5: tumors without mutation. L: size marker.
A wild-type sample of peripheral blood lymphocytes from normal healthy individual and a colorectal cancer sample with codon 12 mutations were used as negative and positive controls, respectively. The controls were included in all experiments. All reactions were performed in duplicate.
Statistical analyses
Cohen's kappa test (κ) was used to evaluate the concordance between the HER2 status by IHC and FISH. This rate was calculated considering negative cases (scores 0 and 1+ and no amplification), and positive cases (score 3+ and amplification). Patients with equivocal IHC or FISH results were not considered for this purpose [25]. Concordance was assessed by Fleiss' equally arbitrary guidelines, which characterize κ values over 0.75 as excellent, 0.40 to 0.75 as fair to good, and below 0.40 as poor [25].
In the remaining analyses, samples with HER2/CEP17>2.2 were classified as presenting HER2 amplification and with scores of 2+ and 3+ as positive HER2 expression.
Logistic regression was used to evaluate the relationship between protein immunoreactivity, gene amplification or mutation, and clinicopathological features. IHC, FISH or PCR-RFLP results, as well as molecular phenotype, were considered dependent variables. Age was not added as a co-variable, since age did not differ between groups (by Student's T-test; data not shown).
A multivariate logistic regression in a forward stepwise approach (condition method) was used to identify variables that may help to predict chemotherapy resistance and to identify risk factors for grade 3 tumors. Age was also added as a dependent variable in the multivariate analyses.
In all analyses, p values less than 0.05 were considered significant. Odds ratio (OR) with 95% confidence intervals are shown.
Results
The protein and gene status and their relationships
For HER2, the percentage of concordant results between IHC and FISH was equal to 93.9%, with a statistically significant κ value of 0.833. The 5 discordant cases were classified as score 1+ by IHC and showed gene amplification by FISH. In addition, 6/32 of tumors classified as score 2+ presented HER2/CEP17 ration ≥2.2 (Table 1).
Table 1. HER2 protein and its gene status in the breast tumors.
| IHC | FISH | Total | ||
| Not amplified | Equivocal | Amplified | ||
| 1+ | 60 (89.55%) | 2 (2.99%) | 5 (7.46%) | 67 (57.76%) |
| 2+ | 23 (71.87%) | 3 (9.38%) | 6 (18.75%) | 32 (27.59%) |
| 3+ | 0 (0%) | 0 (0%) | 17 (100%) | 17 (14.65%) |
| Total | 83 (71.55%) | 5 (4.31%) | 28 (24.14%) | 116 (100%) |
IHC: immunohistochemistry; FISH: fluorescence in situ hybridization.
Table 2 presents the IHC results and Table 3 the FISH results. Figure 2 represents protein immunoreactivity by IHC and gene amplification by FISH assay. No ER-positive case was found without concomitant PR immunoreactivity. Therefore, tumors with ER and PR immunoreactivity were renamed as HR-positive cases for further analyses.
Table 2. Clinicopathological features by protein expression status.
| Factor (N) | HR expression [N(%)] | OR (95% CI) | HER2 expression [N(%)] | OR (95% CI) | MYC expression [N(%)] | OR (95% CI) | |||
| Negative | Positive* | Negative | Positive* | Negative | Positive* | ||||
| Age | |||||||||
| ≤40 (27) | 15 (12.93) | 12 (10.34) | 1.117 (0.470–2.655) | 12 (10.34) | 15 (12.93) | 0.495 (0.207–1.182) | 12 (10.34) | 15 (12.93) | 0.247 (0.100–0.610)** |
| >40 (89)* | 47 (40.52) | 42 (36.21) | 55 (47.41) | 34 (29.31) | 68 (58.62) | 21 (18.10) | |||
| Grade | |||||||||
| 1/2 (96) | 46 (39.66) | 50 (43.10) | 0.230 (0.072–0.738)** | 61 (52.59) | 35 (30.17) | 4.067 (1.433–11.537)** | 67 (57.76) | 29 (25.00) | 1.244 (0.450–3.439) |
| 3 (20)* | 16 (13.79) | 4 (3.45) | 6 (5.17) | 14 (12.07) | 13 (11.21) | 7 (6.03) | |||
| Tumor invasion | |||||||||
| T1/T2 (9) | 4 (3.45) | 5 (4.31) | 0.676 (0.172–2.656) | 6 (5.17) | 3 (2.59) | 1.508 (0.358–6.352) | 7 (6.03) | 2 (1.72) | 1.630 (0.322–8.264) |
| T3/T4 (107)* | 58 (50) | 49 (42.24) | 61 (52.59) | 46 (39.66) | 73 (62.93) | 34 (29.31) | |||
| Lymph node metastasis | |||||||||
| Absent (6) | 6 (5.17) | 0 (0) | <0.001 (0) | 2 (1.72) | 4 (3.45) | 0.346 (0.061–1.971) | 3 (2.59) | 3 (2.59) | 0.429 (0.082–2.235) |
| Present (110)* | 56 (48.28) | 54 (46.55) | 65 (56.03) | 45 (38.79) | 77 (66.38) | 33 (28.45) | |||
| Response to therapy | |||||||||
| Sensitive (50) | 8 (6.90) | 42 (36.21) | 0.042 (0.016–0.113)** | 42 (36.21) | 8 (6.9) | 8.610 (3.483–21.283)** | 41 (35.34) | 9 (7.76) | 3.154 (1.318–7.547)** |
| Resistant (66)* | 54 (46.55) | 12 (10.34) | 25 (21.55) | 41 (35.34) | 39 (33.62) | 27 (23.28) | |||
| HR expression | |||||||||
| Negative (62) | – | – | – | 24 (20.69) | 38 (32.76) | 0.162 (0.070–0.373)** | 37 (31.90) | 25 (21.55) | 0.379 (0.164–0.872)** |
| Positive (54)* | – | – | 43 (37.07) | 11 (9.48) | 43 (37.07) | 11 (9.48) | |||
| HER2 expression | |||||||||
| Negative (67) | – | – | – | – | – | – | 56 (48.28) | 11 (9.48) | 5.303 (2.255–12.473)** |
| Positive (49)* | – | – | – | – | 24 (20.69) | 25 (21.55) | |||
Reference group for logistic regression analysis; ** Differentially expressed between groups, p<0.05. N: number of samples; OR: odds ratio; CI: confidence interval. HR: hormone receptor.
Table 3. Clinicopathological features and protein expression by HER2 and MYC amplification status.
| Factor (N) | HER2 amplification [N(%)] | OR (95% CI) | MYC amplification [N(%)] | OR (95% CI) | MYC duplication or pol8 [N(%)] | OR (95% CI) | High MYC gain [N(%)] | OR (95% CI) | MYC gain [N(%)] | OR (95% CI) | ||||||
| Negative (HER2/CEP17≤2.2) | Positive (HER2/CEP17>2.2)* | Negative (MYC/CEP8≤2.2) | Positive (MYC/CEP8>2.2)* | Negative (MYC/CEP8<1.3) | Positive (MYC/CEP8≥1.3 or pol8) | Negative (MYC/nucleus<5) | Positive (MYC/nucleus≥5)* | Negative (MYC/nucleus<2.5) | Positive (MYC/nucleus≥2.5)* | |||||||
| Age | ||||||||||||||||
| ≤40 (27) | 17 (14.66) | 10 (8.62) | 0.431 (0.169–1.100) | 25 (21.55) | 2 (1.72) | 0.480 (0.366–8.495) | 5 (4.31) | 22 (18.97) | 0.169 (0.059–0.488)** | 24 (20.69) | 3 (2.59)) | 1.622 (0.432–6.085) | 1 (0.86) | 26 (22.41 | 0.093 (0.012–0.723)** | |
| >40 (89)* | 71 (61.21) | 18 (15.52) | 78 (67.24) | 11 (9.48) | 51 (43.97) | 38 (32.76) | 74 (63.79) | 15 (12.93) | 26 (22.41) | 63 (54.31) | ||||||
| Grade | ||||||||||||||||
| 1/2 (96) | 75 (64.66) | 21 (18.10) | 1.923 (0.681–5.432) | 86 (74.14) | 10 (8.62) | 1.518 (0.378–6.100) | 48 (41.38) | 48 (41.38) | 1.500 (0.563–3.997) | 82 (70.69) | 14 (12.07) | 1.464 (0.426–5.028) | 23 (19.83) | 73 (62.93) | 1.260 (0.383–4.150) | |
| 3 (20)* | 13 (11.21) | 7 (6.03) | 17 (14.66) | 3 (2.59) | 8 (6.90) | 12 (10.34) | 16 (13.79) | 4 (3.45) | 4 (3.45) | 16 (13.79) | ||||||
| Tumor invasion | ||||||||||||||||
| T1/T2 (9) | 7 (6.03) | 2 (1.72) | 1.123 (0.220–5.748) | 7 (6.03) | 2 (1.72) | 0.401 (0.074–2.175) | 5 (4.31) | 4 (3.45) | 1.373 (0.349–5.393) | 7 (6.03) | 2 (1.72) | 0.615 (0.117–3.233) | 1 (0.86) | 8 (6.90) | 0.389 (0.046–3.262) | |
| T3/T4 (107)* | 81 (69.83) | 26 (22.41) | 96 (82.76) | 11 (9.48) | 51 (43.97) | 56 (48.28) | 91 (78.45) | 16 (13.79) | 26 (22.41) | 81 (69.83) | ||||||
| Lymph node metastasis | ||||||||||||||||
| Absent (6) | 4 (3.45) | 2 (1.72) | 0.619 (0.107–3.575) | 5 (4.31) | 1 (0.86) | 0.612 (0.066–5.689) | 4 (3.45) | 2 (1.72) | 2.231 (0.392–12.686) | 5 (4.31) | 1 (0.86) | 0.914 (0.100–8.318) | 2 (1.72) | 4 (3.45) | 1.700 (0.294–8.832) | |
| Present (110)* | 84 (72.41) | 26 (22.41) | 98 (84.48) | 12 (10.34) | 52 (44.83) | 58 (50.00) | 93 (80.17) | 17 (14.66) | 25 (21.55) | 85 (73.28) | ||||||
| Response to therapy | ||||||||||||||||
| Sensitive (50) | 46 (39.66) | 4 (3.45) | 6.571 (2.106–20.509)** | 49 (42.24) | 1 (0.86) | 10.889 (1.365–86.840)** | 30 (25.86) | 20 (17.24) | 2.308 (1.089–4.890)** | 48 (41.38) | 2 (1.72) | 7.680 (1.676–35.199)** | 12 (10.34) | 38 (32.76) | 1.074 (0.451–2.557) | |
| Resistant (66)* | 42 (36.21) | 24 (20.69) | 54 (46.55) | 12 (10.34) | 26 (22.41) | 40 (34.48) | 50 (43.10) | 16 (13.79) | 15 (12.93) | 51 (43.97) | ||||||
| HR expression | ||||||||||||||||
| Negative (62) | 39 (33.62) | 23 (19.83) | 0.173 (0.060–0.497)** | 50 (43.10) | 12 (10.34) | 0.079 (0.010–0.627)** | 21 (18.10) | 41 (35.34) | 0.278 (0.129–0.599)** | 46 (39.66) | 16 (13.79) | 0.111 (0.024–0.507)** | 8 (6.90) | 54 (46.55) | 0.273 (0.108–0.691)** | |
| Positive (54)* | 49 (42.24) | 5 (4.31) | 53 (45.69) | 1 (0.86) | 35 (30.17) | 19 (16.38) | 52 (44.83) | 2 (1.72) | 19 (16.38) | 35 (30.17) | ||||||
| HER2 expression | ||||||||||||||||
| Negative (67) | 62 (53.45) | 5 (4.31) | 10.969 (3.761–31.981)** | 65 (56.03) | 2 (1.72) | 9.408 (1.979–44.721)** | 41 (35.34) | 26 (22.41) | 3.574 (1.636–7.808)** | 65 (56.03) | 2 (1.72) | 15.758 (3.417–72.663)** | 20 (17.24) | 47 (40.52) | 2.553 (0.981–6.642)** | |
| Positive (49)* | 26 (22.41) | 23 (19.83) | 38 (32.76) | 11 (9.48) | 15 (12.93) | 34 (29.31) | 33 (28.45) | 16 (13.79) | 7 (6.03) | 42 (36.21) | ||||||
| MYC expression | ||||||||||||||||
| Negative (80) | 77 (66.38) | 3 (2.59) | 58.333 (15.062–225.913)** | 79 (68.10) | 1 (0.86) | 39.500 (4.883–319.520)** | 53 (45.69) | 27 (23.27) | 21.593 (6.067–76.850)** | 78 (67.24) | 2 (1.72) | 31.200 (6.623–146.981)** | 26 (22.41) | 54 (46.55) | 16.852 (2.187–129.871)** | |
| Positive (36)* | 11 (9.48) | 25 (21.55) | 24 (20.70) | 12 (10.34) | 3 (2.59) | 33 (28.45) | 20 (17.24) | 16 (13.79) | 1 (0.86) | 35 (30.17) | ||||||
| HER2 amplification | ||||||||||||||||
| Negative (88) | – | – | – | 87 (75.00) | 1 (0.86) | 65.250 (7.923–537.398)** | 54 (46.55) | 34 (29.31) | 20.647 (4.603–92.614)** | 87 (75.00) | 1 (0.86) | 134.455 (16.267–1111.302)** | 26 (22.41) | 62 (53.45) | 11.323 (1.461–87.758)** | |
| Positive (28)* | – | – | 16 (13.79) | 12 (10.34) | 2 (1.72) | 26 (22.41) | 11 (9.48) | 17 (14.66) | 1 (0.86) | 27 (23.28) | ||||||
Reference group for logistic regression analysis; ** Differentially expressed between groups, p<0.05. N: number of samples; OR: odds ratio; CI: confidence interval. HR: hormone receptor; CEP17: chromosome 17 signals ; CEP8: chromosome 8 signals; pol8: chromosome 8 polysomy.
Figure 2. IHC and FISH analysis in breast tumors.

a) Progesterone immunoreactivity (400x); b) Estrogen immunoreactivity (100x); c) HER2 immunoreactivity, score 3+ (400x); d) Interphase nuclei presenting two or more signals for chromosome 17 centromere (green) and HER2 (red) (1000x); e) MYC immunoreactivity (400x); f) Interphase nuclei presenting two or more chromosome 8 centromere (green) and MYC signals (red) (1000x).
HER2 expression and its amplification were associated with HR (p<0.001, OR: 0.162; 95% CI: 0.070–0.373; p = 0.001, OR: 0.173; 95% CI: 0.060–0.497, respectively) and MYC expression (p<0.001, OR: 5.303, 95% CI: 2.255–12.473; p<0.001, OR: 58.333, 95% CI: 15.062–225.913, respectively) (Table 2 and 3).
MYC expression was also associated with HR (p = 0.023, OR: 0.379, 95% CI: 0.164–0.872) (Table 2). MYC gain was associated with MYC (p<0.05, for all applied cutoffs), HR (p<0.05, for all cutoffs) and HER2 expression (p<0.05, except for the cut point #4), as well as with HER2 amplification (p<0.05, for all cutoffs) (Table 3).
KRAS codon 12 mutation was observed in 9 (7.76%) tumors. KRAS mutation was associated with HER2 (p = 0.033, OR: 4.565, 95% CI: 1.133–18.39) and MYC amplification (p = 0.043, OR: 4.850; 95% CI: 1.049–22.424, for cut point #1) (Table 4).
Table 4. Clinicopathological features and protein expression by KRAS mutation.
| Factor (N) | KRAS mutation [N(%)] | OR (95% CI) | |
| Absent | Present* | ||
| Age | |||
| ≤40 (27) | 23 (19.83) | 4 (3.45) | 0.342 (0.085–1.379) |
| >40 (89)* | 84 (72.41) | 5 (4.31) | |
| Grade | |||
| 1/2 (96) | 91 (78.45) | 5 (4.31) | 4.550 (1.102–18.788)** |
| 3 (20)* | 16 (13.79) | 4 (3.45) | |
| Tumor invasion | |||
| T1/T2 (9) | 8 (6.9) | 1 (0.86) | 0.646 (0.071–5.835) |
| T3/T4 (107)* | 99 (85.34) | 8 (6.9) | |
| Lymph node metastasis | |||
| Absent (6) | 5 (4.31) | 1 (0.86) | 0.392 (0.041–3.775) |
| Present (110)* | 102 (87.93) | 8 (6.9) | |
| Response to therapy | |||
| Sensitive (50) | 48 (41.38) | 2 (1.72) | 2.847 (0.565–14.345) |
| Resistant (66)* | 59 (50.86) | 7 (6.03) | |
| HR expression | |||
| Negative (62) | 55 (47.41) | 7 (6.03) | 0.302 (0.060–1.522) |
| Positive (54)* | 52 (44.83) | 2 (1.72) | |
| HER2 expression | |||
| Negative (107) | 63 (54.31) | 4 (3.45) | 1.790 (0.455–7.043) |
| Positive (9)* | 44 (37.93) | 5 (4.31) | |
| MYC expression | |||
| Negative (80) | 76 (65.52) | 4 (3.45) | 3.065 (0.771–12.176) |
| Positive (36)* | 31 (26.72) | 5 (4.31) | |
| HER2 amplification | |||
| Negative (88) | 84 (72.41) | 4 (3.45) | 4.565 (1.133–18,390)** |
| Positive (28)* | 23 (19.83) | 5 (4.31) | |
| MYC amplification | |||
| MYC/CEP8≤2.2 (103) | 97 (83.62) | 6 (5.17) | 4.850 (1.049–22.424)** |
| MYC/CEP8>2.2 (13)* | 10 (8.62) | 3 (2.59) | |
Reference group for logistic regression analysis; ** Differentially expressed between groups, p<0.05. N: number of samples; OR: odds ratio; CI: confidence interval. HR: hormone receptor; CEP8: chromosome 8 signals.
The impact of MYC and KRAS in the molecular phenotype
Taking in account the HR expression and HER2 amplification to classify tumors by molecular phenotype, 49 (42.2%) of the tumors were deemed luminal A, 5 (4.3%) were luminal B, 23 (19.8%) were HER2 overexpressed, and 39 (33.6%) were triple-negative.
MYC expression was associated with luminal B and HER2 overexpression phenotypes (p = 0.008, OR: 24, 95% CI: 2.329–247.368; p<0.001, OR: 63, 95% CI: 12.021–330.170; respectively) compared to luminal A. The presence of MYC duplication or polysomy 8 (cut point #2) was also associated with the HER2 overexpression subtype (p<0.001, OR: 49.867, 95% CI: 6.143–404.814).
KRAS mutation was detected in 1/49 (2%) luminal A, 1/5 (20%) luminal B, 4/23 (17.4%) HER2 overexpression and 3/39 (7.7%) triple-negative tumors. KRAS mutation was associated with HER2 overexpression phenotype in relation to luminal A (p = 0.044, OR: 10.105, 95% CI: 1.06–96.336).
The impact of protein and gene status on clinicopathological features
MYC expression and MYC gain were more frequently observed in early-onset compared to late-onset tumors (p = 0.002, OR: 0.247, 95% CI: 0.100–0.610; p<0.05, for cutoffs #2 and #4; respectively) (Table 2 and 3).
The expression of HR expression presented a protective effect for grade 3 tumors (p = 0.014, OR: 0.23, 95% CI: 0.072–0.738) (Table 2). On the other hand, HER2 expression and KRAS mutation was a risk factor for grade 3 tumors (p = 0.008, OR: 4.067, 95% CI: 1.433–11.537; p = 0.036, OR: 4.55; 95% CI: 1.102–18.788, respectively) (Table 2 and 4). Since HR and HER2 expression were associated with grade 3 tumors, logistic regression was also performed using the molecular phenotype as dependent variables. Women with HER2 overexpression and triple-negative tumors (p = 0.027, OR: 5.412, 95% CI: 1.216–24.094; p = 0.017, OR: 5.287, 95% CI: 1.342–20.836, respectively) had elevated risk of being diagnosed with grade 3 tumors relative to those with luminal A tumors.
The logistic regression model performed to verify if molecular were together associated with the risk of grade 3 tumors showed that the final model only included KRAS mutation.
The impact of protein and gene status on chemotherapy response
The overall response rate of primary tumor to preoperative chemotherapy was 43%. Among responsive patients, only 4 (8%) patients died at the end of this study (minimum follow-up time of over 12 months). These patients presented metastatic tumors about 2 years after the treatment for primary cancer.
The expression of HR presented a protective effect for treatment-resistance (p<0.001, OR: 0.042, 95% CI: 0.016–0.113) (Table 1). On the other hand, HER2 expression and its gene amplification were a risk factor for chemotherapy resistance (p<0.001, OR: 8.610, 95% CI: 3.483–21.283; p = 0.001, OR: 6.571, 95% CI: 2.106–20.509, respectively) (Table 2 and 3). Additionally, HER2 overexpression (p<0.001, OR: 46.67, 95% CI: 9.229–235.97) and triple-negative (p<0.001, OR: 24.44, 95% CI: 7.887–75.759) subtypes presented an increased risk of being resistant to chemotherapy relative to luminal A.
MYC expression and its gene amplification were a risk factor for chemotherapy resistance (p = 0.01, OR: 3.154, 95% CI: 1.318–7.547; p<0.05, except when the cut point #4 was applied; respectively) (Table 2 and 3).
We conducted a forward stepwise logistic regression model to identify predictors of chemotherapy resistance, entering age, stage, grade, molecular phenotype, KRAS mutation, MYC expression, and the FISH results for detection of its amplification (including the different cutoffs described above for MYC status) as dependent variables. The OR was calculated considering the treatment-resistant group in relation to chemotherapy sensitivity group. The final model included MYC amplification defined as MYC/nucleus ratio of ≥2.5 (cut point #4; p = 0.016, OR: 0.109, 95% CI: 0.018–0.664) as a protective factor. On the other hand, age (p = 0.02, OR: 1.063, 95% CI: 1.01–1.12) was a risk factor. Additionally, luminal B, HER2 overexpression, and triple-negative tumors (p = 0.006, OR: 42.063, 95% CI: 2.956–598.51; p<0.001, OR: 172.754, 95% CI: 15.754–1894.386; p<0.001, OR: 49.008, 95% CI: 8.789–273.268, respectively) presented increased odds of being resistant to chemotherapy relative to luminal A tumors. Moreover, grade 2 tumors presented an increased risk of being resistant to treatment relative to grade 1 (p = 0.042, OR: 10.544, 95% CI: 1.087–102.252). However, grade 3 tumors did not present an increased risk relative to grade 1 in this model.
Discussion
In this study, we evaluated ER and PR expression, HER2 and MYC genes and their protein status, and KRAS mutations in the same set of BC. First, we observed that 24% of tumors presented HER2 amplification, corroborating a previous study (18–20%) [26]. HER2 amplification is the primary mechanism of HER2 overexpression [27]. Although an excellent concordance between IHC and FISH results was detected, 5 cases were scored as 1+ by IHC and presented HER2 amplification. Since standardization of IHC tests may be affected by preanalytical and analytical factors, some groups have suggested the utilization of FISH results for HER2 protein overexpression determination in BC [25]. Therefore, we used only the FISH result for HER2 in the molecular phenotype classification and, then, in the multivariate analyses.
Several definitions for MYC amplification have been used in BC studies. However, these different definitions lead to inconsistent results concerning the role of MYC in breast carcinogenesis. Here, we applied different cutoffs to define MYC amplification as described above, including the acceptance of low MYC gain with or without polysomy 8. The frequency of MYC amplification ranged from 11.2% (cut point #1) to 76.7% (cut point #4) in our sample. Furthermore, MYC overexpression was detected in 31% of BC. Although we found an association between MYC amplification and its expression, as already described in previous studies, our data confirm that mechanisms other than gene amplification are involved in MYC overexpression in BC [9]. However, the assessments of MYC expression by IHC provide variable results depending on the antibody, testing protocol, and scoring system used [10], highlighting that FISH may be an interesting tool in clinical practice due to its reproducibility.
We observed that MYC amplification and expression were more frequent in BC without ER or PR expression, corroborating previous studies [23]. However, some investigations did not find such inverse correlation or even show the opposite correlation (see review [28]). Although literature findings are inconsistent regarding associations between MYC amplification and clinicopathological parameters (in part, probably due to the lack of a unique cutoff for MYC amplification definition), a meta-analysis demonstrated that the correlation of MYC amplification with PR negativity was the only statistically significant association [29].
Here, we detected an association between MYC and HER2, as previously reported [30], [31]. As expected, we also observed an association between MYC expression or its amplification with luminal B or HER2 overexpression in relation to luminal A, confirming the results for HER2 and HR described above. These findings suggest that MYC may be involved in subtype-specific pathways.
MYC expression and gain were more frequently observed in early-onset compared to late-onset BC. To our knowledge, this is the first study to report this association in human primary BC. The exact mechanisms by which MYC may be involved in early-onset BC needs to be elucidated. However, MYC amplification seems to be associated to BRCA1 inactivation in a group of hereditary and sporadic BC [32]. BRCA1 inactivation is usually predisposed to early-onset tumors, with a distinct phenotype characterized by high tumor grade, aneuploidy, high proliferation rate, and ER-negativity [32]. The investigation of MYC targets is still necessary to better understand the heterogeneity of BC.
MYC amplification, as well as HER2 amplification, was associated with KRAS codon 12 mutation. In BC cells, KRAS may be activated by HER2 [1], enhancing the accumulation of MYC activity [11], which may lead to chromosomal instability [33] and contribute to MYC amplification. KRAS mutation was detected in 7.76% of the tumors, corroborating a previous study which reported that 5% of BC presented some KRAS mutation [34]. More than one case of HER2 overexpression and triple-negative subtypes presented KRAS mutation. However, previous studies did not find any KRAS mutation in triple-negative tumors, probably due to its low frequency [35], [36]. Thus, an increased number of tumors are essential to provide evidence of the role of KRAS in human breast carcinogenesis.
In our population, HER2 overexpression and triple-negative were more frequently grade 3 tumors, which is in agreement with the more aggressive phenotype of these tumors [3], [4]. Additionally, we observed that the KRAS mutation was the main predictive factor for grade 3 tumors. Due to its small frequency, further investigations are still necessary to evaluate whether a KRAS codon 12 mutation may predict a worse prognosis in BC patients.
Concerning the response to chemotherapy, we observed that HR predicts chemosensitivity, as previously reported [1]. On the other hand, HER2 amplification or expression predicts resistance to anthracyclines in our sample, highlighting that this group of patients may be suitable for treatment with the monoclonal antibody trastuzumab [37].
Furthermore, MYC expression and amplification (except when accepting low MYC gain) was a risk factor for chemoresistance by univariate logistic regression. However, in the multivariate analysis to identify predictors of resistance to doxorubicin plus cyclophosphamide drugs, we observed that MYC amplification (including low ratio of MYC gain) was a predictor of chemosensibility when adjusted by grade, age, and the molecular phenotyping. This finding is probably due to the significant association of MYC with both HR (good prognosis) and HER2 (worse prognosis).
MYC may have a dual function in cancer cells, i.e. it can promote cell proliferation or induces apoptosis [8] depending on molecular background and tumor microenvironment. Since rapidly proliferating cells are generally more sensitive to chemotherapy, it has been suggested that MYC may sensitive BC cells to apoptosis [38]. Previous in vitro studies demonstrated greater sensitivity of BC cells with MYC amplification to paclitaxel and to doxorubicin compared to those without this amplification [39], [40], [41]. To our knowledge, few studies evaluated the possible role of MYC as a predictor for chemotherapy response in humans. Yasojima et al. reported that MYC was associated with the response to neoadjuvant chemotherapy comprising paclitaxel followed by 5-FU/epirubicin/cyclophosphamide by univariate analysis. However, the multivariate analysis failed to show such association [38]. Without performing a multivariate analysis, Todorovic-Rakovic et al. suggested that patients with MYC amplification treated with chemotherapy (cyclophosphamide, methotrexate and 5-fluorouracil or 5-fluorouracil, adriablastin and cyclophosphamide) had clinical benefits in contrast to patients without amplification [7]. Furthermore, Perez et al. reported that tumors with MYC gain or polysomy 8 appeared to derive more benefits from trastuzumab than tumors without these alterations. The author also reported that patients with MYC/HER2 coamplification benefited significantly more from trastuzumab than patients with only HER2 amplification [23]. Our results and those from the literature suggest that MYC amplification may be used as a predictor factor for chemosensibility and treatment determination. However, it is important to evaluate several markers concomitantly to try to determine a statistical model to identify patients who would best respond to a treatment.
As a result of the increased number of chemotherapy regimens that have been applied in the preoperative setting, it is becoming increasingly important to identify patients who carry a particularly high risk for being unresponsive for a specific treatment. To our knowledge, this is the first study to evaluate the possible prognostic and predictive significance of MYC and KRAS alterations concomitantly with HR and HER2 status, in BC patients treated with neoadjuvant doxorubicin plus cyclophosphamide drugs. We observed an association among the molecular markers investigated. BC with KRAS codon 12 mutations seem to present a worse prognosis. Additionally, MYC amplification may help in the identification of tumors that are sensitive to doxorubicin plus cyclophosphamide. If confirmed in a large set of samples, these markers may be useful for clinical stratification and prognosis.
Funding Statement
Funding for this study was provided as grants and fellowships from the Fundação Amazônia Paraense de Amparo à Pesquisa (FAPESPA/PPSUS 247/2009, #300240/2009), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (to RRB and MACS) and the Fundação de Amparo à Pesquisa do Estado de São Paulo (to MFL). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cleator S, Parton M, Dowsett M (2002) The biology of neoadjuvant chemotherapy for breast cancer. Endocr Relat Cancer 9: 183–195. [DOI] [PubMed] [Google Scholar]
- 2. Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, et al. (2007) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 25: 118–145. [DOI] [PubMed] [Google Scholar]
- 3. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, et al. (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98: 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, et al. (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415: 530–536. [DOI] [PubMed] [Google Scholar]
- 5. Dechaphunkul A, Phukaoloun M, Kanjanapradit K, Graham K, Ghosh S, et al. (2012) Prognostic significance of tissue inhibitor of metalloproteinase-1 in breast cancer. Int J Breast Cancer 2012: 290854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neve RM, Sutterluty H, Pullen N, Lane HA, Daly JM, et al. (2000) Effects of oncogenic ErbB2 on G1 cell cycle regulators in breast tumour cells. Oncogene 19: 1647–1656. [DOI] [PubMed] [Google Scholar]
- 7. Todorovic-Rakovic N, Neskovic-Konstantinovic Z, Nikolic-Vukosavljevic D (2012) C-myc as a predictive marker for chemotherapy in metastatic breast cancer. Clin Exp Med 12: 217–223. [DOI] [PubMed] [Google Scholar]
- 8. Pelengaris S, Khan M (2003) The many faces of c-MYC. Arch Biochem Biophys 416: 129–136. [DOI] [PubMed] [Google Scholar]
- 9. Xu J, Chen Y, Olopade OI (2010) MYC and Breast Cancer. Genes Cancer 1: 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Y, Olopade OI (2008) MYC in breast tumor progression. Expert Rev Anticancer Ther 8: 1689–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sears R, Leone G, DeGregori J, Nevins JR (1999) Ras enhances Myc protein stability. Mol Cell 3: 169–179. [DOI] [PubMed] [Google Scholar]
- 12. Podsypanina K, Politi K, Beverly LJ, Varmus HE (2008) Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci USA 105: 5242–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sobin LH, Wittekind CH (2004) TNM classification of malignant tumours. New York: Wiley-Liss. [Google Scholar]
- 14. Elston CW, Ellis IO (1991) Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 19: 403–410. [DOI] [PubMed] [Google Scholar]
- 15. Saltzman BS, Malone KE, McDougall JA, Daling JR, Li CI (2012) Estrogen receptor, progesterone receptor, and HER2-neu expression in first primary breast cancers and risk of second primary contralateral breast cancer. Breast Cancer Res Treat 135: 849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mukherjee N, Bhattacharya N, Alam N, Roy A, Roychoudhury S, et al. (2012) Subtype-specific alterations of the Wnt signaling pathway in breast cancer: clinical and prognostic significance. Cancer Sci 103: 210–220. [DOI] [PubMed] [Google Scholar]
- 17. Chunder N, Mandal S, Roy A, Roychoudhury S, Panda CK (2004) Differential association of BRCA1 and BRCA2 genes with some breast cancer-associated genes in early and late onset breast tumors. Ann Surg Oncol 11: 1045–1055. [DOI] [PubMed] [Google Scholar]
- 18. Moon HG, Han W, Ahn SK, Cho N, Moon WK, et al. (2013) Breast cancer molecular phenotype and the use of HER2-targeted agents influence the accuracy of breast MRI after neoadjuvant chemotherapy. Ann Surg 257: 133–137. [DOI] [PubMed] [Google Scholar]
- 19. Barinoff J, Hils R, Bender A, Gross J, Kurz C, et al. (2013) Clinicopathological differences between breast cancer in patients with primary metastatic disease and those without: a multicentre study. Eur J Cancer 49: 305–311. [DOI] [PubMed] [Google Scholar]
- 20. da Costa Jde F, Leal MF, Silva TC, Andrade Junior EF, Rezende AP, et al. (2011) Experimental gastric carcinogenesis in Cebus apella nonhuman primates. PLoS One 6: e21988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Silva TC, Leal MF, Calcagno DQ, de Souza CR, Khayat AS, et al. (2012) hTERT, MYC and TP53 deregulation in gastric preneoplastic lesions. BMC Gastroenterol 12: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singhi AD, Cimino-Mathews A, Jenkins RB, Lan F, Fink SR, et al. (2012) MYC gene amplification is often acquired in lethal distant breast cancer metastases of unamplified primary tumors. Mod Pathol 25: 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perez EA, Jenkins RB, Dueck AC, Wiktor AE, Bedroske PP, et al. (2011) C-MYC alterations and association with patient outcome in early-stage HER2-positive breast cancer from the north central cancer treatment group N9831 adjuvant trastuzumab trial. J Clin Oncol 29: 651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dobrzycka B, Terlikowski SJ, Mazurek A, Kowalczuk O, Niklinska W, et al. (2009) Mutations of the KRAS oncogene in endometrial hyperplasia and carcinoma. Folia Histochem Cytobiol 47: 65–68. [DOI] [PubMed] [Google Scholar]
- 25. Martin V, Camponovo A, Ghisletta M, Bongiovanni M, Mazzucchelli L (2012) Internal Quality Assurance Program for ERBB2 (HER2) Testing Improves the Selection of Breast Cancer Patients for Treatment with Trastuzumab. Patholog Res Int 2012: 261857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, et al. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177–182. [DOI] [PubMed] [Google Scholar]
- 27. Akiyama T, Sudo C, Ogawara H, Toyoshima K, Yamamoto T (1986) The product of the human c-erbB-2 gene: a 185-kilodalton glycoprotein with tyrosine kinase activity. Science 232: 1644–1646. [DOI] [PubMed] [Google Scholar]
- 28. Liao DJ, Dickson RB (2000) c-Myc in breast cancer. Endocr Relat Cancer 7: 143–164. [DOI] [PubMed] [Google Scholar]
- 29. Deming SL, Nass SJ, Dickson RB, Trock BJ (2000) C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer 83: 1688–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Al-Kuraya K, Schraml P, Torhorst J, Tapia C, Zaharieva B, et al. (2004) Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res 64: 8534–8540. [DOI] [PubMed] [Google Scholar]
- 31. Park K, Kwak K, Kim J, Lim S, Han S (2005) c-myc amplification is associated with HER2 amplification and closely linked with cell proliferation in tissue microarray of nonselected breast cancers. Hum Pathol 36: 634–639. [DOI] [PubMed] [Google Scholar]
- 32. Grushko TA, Dignam JJ, Das S, Blackwood AM, Perou CM, et al. (2004) MYC is amplified in BRCA1-associated breast cancers. Clin Cancer Res 10: 499–507. [DOI] [PubMed] [Google Scholar]
- 33. Prochownik EV, Li Y (2007) The ever expanding role for c-Myc in promoting genomic instability. Cell Cycle 6: 1024–1029. [DOI] [PubMed] [Google Scholar]
- 34. Karnoub AE, Weinberg RA (2008) Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 9: 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanchez-Munoz A, Gallego E, de Luque V, Perez-Rivas LG, Vicioso L, et al. (2010) Lack of evidence for KRAS oncogenic mutations in triple-negative breast cancer. BMC Cancer 10: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin V, Botta F, Zanellato E, Molinari F, Crippa S, et al. (2012) Molecular characterization of EGFR and EGFR-downstream pathways in triple negative breast carcinomas with basal like features. Histol Histopathol 27: 785–792. [DOI] [PubMed] [Google Scholar]
- 37. Crown J, Dieras V, Kaufmann M, von Minckwitz G, Kaye S, et al. (2002) Chemotherapy for metastatic breast cancer-report of a European expert panel. Lancet Oncol 3: 719–727. [DOI] [PubMed] [Google Scholar]
- 38. Yasojima H, Shimomura A, Naoi Y, Kishi K, Baba Y, et al. (2011) Association between c-myc amplification and pathological complete response to neoadjuvant chemotherapy in breast cancer. Eur J Cancer 47: 1779–1788. [DOI] [PubMed] [Google Scholar]
- 39. Fornari FA Jr, Jarvis DW, Grant S, Orr MS, Randolph JK, et al. (1996) Growth arrest and non-apoptotic cell death associated with the suppression of c-myc expression in MCF-7 breast tumor cells following acute exposure to doxorubicin. Biochem Pharmacol 51: 931–940. [DOI] [PubMed] [Google Scholar]
- 40. Fornari FA Jr, Jarvis WD, Grant S, Orr MS, Randolph JK, et al. (1994) Induction of differentiation and growth arrest associated with nascent (nonoligosomal) DNA fragmentation and reduced c-myc expression in MCF-7 human breast tumor cells after continuous exposure to a sublethal concentration of doxorubicin. Cell Growth Differ 5: 723–733. [PubMed] [Google Scholar]
- 41. Olah E, Csokay B, Prajda N, Kote-Jarai Z, Yeh YA, et al. (1996) Molecular mechanisms in the antiproliferative action of taxol and tiazofurin. Anticancer Res 16: 2469–2477. [PubMed] [Google Scholar]