

Abstract
Human TLRs are critical sensors for microbial components leading to the production of proinflammatory cytokines that are controlled by various mechanisms. Monocytes pretreated with LPS exhibit a state of hyporesponsiveness, referred to as cross-tolerance, to both homologous and heterologous ligands, which play a broader role in innate immunity. To date, LPS-induced cross-tolerance has not been examined regarding microRNA expression kinetics. In this study, THP-1 monocytes treated with various inflammatory ligands showed a continuous amplification of microRNA (miR)-146a over 24 h that is inversely correlated to TNF-α production. In contrast, inhibition of miR-146a showed a reciprocal effect. Thus, the characteristic upregulation of miR-146a in LPS-exposed THP-1 monocytes was studied for cross-tolerance. Strikingly, in LPS-tolerized THP-1 monocytes, only miR-146a showed a continuous overexpression, suggesting its crucial role in cross-tolerance. Similarly, peptidoglycan-primed THP-1 cells showed homologous tolerance associated with miR-146a upregulation. Subsequently, interchangeable differential cross-regulation was observed among non-LPS ligands. TLR2 and TLR5 ligands showed both homologous and heterologous tolerance correlated to miR-146a overexpression. More importantly, inflammatory responses to TLR4, TLR2, and TLR5 ligands were reduced due to knockdown of miR-146a targets IL-1R-associated kinase 1 or TNFR-associated factor 6, suggesting the regulatory effect of miR-146a on these TLRs signaling. Transfection of miR-146a into THP-1 cells caused reduction of TNF-α production, mimicking LPS-induced cross-tolerance. Aside from individual ligands, a whole bacterial challenge in LPS-primed THP-1 monocytes was accompanied by less TNF-α production, which is conversely correlated to miR-146a expression. Our studies have thus demonstrated that miR-146a plays a crucial role for in vitro monocytic cell-based endotoxin-induced cross-tolerance.
Innate immunity is the primary defense mechanism that recognizes, responds to, and resolves invading infectious microbes or their conserved components, known as pathogen-associated molecular patterns (PAMPs). During microbial invasion, danger signals are effectively recognized by the host’s innate immune system through several conserved pattern-recognition receptors. TLRs represent one of the best characterized pathogen-detection systems. Once they are activated by PAMPs, they signal to transcription factors NF-κB to produce proinflammatory cytokines. TLRs possess an extracellular leucine-rich repeat domain (type 1 membrane protein) and cytoplasmic conserved Toll/IL-1R domain. The extracellular domain recognizes microbial ligands (1), whereas the cytoplasmic portion responds by recruiting adaptor kinases to enable signal transduction, most notably through activation of NF-κB transcription factor (2). Therefore, activation of NF-κB by TLRs is a critical event on the road to inflammation, as in LPS-induced systemic inflammation mediated by TLR4.
To date, many TLRs (TLR1–TLR13) have been identified in mammals (3, 4) based on their microbial cell wall specificity or pathogen-specific nucleic acids. TLRs recognize the ligands as being either homodimeric or heterodimeric (2). For example, TLR4 binds to LPS, TLR1/2 or TLR2/6 to lipopeptides, TLR5 to flagellin (subunits of bacterial flagella), and TLR3, TLR7, or TLR9 to nucleic acid. Several PAMPs, including LPS, can stimulate TLR4. However, LPS is the fundamental pathogenic cell wall component of Gram-negative bacteria and is considered one of the most potent immunostimulatory components. The immune system responds to LPS with a systemic production of pro- and anti-inflammatory cytokines (via activating transcription factor NF-κB), such as TNF-α, IL-6, and IL-10, primarily aimed to control growth and dissemination of invaders and subsequently curtail the immune response as needed (5). On the contrary, pathological dysregulation of NF-κB is linked to substantial systemic inflammatory damage that gives rise to sepsis, multiorgan failure, autoimmune diseases, and possibly cancer (6–8). Interestingly, during bacteremia, neutrophils and monocytes from septic patients assume a refractory state to subsequent LPS challenges and no longer produce inflammatory mediators (9, 10). This phenomenon, referred to as endotoxin tolerance (also called LPS hyporesponsiveness or refractoriness), is a host mechanism aimed at limiting inflammatory damage caused by overactivation of the immune system to continuous exposure to Gram-negative bacteria or their products. LPS refractoriness to subsequent LPS challenges (homologous tolerance) has been investigated extensively in vitro using monocytes/macrophage primary cells and cell lines and in vivo using animal models as well as being observed in humans (9, 11–16). Besides homologous tolerance, LPS priming of the immune cells results in diminished cytokine response after subsequent stimulation with non-LPS heterologous TLR ligands (11, 12, 17, 18). This is known as LPS-induced cross-tolerance and has also been observed in association with cells from septic patients (19). Similarly, such other TLR ligands as peptidoglycan (PGN), lipoteichoic acid (LTA), Pam3CSK4CysSerLys4 (Pam3CSK4), and flagellin, plus such cytokines as TNF-α or IL-1β, have been shown to induce homologous tolerance in monocytes/macrophages and, interestingly, they can substitute for each other and sometimes mediate cross-tolerance both in vitro and in vivo (20). LPS-induced tolerance and/or cross-tolerance are thought to play a broader role in host innate immunity, but how it is established is still not completely understood. Therefore, the study of the TLRs regulation in LPS-primed immune cells will help to explicate its role against various microbial insults or whole bacteria, although no detailed or consensus mechanism has been identified to demonstrate how LPS-primed immune cells become hyporesponsive to homologous and/or heterologous ligands, a phenomenon also known as differential cross-regulation (5).
MicroRNAs (miRNAs) have emerged as a new layer of gene expression regulators that act at the posttranscriptional level via the RNA interference mechanism (21). In mammals, the progression of miRNA biogenesis involves the initial transcription of genomic DNA by RNA polymerase II into primary miRNAs, which are sequentially processed by two RNase III enzymes, Drosha (in the nucleus) and Dicer (in the cytoplasm), to become ~23-nucleotide noncoding dsRNA duplexes (22). Eventually, the duplex is loaded into the RNA-induced silencing complex with a guide strand to target mRNAs primarily at their 3′-untranslated regions. This consequence leads to inhibition of translation and/or a decrease in mRNA stability as a result of accelerated decapping and deadenylation (23, 24). miRNAs have been revealed to play important roles in many biological systems, ranging from the development and differentiation of cells to tumors (21), including those in the mammalian immune system (25, 26). During an inflammatory response to microbial insults, many of the miRNAs, including miRNA (miR)-146a induced by TLRs, can negatively regulate the activation of inflammatory pathways in myeloid cells (27–31), although during the course of their expression kinetics, their biological activities in innate immunity are largely unknown. In initial studies on miR-146a expression kinetics, observed in response to cognate ligands for TLRs, the cytoplasmic sensor retinoic acid-inducible gene I (also known as DDX58) and proinflammatory cytokines, including IL-1β, represent the role of miR-146a in innate immunity against pathogenic insults as well as inflammatory diseases (27, 31). Further analysis to determine the biological significance of miR-146a reveals that its expression is NF-κB–dependent, consistent with the presence of an NF-κB binding site in the promoter region, and regulates production of such cytokines as IL-1β and TNF-α in innate immunity by affecting signaling molecules (31). IL-1R–associated kinase-1 (IRAK-1) and TNFR-associated factor 6 (TRAF6) are known to be important signaling adaptor kinases in the downstream of TLR4 signal transduction, and they promote inflammation sustained by proinflammatory cytokines, including TNF-α. Interestingly, these two adaptor kinases are the direct molecular targets for miR-146a, as shown by Taganov et al. (31). Subsequently, our recent in vitro study has demonstrated the mechanistic role of miR-146a in endotoxin tolerance. During this study of LPS tolerance, miR-146a was shown to increase continuously and remain at a high level, exerting negative effects on IRAK-1 and TRAF6 mRNA at the posttranscriptional level (32). Previously Li et al. (33) and Boone et al. (34), respectively, observed LPS tolerance in monocytes due to impairment of IRAK-1 and TRAF6 kinase activity in TLR4 signaling. Interestingly, IRAK-1 and TRAF6 are not only used by TLR4 for signaling, but also by other TLRs, such as TLR2, TLR5, TLR7, TLR8, and TLR9, as well as the IL-1β receptor (35). Therefore, they are considered the common and central adaptor kinases, and should their activity be diminished, cellular refractoriness may happen by other TLRs (except TLR3) signaling. This leads to the speculation that increased miR-146a expression during an LPS-primed state might play a part in a negative feedback pathway for other ligand–TLRs interactions. Considering the ability of miR-146a to regulate TRAF6 and IRAK-1, shared by all TLRs (except TLR3), we hypothesize that it is involved in endotoxin-induced cross-tolerance against various microbial cargo sensed by all other TLRs. Thus, the aim of this study was to investigate the effect of the unique expression pattern of miR-146a in LPS-primed monocytic cells on differential TLR cross-regulation. Our findings suggest that overexpression of miR-146a contributes to controlling proinflammatory cytokine production and confers cross-tolerance to innate immune cells and thus modulates our innate immunity to evade recurrent similar or different bacterial infections or both.
Materials and Methods
Cell culture and innate immune ligand stimulation
Human THP-1 cells, an undifferentiated promonocytic cell line, were obtained from the American Type Culture Collection (Manassas, VA). Cells were maintained by twice weekly passage in RPMI 1640 medium containing 25 mM HEPES and L-glutamine (BioWhittaker, Walkersville, MD), 10% FBS (Mediatech, Manassas, VA), and 100 U/ml penicillin-streptomycin (Mediatech) at 37°C with 5% CO2. Log phase cells were used in all experiments and cultured at the density of 106 cells/ml. To determine the kinetics of ligand-induced cytokine production in vitro, fresh THP-1 monocytes were suspended in complete RPMI 1640 culture medium and seeded at 106 cells/ml in a 24-well plate. Cells were stimulated with the following innate immune ligands: 100 ng/ml LPS (TLR4 ligand) from Salmonella enterica serotype Minnesota Re595 (LPS Se; Sigma-Aldrich, St. Louis, MO), 100 ng/ml Pam3CSK4 (a synthetic bacterial lipoprotein and proinflammatory ligand for TLR2/TLR1), 500 ng/ml PGN (Escherichia coli 0111:B4, TLR2 ligand), 1 μg/ml LPS from Porphyromonas gingivalis (LPS Pg, TLR2 ligand), 1 μg/ml LTA (TLR2 ligand) from Staphylococcus aureus, 100 ng/ml recombinant flagellin (TLR5 ligand) from S. typhimurium, 10 μg/ml muramyl dipeptide (MDP), 10 ng/ml TNF-α, 100 ng/ml IL-1β, and 10 ng/ml IFN-γ (BD Biosciences, Franklin Lakes, NJ). Additionally, to observe the similar TLR ligand stimulatory effect in primary cells, mouse peritoneal-derived macrophages were obtained from 3-mo-old female C57BL/6 mice, which were injected with 0.5 ml 4% sodium thioglycollate, 3 d prior to sacrifice. Primary cells were seeded at 1 × 106/ml in complete DMEM containing 10% FBS in 24-well plates followed by washing after 5 h, using complete growth DMEM to remove nonadherent cells. Adherent macrophages were stimulated with LPS and PGN (0–10 μg/ml) for 6, 12, and 24 h. All of the TLR1, TLR2, and TLR5 ligands were obtained from InvivoGen (San Diego, CA). Ligands were used at concentrations previously reported to induce mediators. Stocks were prepared in tissue culture-grade PBS and preserved at −20°C until needed. Cells were harvested and culture supernatants were collected at various time points over 24 h and stored at −80°C until assayed for cytokines levels. Cell pellets were washed in PBS and stored in RNAlater (Ambion, Austin, TX) at 4°C or frozen at −80°C for total RNA isolation in subsequent analysis.
In vitro induction of homologous tolerance and cross-tolerance
An LPS-induced tolerance and cross-tolerance cell model using monocytic cell line THP-1 was adapted from methods described previously (32, 36, 37), with some minor modifications. Briefly, before starting tolerance and/or cross-tolerance assays, THP-1 cells were cultured for 4 d until cells were in log phase and concentration at 106 cells/ml and viability was checked to be >99% by trypan blue staining. THP-1 cells were transferred to fresh complete medium in new 5- or 25-ml flasks at 5 × 105 cells/ml. Cells were incubated with a low dose of LPS (10 ng/ml) for 18 h. In some cross-tolerance experiments, cells were primed for 18 h with PGN, Pam3CSK4, or flagellin. After two washes with tissue culture-grade PBS, cells in complete culture medium alone (untolerized negative control) or with the same or a different ligand were cultured in a 24-well plate. For observing LPS-induced cross-tolerance against bacteria as a secondary challenge, E. coli DH10B and P. gingivalis strain 33277 were grown until mid-log phase, then harvested and placed in sterile saline, followed by killing at 65°C for 30 min as described previously (38). Heat-killed bacteria were then washed three times with sterile saline and used at a final concentration equivalent to 1 × 106 CFU/ml. Similarly, to observe the TLR ligand tolerance efficiency in primary cells, mouse peritoneal macrophages were primed with LPS or PGN (100 ng/ml) in 24-well plates followed by washing and challenged with PGN, Pam3CSK4, or LPS. After incubation for 5 h at 37°C in 5% CO2, supernatants were harvested by centrifugation (1500 × g at 4°C, 5 min) and immediately stored at −80°C until assayed for inflammatory mediators TNF-α, IL-1β, or IL-6.
Quantification of miRNA and mRNA expression level by quantitative real-time PCR
Total RNA of TLR ligands in treated and untreated THP-1 cells were prepared using the mirVana miRNA isolation kit (Ambion) following the manufacturer’s protocol. RNA yield and purity were determined using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technology, Wilmington, DE), and equal amounts of each RNA (A260/A280 ≈ 2.0, 6.7 ng for miRNA) were used for quantitative stem-loop reverse transcription and real-time PCR (qRT-PCR) analysis. Quantification of mature miRNAs expression was performed using the TaqMan microRNA reverse transcription kit, TaqMan Universal PCR Master mix, and TaqMan microRNA assay primers of interest for human miRNAs (Applied Biosystems, Foster City, CA). For mRNA analysis, a High Capacity cDNA RT kit (Applied Biosystems) and TaqMan mRNA assay primers for IRAK-1, TRAF6, and TLR4 were used with 33 ng total RNA per reaction. The cycle threshold (Ct) values, corresponding to the PCR cycle number at which fluorescence emission reaches a threshold above baseline emission, were determined, and miRNA expression values were calculated using the abundant and virtually pure RNU44 as an endogenous control (Applied Biosystems) following the 2−ΔΔCt method (39). mRNA for gene expression values were quantified in the same way after normalization to mammalian 18S rRNA.
THP-1 cell transfection
miR-146a functional analyses were performed using synthetic miR-146a mimic and miR-146a inhibitor (anti-miRNA inhibitor) obtained from Ambion and reconstituted in nuclease-free water at a concentration of 20 μM. Stocks were stored in aliquots at −80°C prior to use. One day before the transfection, cells were transferred to fresh culture medium at a concentration of 5 × 105 cells/ml. The following day, THP-1 cells adjusted to 5 × 105cells/well were transfected with miR-146a mimic (20 nM) or inhibitor (40 nM) using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. miR-146a mimic-transfected THP-1 cells were incubated for 24 h, followed by washing twice with complete growth medium. The washed cells were treated with different ligands at the abovementioned concentration for 5 h. For miR-146a inhibitor experiments, transfected THP-1 cells were incubated for 24 h, followed by washing with complete growth medium. Cells were then challenged with various ligands for 5 h. Supernatants from cell cultures were collected and assayed for cytokines secretion, and cell pellets were used for RNA isolation and qRT-PCR analysis.
ELISA for cytokine assay
Supernatants were collected from cell cultures at different time points after being induced with various stimuli. Secreted cytokines TNF-α, IL-1β, and IL-6 in the supernatants were measured by ELISA using OptEIA cytokine kits as recommended by the manufacturer (BD Biosciences). Absorbance was measured at 405 nm using a microplate reader (model 680; Bio-Rad, Hercules, CA). A405 was converted to protein concentrations (pg/ml) using standard curves of recombinant human or mouse cytokines.
Western blot analysis
LPS-primed and unprimed THP-1 cells (5 × 106/condition) were collected 2 h after LPS Se, LTA, Pam3CSK4, PGN, LPS Pg, and flagellin challenge, pelleted at 1000 × g for 10 min, and lysed on ice for 10 min in 1 ml lysis buffer (50 mM HEPES [pH 7.6], 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 20 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM NaF, 1 mM benz-amidine, 5 mM para-nitrophenyl phosphate, 1 mM DTT, 1 mM PMSF, and complete protease inhibitor mixture from Roche Diagnostic, Indianapolis, IN). Supernatants were collected after centrifugation at 13,000 rpm for 20 min at 4°C. Similarly, PGN- and Pam3CSK4-primed THP-1 cell lysates were prepared 2 h after homologous or LPS challenge. Soluble lysates were quantitated for protein concentration using a Bio-Rad protein assay kit, separated by SDS-PAGE (10% acrylamide; Bio-Rad) along with Precision Plus Protein standards (Bio-Rad), and electrotransfered to a polyvinylidene difluoride membrane (Bio-Rad). The membranes were blocked for 1 h at room temperature or overnight at 4°C with 5% nonfat milk in PBS/0.05% Tween 20 (PBS-T) and were probed with primary rabbit anti–IRAK-1 or anti-TRAF6 Ab at a concentration of 1:200 (Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were washed three times with PBS-T and incubated for 1 h with goat anti-rabbit IgG-HRP at a concentration of 1:5000 (SouthernBiotech, Birmingham, AL). After washing in PBS-T, reactive protein bands were visualized by SuperSignal Pico chemiluminescent reagent (Pierce).
Statistical analysis
Data are presented in figures as mean ± SD. For multiple group comparisons, one-way ANOVA (p < 0.05) was performed, followed by the two-sided, unpaired Student t test as described by Shaffer (40). An unpaired, two-tailed Student t test was used to compare two independent groups. For all statistical analysis, Prism for Windows, version 5.0 (GraphPad Software, San Diego, CA) was used, and p < 0.05 was considered statistically significant.
Results
Analysis of innate immune ligand-induced TNF-α secretion and miRNA expression kinetics in monocytic THP-1 cells
Microbial ligands (or PAMPs) are recognized by innate immune receptors, such as lipopeptides and lipoproteins by TLR2 (41, 42), double-stranded viral RNA by TLR3 (43), LPS by TLR4 (41), and bacterial flagellin from both Gram-positive and Gram-negative bacteria by TLR5 (44). These ligands have been shown to produce a diverse array of inflammatory mediators, including TNF-α in vitro, as well as in vivo or ex vivo conditions (12, 36, 45–47). To observe TNF-α production in vitro, the most commonly employed acute monocytic leukemia cell line THP-1 (6) was used in the current study. After TLR ligand stimulation of THP-1 cells, kinetics of TNF-α production were examined. As shown in Fig. 1A, log phase THP-1 cells were treated with agonists for TLR2 (or TLR1), TLR4, TLR5, and NOD2, or cytokines IL-1β and IFN-γ, and TNF-α production in supernatants at 8 or 24 h was assessed by ELISA. In supernatants from ligand-stimulated THP-1 cell culture, TNF-α was detected at a significant level in response to all ligands except MDP, IL-1β, and IFN-γ (Fig. 1A). TNF-α protein levels at 8 and 24 h were also correlated with mRNA levels (data not shown).
FIGURE 1.

TNF-α protein secretion and miRNA expression kinetics in response to a panel of innate immune ligands in THP-1 monocytes. THP-1 cells were incubated with various ligands for 8 or 24 h, as indicated at the foot of the graphs. TNF-α in culture supernatants was measured by ELISA (A). Total RNAs were purified from the respective cell pellets, and the expressions of miR-146a (B), miR-155 (C), miR-132 (D), and miR-16 (E) were analyzed by qRT-PCR. miRNA expressions were normalized with RNU44. All results are expressed as mean ± SD from three independent experiments. *p < 0.05; **p < 0.01 compared with untreated cells.
LPS induces upregulation of miR-146a in THP-1 monocytes, as described by Taganov et al. (31) and Bazzoni et al. (48), through microarray analysis and independently confirmed by our laboratory (32). miR-146a is also induced in response to various other components from both Gram-positive and Gram-negative bacteria, as well as proinflammatory cytokines (31). Following these observations and considering the subsequent experimental purposes of this study, expression of miR-146a, miR-155, miR-132, and miR-16 were determined by qRT-PCR analysis on the same RNA samples (Fig. 1B–E). The fold changes in miRNA expression were calculated by comparing the value of TLR ligand-treated cells to that of untreated samples cultured in parallel. miR-146a showed significant expression after 8 h exposure of THP-1 cells to LPS Se, PGN, Pam3CSK4 (TLR2-TLR1 ligand), and to LPS Pg, flagellin (a major component of the bacterial flagellar filament and agonist of the TLR5 receptor), and IL-1β. In contrast, stimulation of LTA, NOD2 ligand MDP, or IFN-γ induced little or no miR-146a expression. Interestingly, there was a substantial increase at 24 h that reached up to 26-fold for LPS Se, 15- to 17-fold for Pam3CSK4, PGN, and LPS Pg, 10-fold for flagellin, and 7-fold for IL-1β (Fig. 1B). In contrast, after 8 h, miR-155 showed an increase of 3-fold for LPS Se, 4- to 5-fold for Pam3CSK4, PGN, and LPS Pg, and 3-fold for IL-1β (Fig. 1C). Similarly, after 8 h, miR-132 expression showed increases of 10-fold for LPS Se, 15-fold for Pam3CSK4 and PGN, 6-fold for LPS Pg, and 3-fold for flagellin (Fig. 1D). No significant change in the expression of miR-16 was observed (Fig. 1E). This last result could serve as a control miRNA that is regulated independently from TLR ligand stimulation together with the internal control RNU44. Notably, in response to the TLR ligands and IL-1β, among those examined, only miR-146a showed an increase between 8 and 24 h (Fig. 1B) and this was negatively correlated to TNF-α release.
Primary mouse peritoneal macrophages were also used to observe the relationship between TNF-α and miR-146a expression stimulated by different doses of LPS or PGN for 6, 12, and 24 h (Supplemental Fig. 1A–D). Interestingly, LPS- or PGN-treated primary cells showed dramatic TNF-α production at 6 h, either peaking at 6 or 12 h, and reduced by 24 h (Supplemental Fig. 1A, 1C). In contrast, miR-146a showed gradual increase for all the time points shown (Supplemental Fig. 1B, 1D). Note that at the highest concentration of 10 μg/ml LPS or PGN, the levels of miR-146a were actually lower than when 1 μg/ml LPS or PGN was used, indicating that an upper limit could be reached with these primary cells.
Inhibition of miR-146a increases TNF-α production in TLR ligand-stimulated THP-1 cells
As shown in Fig. 1A versus Fig. 1B, there is a general inverse correlation between miR-146a expression and TNF-α production. To examine whether miR-146a has any direct effect on TNF-α production, THP-1 cells were transfected with miR-146a inhibitor and then 24 h later stimulated with various TLR ligands. Because the level of miR-146a is very low in unstimulated THP-1 cells, the knockdown effect in the cells transfected with the miR-146a inhibitor might not be easily appreciated (32). Results shown in Fig. 2A confirm that transfection of miR-146a inhibitor prior to LPS stimulation could efficiently block and/or downregulate miR-146a expression by >90% compared with mock-transfected cells primed with LPS. Fig. 2B illustrates how THP-1 cells transfected with miR-146a inhibitor showed a 52 and 60% increase in the level of TRAF6 and IRAK-1 mRNA, respectively, whereas an unrelated gene, lamin A/C, was unaffected. Increases of IRAK-1 and TRAF6 protein levels were confirmed by Western blot analysis (Fig. 2C), consistent with the changes in mRNA levels and previous reports (27, 32). Cells transfected with miR-146a inhibitor showed increased TNF-α production in response to LPS Se (62%), Pam3CSK4 (53%) PGN (52%), LPS Pg (42%), and flagellin (40%) (Fig. 2D). These data show the level to which miR-146a and adaptor kinases TRAF6 and IRAK-1 affected TNF-α production in response to LPS, Pam3CSK4, PGN, and flagellin, but not LTA and other ligands known to not affect miR-146a production.
FIGURE 2.
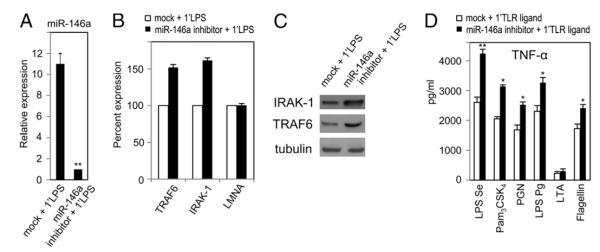
Inhibition of miR-146a expression increases TNF-α response to TLR ligands. THP-1 cells transfected with 40 nM of miR-146a inhibitor for 24 h, along with mock-transfected controls, were washed with complete growth medium. Cells were then challenged with the various ligands for 5 h. A. Total RNA from cell pellets of mock- and miR-146a inhibitor-transfected cells were analyzed by qRT-PCR for miR-146a expression. B, Total RNAs were analyzed for TRAF6, IRAK-1, and unrelated lamin A/C (LMNA, negative control) mRNA expression in both mock- and miR-146a inhibitor-transfected cells by qRT-PCR. C, Western blot analysis for IRAK-1, TRAF6, and tubulin in the cell lysates collected 24 h after with or without (mock) miR-146a inhibitor transfection. D, TNF-α protein in supernatants of mock-transfected and miR-146a inhibitor-transfected cells and challenged with the various ligands shown at the foot of the graph were measured using ELISA. Data are expressed as mean ± SD of three independent experiments. *p < 0.05; **p < 0.01 compared with mock-transfected cells.
LPS-induced miR-146a may account for cross-tolerance to a panel of innate immune ligands in THP-1 cells
Involvement of LPS in homologous tolerance (32) as well as cross-tolerance to other TLR stimuli (12, 49–51) has been shown in vitro and/or in vivo or ex vivo. Fig. 3 shows the extent of LPS-induced cross-tolerance to a panel of innate immune ligands associated with miRNA expression in terms of proinflammatory cytokines using the THP-1 cell model. THP-1 monocytes were primed with 10 ng/ml LPS for 18 h, followed by washing with PBS and challenging with various agonists, as described in Materials and Methods. After 5 h incubation with challenged ligands, TNF-α protein level was analyzed by ELISA (Fig. 3A). TNF-α levels decreased significantly (p < 0.01) after secondary challenges with Pam3CSK4, PGN, LPS Pg, and flagellin in comparison with the same stimulation of naive THP-1 cells. MDP (NOD2 ligand), IL-1β, and IFN-γ did not induce TNF-α (Fig. 1A) regardless of the LPS priming. Analysis of TNF-α mRNA levels by qRT-PCR (Fig. 3B) showed levels consistent with the changes in protein data of cross-tolerized THP-1 cells, and reduction of TNF-α mRNA was significant compared with the untolerized control. Previously, LaRue and McCall (52) reported that neutrophils from septic patients showed less LPS-induced IL-1β mRNA and protein synthesis, which was supported by their in vitro studies using THP-1 monocytes. Production of other proinflammatory cytokines, such as IL-6, is also known to be reduced in LPS-primed cells (53). Consistent with these reports, LPS-primed cells showed reduced IL-6 and IL-1β production in response to the innate immune stimuli, as shown in Fig. 3C and 3D. The reduced production of IL-6 and IL-1β was similar to the pattern observed for TNF-α production.
FIGURE 3.

High levels of LPS-induced miR-146a may account for cross-tolerance to non-LPS agonists in the THP-1 cell model. THP-1 cells primed with 10 ng/ml LPS for 18 h (cross-tolerized, filled bars) and untreated controls incubated for the same time period (untolerized, open bars) were washed twice with PBS, then challenged with various ligands or medium alone for 5 h. Culture supernatants and total RNA were analyzed for cytokines using ELISA (A, C, D), or miRNA (E, F, J) and mRNA expression (B, G, H) by qRT-PCR, as described in Materials and Methods. Changes in IRAK-1 and TRAF6 protein levels in LPS-primed and unprimed THP-1 cells challenged with various TLR ligands or medium alone for 2 h were analyzed by Western blot with tubulin expression shown as loading controls (I). Serial dilutions of untreated THP-1 cell lysates (100, 50, 25%) were included in the Western blot to document the semiquantitative measurement for IRAK-1 and TRAF6 expression. THP-1 monocytes were cultured with or without 100 ng/ml PGN continuously for 18 h followed by washing and cultured in complete medium for another 0, 12, or 22 h (PGN withdrawal). At each time point, 6 × 105 cells were challenged with 500 ng/ml PGN for 3 h prior to analysis of TNF-α production by ELISA (J) and miR-146a expression by qRT-PCR analysis (K). All results are expressed as mean ± SD from three independent experiments. *p < 0.05; **p < 0.01 compared with untolerized THP-1 cells.
Because miR-146a was shown to be responsible for LPS tolerance (32), its levels in the cross-tolerized conditions were confirmed as it was postulated to have a role in LPS-induced cross-tolerance (Fig. 3E). As expected, miR-146a in the cross-tolerized sample showed higher expression during 18 h initial LPS-priming incubation plus 5 h ligand challenge, but little or no changes in the expression of miR-16 (Fig. 3F) were observed compared with unprimed controls. For Fig. 3E, the open bars (untolerized) depict miR-146a levels challenged by different ligands for a total of only 5 h, as there was no priming at 0–18 h. These results are consistent with data already presented in Fig. 1B for exposure of 8 and 24 h. The filled bars (tolerized) represent miR-146a levels after 18 h LPS priming and 5 h challenge with different ligands. There are no statistical differences among the filled bars for all LPS-primed cells, but they are clearly different from untreated cells.
Figs. 3G and 3H show the mRNA expression of known miR-146a targets IRAK-1 and TRAF6 in both cross-tolerized and untolerized cells, respectively. The mRNA levels of IRAK-1 and TRAF6 were not affected significantly between cross-tolerized and untolerized cells; this is not unexpected, as miR-146a regulates these adaptor molecules via primarily translational repression (31, 32). Fig. 3I shows Western blot analysis of IRAK-1 and TRAF6 protein levels comparing LPS cross-tolerized cells to control untolerized cells demonstrating moderate reductions of ~50–75% in all cases, as expected. This level of reduction in IRAK-1 and TRAF6 was consistent with “medium alone” control (untolerized cells versus cross-tolerized cells) when cells were tolerized by LPS priming but not exposed to secondary challenge. Consequently, miR-146a may play an important role in LPS-induced cross-tolerance based on the inverse correlation with proinflammatory cytokine production and the repressed levels of IRAK-1 and TRAF6.
To support that the THP-1 cell-based model of endotoxin cross-tolerance can be observed in primary cells, mouse primary macrophages were also tolerized with LPS or PGN. Supplemental Fig. 1E shows decrease in TNF-α production by 50% in LPS–LPS homologous tolerance (priming challenge) and by 75% in PGN–PGN homologous tolerance. Heterologous tolerance varies between 40% reduction in TNF-α for PGN–LPS to 52% TNF-α reduction for LPS–PGN. Supplemental Fig. 1F shows a 5- to 10-fold increase in expression of miR-146a in LPS-tolerized cells, consistent with its potential role in endotoxin tolerance in primary cells as well.
Previously, highly elevated miR-146a expression was shown to be dependent on the continuous presence of LPS in the medium as demonstrated by its decreasing levels after 22 h LPS withdrawal (32). Accordingly, THP-1 monocytes were primed with PGN continuously for 18 h, then washed twice with PBS, and cultured in complete growth medium for an additional 0, 12, or 22 h (PGN withdrawal). With 18 h continuous PGN priming and 5 h PGN challenge, the same condition used in Fig. 3A, negative correlation between TNF-α secretion and miR-146a expression, was observed as expected (Fig. 3J, 3K). However, after 12 h PGN withdrawal, cells started to regain PGN responsiveness and almost completely recovered from tolerance after 22 h PGN withdrawal (Fig. 3J). Thus, upregulated miR-146a from PGN priming, similar to LPS priming (32), is important for maintaining tolerance and cross-tolerance. The t1/2 of miR-146a estimated from this expertiment was ~9–10 h.
LPS- or PGN-primed THP-1 monocytes show effective homologous tolerance with higher priming dose correlating with higher miR-146a levels
The effectiveness of endotoxin tolerance in THP-1 cells correlated with an increase in priming dose up to 1000 ng/ml LPS was reported in our earlier study with secondary challenge LPS doses of up to 1000 ng/ml (32). Thus, to further advance our understanding of whether homologous tolerance can be achieved with even higher doses of LPS, a range of challenge concentrations of LPS up to 10,000 ng/ml (or PGN up to 2,000 ng/ml) was employed to primed THP-1 cells. Priming with 10 ng/ml LPS or higher concentrations resulted in an 80–90% decrease (p < 0.01) in TNF-α production, even at the highest dose of LPS challenge (10,000 ng/ml, Fig. 4A). Fig. 4B shows the results of gradual augmentation of miR-146a correlated with degree of tolerance observed in Fig. 4A. These data are uniformly consistent with our previous report, which found that the increase in miR-146a expression and reduction in its target IRAK-1 and TRAF6 protein levels were LPS dose-dependent (32), with the exception that the current experiment further reinforces the previous conclusion that LPS tolerance was maintained even when up to a 10 times higher secondary challenge dose of LPS was employed. A similar homologous tolerance phenomenon was observed for the TLR2 ligand PGN. Fig. 4C illustrates how THP-1 cells primed with 10 ng/ml or a higher concentration of PGN were significantly resistant to secondary PGN challenges of up to 2000 ng/ml, and this homologous tolerance was correlated with respective priming doses and upregulation of miR-146a (Fig. 4D).
FIGURE 4.
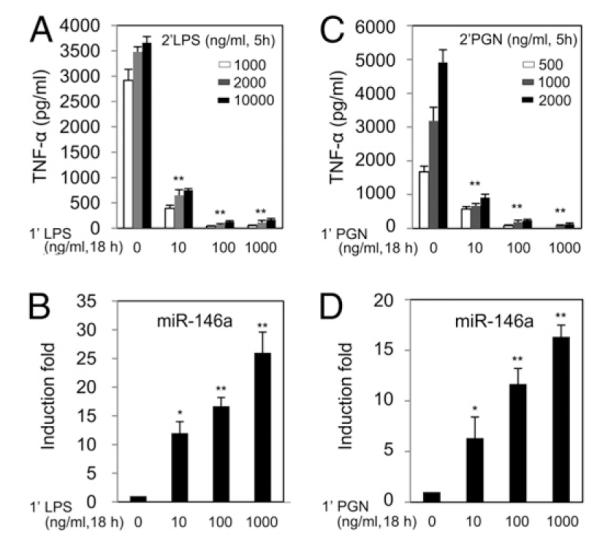
LPS- and PGN-tolerized THP-1 cells show efficient tolerance to high doses of respective ligands with dependency on priming dose and are inversely correlated to induced levels of miR-146a. THP-1 cells primed with 0, 10, 100, or 1000 ng/ml LPS Se (1’LPS) or PGN (1’PGN) continuously for 18 h were washed twice with PBS, then challenged with high doses of LPS Se up to 10,000 ng/ml (2’LPS) or PGN up to 2,000 ng/ml (2’PGN). Supernatants and cell pellets were collected 5 h after start of challenge to measure TNF-α protein by ELISA (A, C) and miR-146a expression analysis by qRT-PCR (B, D). Data points and error bars represent mean ± SD of three independent experiments. *p < 0.05; **p < 0.01 compared with unprimed THP-1 cells.
Cross-regulation between TLR2, TLR4, and TLR5 ligands in THP-1 cells
Fig. 5A shows that priming with 100 ng/ml PGN (TLR2 ligand) resulted in significant hyporesponsiveness to PGN (500 ng/ml), as well as to heterologous ligand LPS (100 ng/ml) where TNF-α protein production dropped by >90% compared with unprimed controls observed in ELISA. In Fig. 5F, qRT-PCR analysis showed an ~12-fold increase of miR-146a expression in the PGN-primed cells (similar to Fig. 1B), indicating again a negative correlation with TNF-α production, as shown in Fig. 5A. Similar cross-tolerance and correlation of TNF-α and miR-146a expression was observed for another TLR2 ligand, Pam3CSK4 (Fig. 5B, 5G). Flagellin is a potent simulator of innate immunity and is recognized by TLR5, which triggers defense responses both at epithelial surfaces and systemically. Fig. 5C showed that priming with 100 ng/ml flagellin diminished TNF-α production by 95% in a flagellin secondary challenge, as well as in challenges with heterologous ligands Pam3CSK4 (50%) and PGN (70%). In contrast to these results, there was no significant change of TNF-α production between flagellin-primed and control unprimed THP-1 cells against LPS (Fig. 5C), and this observation is consistent with data reported by Mizel and Snipes (47). Note that miR-146a expression was induced in flagellin-primed cells (Fig. 5D). To obtain further understanding of why flagellin-primed THP-1 cells did not show hyporesponsiveness to LPS, it was postulated that an increased expression of TLR4 in stimulated cells, as reported by Siedlar et al. (54), might override miR-146a activity. Fig. 5E shows that TLR4 mRNA expression measured by qRT-PCR was higher in flagellin-primed cells than in unprimed controls. A moderate reduction of IRAK-1 and even less for TRAF6 was observed by Western blot analysis in PGN- and Pam3CSK4-primed THP-1 monocytes (Fig. 5H, 5I). Consequently, these in vitro data suggest cross-talking among TLRs, and may be attributed in part to miR-146a overexpression, affecting TNF-α production in primed cells against subsequent stimulation by homologous and heterologous ligands. The fact that flagellin-primed THP-1 cells did not affect TNF-α production in the LPS challenges served as an interesting control for observed specificity; the mechanism underlying it remains unclear even with the observed increase level of TLR4 and will be a subject of future studies.
FIGURE 5.

Peptidoglycan (PGN)-, Pam3CSK4 (Pam)-, and flagellin (Flag)-induced miR-146a contributes to cross-tolerance in THP-1 cells. THP-1 cells were primed with 100 ng/ml PGN, Pam3CSK4, or flagellin for 18 h, washed twice with PBS, then challenged with LPS Se (100 ng/ml), PGN (500 ng/ml), Pam3CSK4 (100 ng/ml), or flagellin (100 ng/ml). Supernatants and cell pellets were collected 5 h later for analysis of TNF-α protein by ELISA (A–C), miR-146a (D, F, G), and TLR4 mRNA expression in total RNA by qRT-PCR (E). Data points and error bars represent mean ± SD of three independent experiments. *p < 0.05; **p < 0.01 compared with untolerized THP-1 cells. PGN- and Pam3CSK4-primed THP-1 cells were challenged with various homologous or LPS for 2 h, and then cell lysates were analyzed for IRAK-1, TRAF6, and tubulin expression compared with control by Western blot (H, I).
IRAK-1 and TRAF6 knockdowns reduce inflammatory response to TLR ligands
IRAK-1 and TRAF6, the known molecular targets for miR-146a, are important adaptor kinases that help to amplify the initial signal transduction response by all TLRs except TLR3. The absence and/or inhibition of these kinases cause reduced inflammatory response by TLR ligands both in vivo and in vitro, indicating that these molecules mediate major mechanism of endotoxin tolerance (16, 32, 33, 36, 54, 55). To directly examine the importance of IRAK-1 and TRAF6 for other TLRs signal transduction in THP-1 monocyte activation, IRAK-1 and TRAF6 mRNA expression was targeted by specific siRNA transfection (Fig. 6). Twenty-four hours after transfection of THP-1 cells with 40 nM small interfering (si) RNA targeting IRAK-1 (siIRAK-1) resulted in a 60% decrease in IRAK-1 mRNA level but no affect on the mRNA of TRAF6 or lamin A/C (Fig. 6A). Similarly, the TRAF6 mRNA level was reduced by 80% following transfection with siRNA targeting TRAF6 (siTRAF6, Fig. 6B) confirming successful transfection and knockdown. Transfected cells were treated with various ligands, and culture supernatants were collected after 24 h incubation for cytokine production analysis. THP-1 cells deficient in IRAK-1 showed a reduction in TNF-α production in response to LPS Se (80%), Pam3CSK4 (50%), PGN (57%), LPS Pg (78%), and flagellin (60%) compared with mock-transfected cells, with LTA serving as a negative control (Fig. 6C). Similarly, Fig. 6C also shows that TRAF6 knockdown diminished TNF-α production by THP-1 cells in response to LPS Se (70%), Pam3CSK4 (52%), PGN (33%), LPS Pg (53%), and flagellin (35%). Similar effects were observed for IL-1β (Fig. 6D) and IL-6 production (Fig. 6E). These data are consistent with the report by Baltimore et al. (25) that IRAK-1 and TRAF6 are the central adaptor kinases, the absence of which greatly affects the ability to transmit subsequent TLRs signal transduction.
FIGURE 6.

IRAK-1 and TRAF6 knockdown results in decreased inflammatory response to microbial ligands. siRNA targeting IRAK-1 (A) or TRAF6 (B) were transfected into THP-1 cells, and knockdown efficiency was determined using qRT-PCR; lamin A/C (LMNA) was used as an unrelated mRNA control. Twenty-four hours after transfection, mock-transfected, IRAK-1–deficient, or TRAF6-deficient THP-1 cells were stimulated for 24 h with the indicated ligands shown at the foot of the graph (C–E). Cytokines released in the culture supernatants were determined by ELISA. Data are representative of three independent experiments and expressed as mean ± SD. *p < 0.05; **p < 0.01 compared with mock-transfected cell.
Upregulation of miR-146a alone can mimic LPS priming to induce cross-tolerance
LPS has been shown to induce the expression of a few regulatory miRNAs, including miR-146a (31, 48, 55), which is critical for endotoxin-induced tolerance (32). LPS-pretreated THP-1 cells showed cross-tolerance to a panel of innate immune ligands linked to miR-146a expression (Fig. 3A). Following this, to determine the direct functional role of miR-146a in endotoxin-induced cross-tolerance, THP-1 cells were transfected with 20 nM miR-146a mimic followed by challenges with various TLR ligands. After 24 h, miR-146a showed significantly higher expression (p < 0.01) in miR-146a mimic-transfected cells compared with mock-transfected cells, confirming the successful transfection (Fig. 7A). In Fig. 7B, TNF-α production by transfected THP-1 cells 5 h after challenges by LPS Se (76%), Pam3CSK4 (54%), PGN (30%), LPS Pg (64%), and flagellin (43%) was significantly reduced compared with the mock-transfected cells. The effect of overexpression of miR-146a on TRAF6 and IRAK-1 was validated by monitoring their mRNA levels (Fig. 7C), which were reduced by 60 and 55%, respectively, compared with mock-transfected cells. An unrelated human gene, lamin A/C, showed no significant changes in expression level between miR-146a mimic-transfected and mock-transfected controls. Reduction of TRAF6 and IRAK-1 protein levels was documented by Western blot analysis (Fig. 7D), consistent with previous reports (27, 32). Taken together, these data demonstrate that LPS-induced miR-146a upregulation plays a pivotal role in providing cross-tolerance against a range of innate immune ligands.
FIGURE 7.
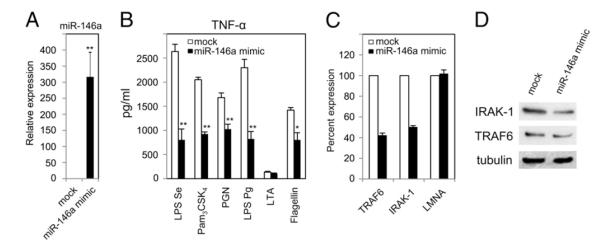
Transfected miR-146a alone mimics LPS-induced cross-tolerance to various innate immune TLR ligands. THP-1 cells were transfected with 20 nM miR-146a mimic. They and mock-transfected cells were incubated for 24 h, washed with complete growth medium, and then challenged with indicated ligands for another 3 h. A, RNA isolates were prepared from cell pellets of mock and transfected cells followed by qRT-PCR for miR-146a expression normalized to RNU44. B, TNF-α protein measured using ELISA in supernatants from mock- and miR-146a-mimic-transfected cells. C, The RNA samples were also analyzed by qRT-PCR for mRNA expression of miR-146a targets TRAF6, IRAK-1, and unrelated lamin A/C (LMNA, negative control). Data are representative of three independent experiments and expressed as mean ± SD. *p < 0.05; **p < 0.01 compared with mock-transfected cells. Western blot analysis for IRAK-1, TRAF6, and tubulin in the cell lysates collected 24 h after with miR-146a-mimic transfection (D) or without (mock).
miR-146a expression links to cross-tolerance against heat-killed whole bacteria
In the preceding experiments, LPS-induced cross-tolerance was observed in TLR1, TLR2, and TLR5 ligands where miR-146a played a dominant role. However, in nature, individuals are exposed to whole bacteria rather than a single microbial ligand. Fig. 8 shows LPS-induced cross-tolerance demonstrated using heat-killed whole bacteria. Priming THP-1 cells with low-dose LPS (10 ng/ml) resulted in significant (p < 0.01) reduction of TNF-α production following the secondary challenge with heat-killed E. coli (75%) or P. gingivalis (57%) at 1 × 106 CFU/ml (Fig. 8A). Similar hyporesponsiveness by human leukocytes has been observed by de Vos et al. (12). To demonstrate the role of miR-146a alone in this cross-tolerance, TNF-α production by miR-146a mimic-transfected THP-1 cells that were subsequently challenged with heat-killed E. coli at 1 × 106 CFU/ml for 5 h was examined. In Fig. 8B, miR-146a mimic-transfected THP-1 cells showed hyporesponsiveness to heat-killed E. coli, and this sensitivity is miR-146a mimic dose-dependent. Thus, miR-146a plays an important role in providing cross-tolerance in the in vitro THP-1 cell model against various TLRs ligands, as well as whole heat-killed bacteria.
FIGURE 8.
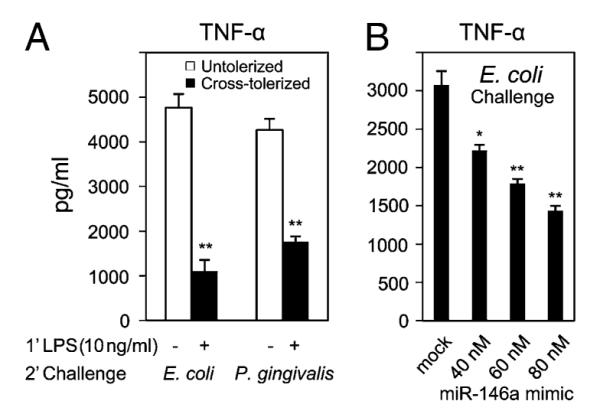
Elevated levels of LPS-induced miR-146a contribute to resistance to heat-killed bacteria in the THP-1 cell model. A, THP-1 cells primed with 10 ng/ml LPS Se for 18 h (cross-tolerized, filled bars) and untreated controls incubated for the same time period (untolerized, open bars) were washed twice with PBS, then challenged with heat-killed E. coli or P. gingivalis at 1 × 106 CFU/ml for 3 h. B, THP-1 cells transfected with miR-146a mimic (40, 60, or 80 nM), followed by incubation for 24 h, and mock-transfected cells were washed with complete growth medium and challenged with heat-killed E. coli (1 × 106 CFU/ml). TNF-α protein in supernatants from mock-transfected and miR-146a mimic-transfected cells was measured using ELISA. Data are representative of three independent experiments and expressed as mean ± SD. *p < 0.05; **p < 0.01 compared with untolerized or mock-transfected cells.
Discussion
While involvement of miR-146a in innate immunity has previously been reported in response to various microbial components and cytokines, much is still not known about its biological significance. In this study, the kinetics of TLR ligand-induced miR-146a expression were analyzed to elucidate its broader role in innate immunity. Consistent with previous findings, exposure of THP-1 monocytes to various bacterial inflammatory insults, such as LPS, PGN, and flagellin, resulted in rapid and continuous expression of mature miR-146a. Similar kinetics were not observed, however, for other LPS-induced miRNAs, such as miR-155 and miR-132 (Fig. 1). Owing to low basal levels of miR-146a in THP-1 monocytes, production of proinflammatory cytokines such as TNF-α takes place continuously once LPS stimulation occurs. As miR-146a expression starts to rise, the expression of IRAK-1 and TRAF6 are putatively inhibited via translational repression and mRNA degradation and thus remove the amplification effectors in TNF-α production (32). A similar negative correlation in TLR2, TLR4, and TLR5 microbial ligands-induced cytokine release and miR-146a overexpression was also observed in this study. TNF-α production was increased in response to the same ligands after blocking miR-146a activity, indicating its direct or indirect effect on TLR signaling. The new data in this article are thus focused on defining the role of miR-146a in endotoxin-induced cross-tolerance to those ligands, many of which, except LTA, MDP, and IFN-γ, also induce miR-146a upregulation.
Endotoxin tolerance is known to prevent inflammatory response to homologous or heterologous ligand challenge. Upon pre-exposure to other TLR ligands from Gram-positive or Gram-negative bacteria, monocytes acquire similar hyporesponsiveness to unrelated ligands. In the case of LPS-induced tolerance, a number of mechanisms have been proposed, including downregulation of TLR4 expression (41), degradation of such adaptor proteins as IRAK-1 or TRAF6 (32, 33, 36) decreasing the association of TLR4 and MyD88 (56), suppression by IRAK-M and TRIM30α (57, 58), downregulation of NF-κB activity, disruption of chromatin remodeling, expression of anti-inflammatory cytokines, and, more recently, by the post-transcriptional silencing of adaptor kinase mRNA through the action of miR-146a (32). In most cases, LPS-induced cross-tolerance is thought to occur by way of a similar mechanism involved in homologous tolerance. The key regulator of cross-tolerance is poorly understood, unfortunately, and thus our study of the mechanistic role of miR-146a in LPS-induced cross-tolerance is novel.
Our proposed model for the mechanistic role of miR-146a in LPS-induced cross-tolerance in THP-1 monocytes is as follows. In this THP-1 cell model, LPS/TLR4 signaling is facilitated by LPS-binding protein and CD14 and is mediated by a TLR4/MD-2 receptor complex. The downstream of the TLR4 signaling cascade is initiated after the binding of the Toll/IL-1R domain with adaptor protein MyD88. This binding acts as a bridge between TLR4 and incoming IRAK-1 adaptor kinase, which further recruits TRAF6. This series of interactions triggers overall signal amplification, activation, and translocation of the transcription factor NF-κB, which quickly initiates the expression of immune-responsive genes and such proinflammatory cytokines as TNF-α at mRNA and protein levels peaking at 2 and 4 h, respectively (32). Although miR-146a transcription is also known to be regulated by NF-κB (31), there appears to be a delay of between 2 and 4 h in its expression, with the level clearly elevated at 8 h (32), and it remains at a high level (from 30- to 100-fold) for up to 72 h (data not shown). The increase in the miR-146a level has negative effects on mRNA expression of IRAK-1 and TRAF6, which are shared in the TLR4, TLR1/6, TLR2, and TLR5 pathway. Thus, the progression of these signal transductions are controlled or inhibited by miR-146a upregulation in LPS-primed THP-1 cells. In nature, individuals are exposed to various doses of different TLR ligands at the primary or secondary level. Notably, LPS-tolerized cells can resist 100-fold higher doses of homologous secondary challenges as measured by the reduction in TNF-α production (32). Similarly, TLR2 ligands such as PGN showed hyporesponsiveness to higher doses of homologous secondary challenges where miR-146a was upregulated while adaptor kinases were downregulated. Thus, once miR-146a upregulation takes place, THP-1 monocytes then become hyporesponsive to higher doses of secondary challenges of many known TLR ligands.
Biological significance of LPS-induced cross-tolerance contributed by miR-146a
In this article, the ability of LPS-primed THP-1 monocytes to induce cross-tolerance to other TLR agonists is congruent with previous in vitro and in vivo findings (12, 50). Monocytes or macrophages pretreated with LPS showed hyporesponsiveness to TLR2 ligands (LTA, PGN, Pam3CSK4, LPS from P. gingivalis) (36, 59, 60) or flagellin, as reported in this study and by others (47). Similarly, in this study, Pam3CSK4, PGN, and flagellin demonstrated both self-tolerance and cross-tolerance to LPS, which is consistent with previous findings (20, 36, 61). Jacinto et al. (36) observed that LPS priming caused disruption of IRAK kinases and development of cross-tolerance to LTA. However, LTA did not show cross-tolerance to LPS at a significant level; the present studies suggest this is true in part because LTA does not induce high levels of miR-146a. Consistent with these in vitro findings, Lehner et al. (61) and Dalpke et al. (20) observed that LPS-primed mice were hyporesponsive to LTA and CpG DNA. Although LPS cross-tolerized cells to flagellin (Fig. 3E), flagellin did not prevent a strong response to LPS (Fig. 5C). This finding raises the possibility that flagellin and LPS tolerance may be mediated by different, distinct mechanisms. Regarding this idea, Mizel and Snipes (47) observed that flagellin tolerance was not due to decreased surface expression of TLR5 or IRAK-1 degradation. Interestingly, flagellin-primed cells showed an increase of TLR4 expression (Fig. 4E), a finding corroborated by van Aubel et al. (62). Although it is not still clear why flagellin does not induce cross-tolerance against LPS, the latter finding provides additional evidence that there is a difference between flagellin and LPS tolerance.
Note that cross-tolerization also occurs between LPS and cytokines such as TNF-α and IL-1. Repeated injections or infusions of TNF-α in rats were shown to protect against an LD50 injection of LPS (63), suppressing LPS-induced fever (64) and cytokine production by bone marrow-derived adherent cells (65). TNF-α and LPS do not induce reciprocal tolerance in murine macrophages (16). However, THP-1 cells pretreated with TNF-α showed a partial repression of LPS-induced cell signaling, as reported by Ferlito et al. (49). This may be linked to the miR-146a expression observed in this study. The difference between murine macrophages and human cells, including THP-1 monocytes, to various ligands has been elucidated by Bowie (58). Following this observation, Bowie concluded that TLR expression patterns are not entirely equivalent in humans and mice. Previously, however, De Nardo et al. (66) observed that LPS-primed primary mouse macrophages showed tolerance due to IRAK-1 protein degradation. Accordingly, in this study, LPS- or PGN-primed mouse peritoneal macrophages showed homologous or heterologous tolerance where miR-146a showed elevated expression in LPS-tolerized cells that can regulate the adaptor kinases. IL-1β was shown to be involved in the induction of homologous tolerance and in limiting the response of THP-1 cells to LPS, as shown by others both in vivo and in vitro (67). This new finding regarding cross-tolerance (except flagellin to LPS) is linked to miR-146a overexpression. As expected, IRAK-1 or TRAF6 adaptor kinase expression was inhibited by PGN, and Pam3CSK4-tolerized cells provide additional support for miR-146a involvement in cross-tolerance. Interestingly, our study (data not shown) and Perry et al. (68) both found continuous miR-146a expression in IL-1β treated A549 cells, which showed negative effect on IRAK-1 and TRAF6 at the posttranscriptional level. In their study, miR-146a alone has been shown to negatively regulate the release of proinflammatory chemokines, including IL-8 and RANTES in IL-1β–treated A549 cells.
IRAK-1 and TRAF6, known as the proximal protein kinases in TLR signaling pathways (69), are the molecular targets for miR-146a. TLR signaling is impaired due to the degradation or translation inhibition of these adaptor kinases during innate immune activation, giving rise to endotoxin tolerance (16, 32, 36, 54, 70). Similarly, silencing of adaptor kinases rendered THP-1 monocytes hyporesponsive to a set of innate immune ligands and resulted in lower cytokine production, namely TNF-α, IL-1β, or IL-6. However, silencing of either IRAK-1 or TRAF6 did not cause complete suppression of cytokines in response to the TLR ligands used in this study (Fig. 6). A similar pattern of LPS response was observed after knockdown either of the adaptor kinases in monocytes or macrophages, as reported by others (66, 71). These findings imply that IRAK-1 and TRAF6 translational inhibition by miR-146a in LPS-primed cells contributes to homologous and heterologous tolerance at a significant level, but not in a complete manner. Such a conclusion is consistent with the failure of complete resistance to LPS-induced death in IRAK-deficient mice (66). Additionally, a deficiency of IRAK-1 impairs, but does not completely eliminate, IL-1 actions as reported by Thomas et al. (72). This suggests that other members of the IRAK family may compensate in part for the functions of IRAK-1.
Notably, during LPS stimulation of THP-1 cells, a few miRNAs, including miR-146a, are upregulated. The biological significance of such miRNAs as miR-132 (31), miR-9 (48), and miR-155 (31) to LPS-induced cross-tolerance is not clear. They may participate directly or indirectly to LPS-induced cross-tolerance. The activation of NF-κB by TLRs is critical to inflammation. Among the LPS-induced miRNAs, only miR-146a has been shown to be NF-kB–dependent, and inhibition of NF-κB by PDTC, an NF-κB inhibitor, decreased miR-146a expression (27, 31). Consequently, in this report, miR-146a overexpression by miR-146a mimic transfection caused THP-1 monocytes to be less responsive to various ligands, including LPS. Krützfeldt et al. (73, 74) introduced the usage of chemically modified miRNA inhibitors, known as antagomirs, to define the biological functions of miRNAs. In this study, miR-146a knockdown in THP-1 cells showed more inflammatory response to non-LPS ligands. Interestingly, PGN-primed THP-1 monocytes recovered responsiveness correlating with the reduced levels of miR-146a in the PGN withdrawal experiment (Fig. 3J, 3K) demonstrated its role to maintain immune homeostasis.
In addition to purified ligands, LPS-induced cross-tolerance against whole bacteria was also shown in vitro (Fig. 8). Accordingly, Cavaillon et al. (75) first reported that LPS-tolerized human monocytes were hyporeactive in TNF-α production, to heat-killed Streptococcus pyogenes, S. aureus, and zymosan (a TLR2 ligand). Reduced ex vivo responsiveness to whole heat-killed Gram-positive bacteria, such as S. pyogenes and S. aureus (75), has been also reported in severe sepsis. Similarly, in this report, LPS-primed monocytes showed significant reduction of TNF-α after challenging with E. coli and P. gingivalis. Interestingly, miR-146a expression showed a negative correlation with TNF-α production, indicating its role in hyporesponsiveness to whole bacteria. This has also been supported by transfection of miR-146a mimic alone. Thus, this finding fully supports the dominant role of miR-146a in LPS-induced cross-tolerance compared with other negative regulators previously suggested.
Implication of differential cross-regulation in innate immunity
Various PAMPs from both Gram-positive and Gram-negative bacteria have been shown to interact with particular TLRs. Thus, while sepsis is more commonly caused by Gram-positive bacteria, it can also be caused by Gram-negative bacteria. Moreover, LPS from Gram-negatives is one of the best studied immunostimulatory components of bacteria and can induce systemic inflammation and sepsis if excessive signals occur without proper regulation. In our previous report, miR-146a was upregulated in LPS-stimulated THP-1 cells, showing it to be a regulator of proinflammatory cytokine production by establishing tolerance against LPS (32). Interestingly, both we and Taganov et al. (31) observed elevated miR-146a expression in response to TLR2 or to TLR5 stimulation by bacterial and fungal components or following exposure to TNF-α or IL-1β. Following this, the ligands that showed miR-146a expression were primarily considered in our LPS-induced cross-tolerance study. This upregulated miR-146a caused by LPS provided cross-tolerance in THP-1 cells. Analogous to LPS, both TLR2 and TLR5 ligands showed cross-tolerance to unrelated ligands provided by miR-146a expression. miR-146a induction, however, was not observed in response to a nucleic acid analog, such as polyinosinic-polycytidylic acid (31), although, in a recent study, Hou et al. (27) observed overexpression of miR-146a in response to vesicular stomatitis virus infection in a mouse infection model. In that study, miR-146a was shown to regulate type 1 IFN by inhibiting retinoic acid-inducible gene I signaling molecules IRAK-1, IRAK-2, and TRAF-6 (27). Interestingly, a report by Tang et al. (76) showed the downregulation of miR-146a in a subset of human lupus patients with elevated expression of type 1 IFN. In THP-1 monocytes, NOD2 is highly expressed and is a receptor for PGN or its break-down product MDP. In our study, MDP alone was unable to stimulate proinflammatory cytokine production in THP-1 cells, but, in combination with LPS, produced a synergistic effect, as shown by others (77). After 18 h MDP treatment, THP-1 cells did not show cross-tolerance to TLR2 or TLR4 ligands (data not shown), although after prolonged treatment of primary monocyte-derived macrophages with MDP, Hedl et al. (78) observed cross-tolerance against Pam3CSK4 and lipid A (TLR4 ligand). This discrepancy might be due to cell type differences or treatment period or both. In another study, Gutierrez et al. (79) observed that NOD2-induced NF-κB activation involving IRAK-1 is likely to activate cytokine production. Thus, miR-146a is likely to downregulate NOD2 activity by affecting IRAK-1 and contributes to maintaining a state of hyporesponsiveness toward commensal microflora. In any case, TNF-α can be produced through activation of all TLRs, except TLR3, and all of these pathways involve miR-146a target molecules TRAF6 and IRAK-1 adaptor kinases (35). Recently, de Vos et al. (12) observed LPS-induced cross-tolerance against all TLR ligands, including TLR3 and TLR7, in human volunteers. Similarly, LPS-induced cross-tolerance was observed in THP-1 cells against TLR3 ligands, such as polyinosinic-polycytidylic acid, in terms of TNF-α production (data not shown). Whether innate immunity involving miRNAs, including miR-146a, directly or indirectly regulates TLR3 and TLR7 signaling remains to be determined. Therefore, endotoxin-induced cross-tolerance associated with upregulated miR-146a may have a broader role in regulating TNF-α, a key player in the cytokine network, induced by a number of pattern recognition receptors in innate immunity. In the future, however, in vivo investigations, such as the phenotypic analysis of mice with targeted deletion of miR-146a, will be necessary to fully explore the role of this miRNA in innate immunity. It is interesting to speculate whether all of these TLRs involving TRAF6 and IRAK-1 were regulated by miR-146a in a comparable manner. During LPS induction, some of the miRNAs have been reported to be downregulated, including let-7i, which is thought to target TLR4 itself (80), and miR-125b, which is known to target TNF-α (81). Sometimes miRNAs are dysregulated in cancer (82) or aberrantly expressed in such inflammatory diseases as rheumatoid arthritis (83). Thus, miRNAs may form a key link between inflammation and cancer; however, the induction of specific miRNAs by TLRs, including miR-146a, as a key step in tumor progression is still unclear.
In summary, multiple lines of evidence reported in this study substantiate the indispensable role of miR-146a in endotoxin-induced cross-tolerance. miR-146a expression involved in LPS-induced cross-tolerance operates as a negative regulatory feedback (or tuning) mechanism to prevent the destructive consequences of uncontrolled inflammatory reaction caused by overactivation of TLR signaling. Modulation of miR-146a level can have an opposite effect, notably on the levels of adaptor kinases, leading to the attenuation of TLRs signaling. This finding highlights the importance of further investigations on whether miR-146a can be used as a therapeutic intervention for boosting or limiting TLR activation to maintain a controlled immune response.
Supplementary Material
Acknowledgments
E. coli DH10B and P. gingivalis strain 33277 were gifts from the laboratories of Dr. Robert Burne and Dr. Richard Lamont, respectively, at the University of Florida. We acknowledge and thank Steven J. Ross for help with early editing.
This work was supported in part by a grant from the Lupus Research Institute and by National Institutes of Health Grant AI47859. M.A.N. was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases Rheumatology Training Grant T32 AR007603.
Abbreviations used in this article
- IRAK-1
IL-1R–associated kinase 1
- LPS Pg
LPS from Porphyromonas gingivalis
- LPS Se
LPS from Salmonella enterica
- LTA
lipoteichoic acid
- MDP
muramyl dipeptide
- miR
microRNA
- miRNA
microRNA
- Pam3CSK4
Pam3CSK4CysSerLys4
- PAMP
pathogen-associated molecular pattern
- PGN
peptidoglycan
- qRT-PCR
quantitative real-time PCR
- si
small interfering
- TRAF6
TNFR-associated factor 6
Footnotes
Disclosures The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akira S, Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 4.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of Toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 6.Beutler B, Cerami A. Cachectin: more than a tumor necrosis factor. N. Engl. J. Med. 1987;316:379–385. doi: 10.1056/NEJM198702123160705. [DOI] [PubMed] [Google Scholar]
- 7.Danner RL, Elin RJ, Hosseini JM, Wesley RA, Reilly JM, Parillo JE. Endotoxemia in human septic shock. Chest. 1991;99:169–175. doi: 10.1378/chest.99.1.169. [DOI] [PubMed] [Google Scholar]
- 8.Shishodia S, Aggarwal BB. Nuclear factor-kB activation: a question of life or death. J. Biochem. Mol. Biol. 2002;35:28–40. doi: 10.5483/bmbrep.2002.35.1.028. [DOI] [PubMed] [Google Scholar]
- 9.Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit. Care. 2006;10:233. doi: 10.1186/cc5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCall CE, Grosso-Wilmoth LM, LaRue K, Guzman RN, Cousart SL. Tolerance to endotoxin-induced expression of the interleukin-1β gene in blood neutrophils of humans with the sepsis syndrome. J. Clin. Invest. 1993;91:853–861. doi: 10.1172/JCI116306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biswas SK, Bist P, Dhillon MK, Kajiji T, Del Fresno C, Yamamoto M, Lopez-Collazo E, Akira S, Tergaonkar V. Role for MyD88-independent, TRIF pathway in lipid A/TLR4-induced endotoxin tolerance. J. Immunol. 2007;179:4083–4092. doi: 10.4049/jimmunol.179.6.4083. [DOI] [PubMed] [Google Scholar]
- 12.de Vos AF, Pater JM, van den Pangaart PS, de Kruif MD, van ’t Veer C, van der Poll T. In vivo lipopolysaccharide exposure of human blood leukocytes induces cross-tolerance to multiple TLR ligands. J. Immunol. 2009;183:533–542. doi: 10.4049/jimmunol.0802189. [DOI] [PubMed] [Google Scholar]
- 13.del Fresno C, García-Rio F, Gómez-Piña V, Soares-Schanoski A, Fernández-Ruíz I, Jurado T, Kajiji T, Shu C, Marín E, Gutierrez delArroyo A, et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J. Immunol. 2009;182:6494–6507. doi: 10.4049/jimmunol.0803350. [DOI] [PubMed] [Google Scholar]
- 14.Dobrovolskaia MA, Vogel SN. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002;4:903–914. doi: 10.1016/s1286-4579(02)01613-1. [DOI] [PubMed] [Google Scholar]
- 15.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 16.Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and Toll-like receptor 2 and 4 gene expression. J. Immunol. 2000;164:5564–5574. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- 17.Beutler B. SHIP, TGF-β, and endotoxin tolerance. Immunity. 2004;21:134–135. doi: 10.1016/j.immuni.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 18.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 19.Adib-Conquy M, Cavaillon JM. Compensatory anti-inflammatory response syndrome. Thromb. Haemost. 2009;101:36–47. [PubMed] [Google Scholar]
- 20.Dalpke AH, Lehner MD, Hartung T, Heeg K. Differential effects of CpG-DNA in Toll-like receptor-2/-4/-9 tolerance and cross-tolerance. Immunology. 2005;116:203–212. doi: 10.1111/j.1365-2567.2005.02211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 22.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 23.Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009;21:452–460. doi: 10.1016/j.ceb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 25.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat. Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 26.Lodish HF, Zhou B, Liu G, Chen CZ. Micromanagement of the immune system by microRNAs. Nat. Rev. Immunol. 2008;8:120–130. doi: 10.1038/nri2252. [DOI] [PubMed] [Google Scholar]
- 27.Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. MicroRNA-146a feedback inhibits RIG-I-dependent type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 2009;183:2150–2158. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- 28.Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc. Natl. Acad. Sci. USA. 2009;106:15819–15824. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruggiero T, Trabucchi M, DeSanta F, Zupo S, Harfe BD, McManus MT, Rosenfeld MG, Briata P, Gherzi R. LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. FASEB J. 2009;23:2898–2908. doi: 10.1096/fj.09-131342. [DOI] [PubMed] [Google Scholar]
- 30.Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LA. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010;11:141–147. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- 31.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nahid MA, Pauley KM, Satoh M, Chan EKL. miR-146a is critical for endotoxin-induced tolerance: implication in innate immunity. J. Biol. Chem. 2009;284:34590–34599. doi: 10.1074/jbc.M109.056317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Cousart S, Hu J, McCall CE. Characterization of interleukin-1 receptor-associated kinase in normal and endotoxin-tolerant cells. J. Biol. Chem. 2000;275:23340–23345. doi: 10.1074/jbc.M001950200. [DOI] [PubMed] [Google Scholar]
- 34.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 35.Krishnan J, Selvarajoo K, Tsuchiya M, Lee G, Choi S. Toll-like receptor signal transduction. Exp. Mol. Med. 2007;39:421–438. doi: 10.1038/emm.2007.47. [DOI] [PubMed] [Google Scholar]
- 36.Jacinto R, Hartung T, McCall C, Li L. Lipopolysaccharide- and lipoteichoic acid-induced tolerance and cross-tolerance: distinct alterations in IL-1 receptor-associated kinase. J. Immunol. 2002;168:6136–6141. doi: 10.4049/jimmunol.168.12.6136. [DOI] [PubMed] [Google Scholar]
- 37.Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, Shiratori Y, Omata M. Distinct mechanism of Helicobacter pylori-mediated NF-κB activation between gastric cancer cells and monocytic cells. J. Biol. Chem. 2001;276:44856–44864. doi: 10.1074/jbc.M105381200. [DOI] [PubMed] [Google Scholar]
- 38.Nahid AM, Sugii S. Binding of porcine ficolin-α to lipopolysaccharides from Gram-negative bacteria and lipoteichoic acids from Gram-positive bacteria. Dev. Comp. Immunol. 2006;30:335–343. doi: 10.1016/j.dci.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.Shaffer JP. Modified sequentially rejective multiple test procedures. J. Am. Statist. Assoc. 1986;81:826–831. [Google Scholar]
- 41.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 42.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 43.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 44.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh TK, Mickelson DJ, Fink J, Solberg JC, Inglefield JR, Hook D, Gupta SK, Gibson S, Alkan SS. Toll-like receptor (TLR) 2–9 agonists-induced cytokines and chemokines, I: Comparison with T cell receptor-induced responses. Cell. Immunol. 2006;243:48–57. doi: 10.1016/j.cellimm.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 46.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell. Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 47.Mizel SB, Snipes JA. Gram-negative flagellin-induced self-tolerance is associated with a block in interleukin-1 receptor-associated kinase release from Toll-like receptor 5. J. Biol. Chem. 2002;277:22414–22420. doi: 10.1074/jbc.M201762200. [DOI] [PubMed] [Google Scholar]
- 48.Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA. 2009;106:5282–5287. doi: 10.1073/pnas.0810909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferlito M, Romanenko OG, Ashton S, Squadrito F, Halushka PV, Cook JA. Effect of cross-tolerance between endotoxin and TNF-α or IL-1β on cellular signaling and mediator production. J. Leukoc. Biol. 2001;70:821–829. [PubMed] [Google Scholar]
- 50.Wang JH, Doyle M, Manning BJ, Di Wu Q, Blankson S, Redmond HP. Induction of bacterial lipoprotein tolerance is associated with suppression of Toll-like receptor 2 expression. J. Biol. Chem. 2002;277:36068–36075. doi: 10.1074/jbc.M205584200. [DOI] [PubMed] [Google Scholar]
- 51.Sato S, Nomura F, Kawai T, Takeuchi O, Mühlradt PF, Takeda K, Akira S. Synergy and cross-tolerance between toll-like receptor (TLR) 2-and TLR4-mediated signaling pathways. J. Immunol. 2000;165:7096–7101. doi: 10.4049/jimmunol.165.12.7096. [DOI] [PubMed] [Google Scholar]
- 52.LaRue KE, McCall CE. A labile transcriptional repressor modulates endotoxin tolerance. J. Exp. Med. 1994;180:2269–2275. doi: 10.1084/jem.180.6.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.West MA, Heagy W. Endotoxin tolerance: a review. Crit. Care Med. 2002;30:S64–S73. [PubMed] [Google Scholar]
- 54.Siedlar M, Frankenberger M, Benkhart E, Espevik T, Quirling M, Brand K, Zembala M, Ziegler-Heitbrock L. Tolerance induced by the lipopeptide Pam3Cys is due to ablation of IL-1R-associated kinase-1. J. Immunol. 2004;173:2736–2745. doi: 10.4049/jimmunol.173.4.2736. [DOI] [PubMed] [Google Scholar]
- 55.Yeo SJ, Yoon JG, Hong SC, Yi AK. CpG DNA induces self and cross-hyporesponsiveness of RAW264.7 cells in response to CpG DNA and lipopolysaccharide: alterations in IL-1 receptor-associated kinase expression. J. Immunol. 2003;170:1052–1061. doi: 10.4049/jimmunol.170.2.1052. [DOI] [PubMed] [Google Scholar]
- 56.Medvedev AE, Lentschat A, Wahl LM, Golenbock DT, Vogel SN. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J. Immunol. 2002;169:5209–5216. doi: 10.4049/jimmunol.169.9.5209. [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi K, Hernandez LD, Galán JE, Janeway CA, Jr., Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 58.Bowie AG. TRIM-ing down Tolls. Nat. Immunol. 2008;9:348–350. doi: 10.1038/ni0408-348. [DOI] [PubMed] [Google Scholar]
- 59.Burkart V, Kim YE, Hartmann B, Ghiea I, Syldath U, Kauer M, Fingberg W, Hanifi-Moghaddam P, Müller S, Kolb H. Cholera toxin B pretreatment of macrophages and monocytes diminishes their proinflammatory responsiveness to lipopolysaccharide. J. Immunol. 2002;168:1730–1737. doi: 10.4049/jimmunol.168.4.1730. [DOI] [PubMed] [Google Scholar]
- 60.Hajishengallis G, Martin M, Sojar HT, Sharma A, Schifferle RE, DeNardin E, Russell MW, Genco RJ. Dependence of bacterial protein adhesins on Toll-like receptors for proinflammatory cytokine induction. Clin. Diagn. Lab. Immunol. 2002;9:403–411. doi: 10.1128/CDLI.9.2.403-411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lehner MD, Morath S, Michelsen KS, Schumann RR, Hartung T. Induction of cross-tolerance by lipopolysaccharide and highly purified lipoteichoic acid via different Toll-like receptors independent of paracrine mediators. J. Immunol. 2001;166:5161–5167. doi: 10.4049/jimmunol.166.8.5161. [DOI] [PubMed] [Google Scholar]
- 62.van Aubel RA, Keestra AM, Krooshoop DJ, van Eden W, van Putten JP. Ligand-induced differential cross-regulation of Toll-like receptors 2, 4 and 5 in intestinal epithelial cells. Mol. Immunol. 2007;44:3702–3714. doi: 10.1016/j.molimm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 63.Fraker DL, Stovroff MC, Merino MJ, Norton JA. Tolerance to tumor necrosis factor in rats and the relationship to endotoxin tolerance and toxicity. J. Exp. Med. 1988;168:95–105. doi: 10.1084/jem.168.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klir JJ, McClellan JL, Kozak W, Szelényi Z, Wong GH, Kluger MJ. Systemic but not central administration of tumor necrosis factor-a attenuates LPS-induced fever in rats. Am. J. Physiol. 1995;268:R480–R486. doi: 10.1152/ajpregu.1995.268.2.R480. [DOI] [PubMed] [Google Scholar]
- 65.Ogle CK, Guo X, Chance WT, Ogle JD. Induction of endotoxin tolerance in rat bone marrow cells by in vivo infusion of tumor necrosis factor. Crit. Care Med. 1997;25:827–833. doi: 10.1097/00003246-199705000-00019. [DOI] [PubMed] [Google Scholar]
- 66.De Nardo D, Nguyen T, Hamilton JA, Scholz GM. Down-regulation of IRAK-4 is a component of LPS- and CpG DNA-induced tolerance in macrophages. Cell. Signal. 2009;21:246–252. doi: 10.1016/j.cellsig.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 67.Henricson BE, Neta R, Vogel SN. An interleukin-1 receptor antagonist blocks lipopolysaccharide-induced colony-stimulating factor production and early endotoxin tolerance. Infect. Immun. 1991;59:1188–1191. doi: 10.1128/iai.59.3.1188-1191.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in microRNA-146a expression negatively regulate the IL-1β-induced inflammatory response in human lung alveolar epithelial cells. J. Immunol. 2008;180:5689–5698. doi: 10.4049/jimmunol.180.8.5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 70.Li CH, Wang JH, Redmond HP. Bacterial lipoprotein-induced self-tolerance and cross-tolerance to LPS are associated with reduced IRAK-1 expression and MyD88-IRAK complex formation. J. Leukoc. Biol. 2006;79:867–875. doi: 10.1189/jlb.0905505. [DOI] [PubMed] [Google Scholar]
- 71.Pauley KM, Satoh M, Pauley BA, Dominguez-Gutierrez PR, Wallet SM, Holliday LS, Cha S, Reeves WH, Chan EK. Formation of GW/P bodies as marker for microRNA-mediated regulation of innate immune signaling in THP-1 cells. Immunol. Cell Biol. 2010;88:205–212. doi: 10.1038/icb.2009.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thomas JA, Allen JL, Tsen M, Dubnicoff T, Danao J, Liao XC, Cao Z, Wasserman SA. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J. Immunol. 1999;163:978–984. [PubMed] [Google Scholar]
- 73.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with “antagomirs”. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 74.Krützfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–2892. doi: 10.1093/nar/gkm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Endotoxin tolerance: is there a clinical relevance? J. Endotoxin Res. 2003;9:101–107. doi: 10.1179/096805103125001487. [DOI] [PubMed] [Google Scholar]
- 76.Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, Huang X, Zhou H, de Vries N, Tak PP, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60:1065–1075. doi: 10.1002/art.24436. [DOI] [PubMed] [Google Scholar]
- 77.Yang S, Tamai R, Akashi S, Takeuchi O, Akira S, Sugawara S, Takada H. Synergistic effect of muramyldipeptide with lipopolysaccharide or lipoteichoic acid to induce inflammatory cytokines in human monocytic cells in culture. Infect. Immun. 2001;69:2045–2053. doi: 10.1128/IAI.69.4.2045-2053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hedl M, Li J, Cho JH, Abraham C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc. Natl. Acad. Sci. USA. 2007;104:19440–19445. doi: 10.1073/pnas.0706097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, Nunez G, Fernandez-Luna JL. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-κB activation. J. Biol. Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 80.Chen XM, Splinter PL, O’Hara SP, LaRusso NF. A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J. Biol. Chem. 2007;282:28929–28938. doi: 10.1074/jbc.M702633200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-α stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 82.Li Y, Vandenboom TG, II, Wang Z, Kong D, Ali S, Philip PA, Sarkar FH. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70:1486–1495. doi: 10.1158/0008-5472.CAN-09-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chan EKL, Satoh M, Pauley KM. Contrast in aberrant microRNA expression in systemic lupus erythematosus and rheumatoid arthritis: is microRNA-146 all we need? Arthritis Rheum. 2009;60:912–915. doi: 10.1002/art.24421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.