

Abstract
Asthma is a common inflammatory disease involving crosstalk between innate and adaptive immunity. We reveal that antibacterial innate immunity protein, peptidoglycan recognition protein 1 (Pglyrp1), is involved in the development of allergic asthma. Pglyrp1−/− mice developed less severe asthma than wild type (WT) mice following sensitization with house dust mite (HDM) allergen. HDM-sensitized Pglyrp1−/− mice, compared with WT mice, had diminished: bronchial hyper-responsiveness (lung airway resistance); numbers of eosinophils, neutrophils, lymphocytes, and macrophages in bronchoalveolar lavage fluid and lungs; inflammatory cell infiltrates in the lungs around bronchi, bronchioles, and pulmonary arteries and veins; lung remodeling (mucin-producing goblet cell hyperplasia and metaplasia and smooth muscle hypertrophy and fibrosis); levels of IgE, eotaxins, IL-4, IL-5, and IL-17 in the lungs; and numbers of Th2 and Th17 cells and expression of their marker genes in the lungs. The mechanism underlying this decreased sensitivity of Pglyrp1−/− mice to asthma was increased generation and activation of CD8α+β+ and CD8α+β− plasmacytoid dendritic cells (pDC) and increased recruitment and activity of regulatory T (Treg) cells in the lungs. In vivo depletion of pDC in HDM-sensitized Pglyrp1−/− mice reversed the low responsive asthma phenotype of Pglyrp1−/− mice to resemble the more severe WT phenotype. Thus, Pglyrp1−/− mice efficiently control allergic asthma by up-regulating pDC and Treg cells in the lungs, whereas in WT mice Pglyrp1 is proinflammatory and decreases pDC and Treg, and increases pro-asthmatic Th2 and Th17 responses. Blocking Pglyrp1 or enhancing pDC in the lungs may be beneficial for prevention and treatment of asthma.
Introduction
Asthma is a common inflammatory disease of the airways characterized by airway hyper-responsiveness, increased mucus secretion, chronic inflammation, and airway remodeling response. Asthma affects 7% of the USA population with sharply increasing prevalence and $20 billion annual healthcare costs. Sensitivity to asthma is determined by multigenic predisposition, aberrant immune response, and environmental factors, as ~50% of asthma cases are caused by allergy to environmental allergens. Thus, asthma may be classified as atopic (allergic or extrinsic) and non-atopic (non-allergic or intrinsic) (1).
Prevention of asthma is based on avoidance of allergens and the use of anti-inflammatory corticosteroids, whereas treatment of acute symptoms is usually accomplished with inhaled short-acting beta-2 agonists. Because both of these approaches have been used for decades with only partial success, new approaches to the prevention and treatment of asthma are needed, which can only come from better understanding of the etiology and pathogenesis of asthma (1).
Recent data indicate that innate immunity (in addition to acquired immunity) is crucial for triggering and maintaining allergic diseases, including asthma (2–5). One of the most frequent allergens involved in asthma is house dust mite (HDM) allergen, as 10% of individuals with asthma are allergic to HDM (6). HDM can be used to induce experimental asthma in a mouse model that closely resembles human asthma (7–9). Recent genome-wide profiling of the lung transcriptome of mice with HDM-induced experimental asthma identified several genes that were involved in asthma (9). One of these genes was Pglyrp1 (9), which codes for antibacterial protein, peptidoglycan recognition protein 1.
Pglyrp1 belongs to a family of evolutionary conserved innate immunity proteins, which in mammals also include Pglyrp2, Pglyrp3, and Pglyrp4. Pglyrp1, Pglyrp3, and Pglyrp4 are directly bactericidal (10–13) and bind to intact peptidoglycan (the main component of bacterial cell wall) and peptidoglycan fragments, and also to other microbial cell wall components, including LPS and lipoteichoic acid (10–18). These Pglyrps kill bacteria by over-activating bacterial stress-response two-component systems and inducing lethal membrane depolarization and oxidative stress in bacteria (13). Pglyrp2 is an N-acetylmuramoyl-L-alanine amidase that hydrolyzes bacterial peptidoglycan and is also bactericidal (19). All mammalian Pglyrps are secreted proteins (15,16). Pglyrp1 is highly expressed in neutrophils’ and eosinophils’ granules and to a lower extent in epithelial and other cells (11,14,20,21). Other Pglyrps are expressed in epithelial cells in the skin and mucous membranes, and Pglyrp2 is also expressed in the liver (10,20,22).
Due to their antibacterial activity Pglyrps maintain beneficial healthy intestinal microbiome, which protects mice from experimentally induced ulcerative colitis (22). This protective effect is non-redundant, i.e., each of the single Pglyrp knockout mice (Pglyrp1−/−, or Pglyrp2−/−, or Pglyrp3−/−, or Pglyrp4−/−) is more sensitive to colitis, likely because of the unique effect of each Pglyrp deficiency on gut microbiome (22). Mammalian Pglyrps also have unique non-redundant effects on intestinal, skin, and joint inflammation. All Pglyrps are anti-inflammatory in the intestine (22,23), Pglyrp2 protects against psoriasis-like skin inflammation (24) and is required for the development of experimental arthritis (25), whereas Pglyrp3 and Pglyrp4 protect against atopic dermatitis (26). By contrast, Pglyrp1 has a pro-inflammatory effect in three mouse models of inflammatory skin diseases (atopic dermatitis, contact dermatitis, and psoriasis) (24,26). Pglyrp1 also has anti-inflammatory effect in experimentally induced arthritis (25). Thus, Pglyrp1 is often pro-inflammatory, whereas other Pglyrps, depending on the type of inflammation and the type of Pglyrp, can have anti- or pro-inflammatory effects.
Many genes are associated with predisposition to allergic and inflammatory diseases, such as asthma, atopic dermatitis, psoriasis, and inflammatory bowel disease, and genetic predisposition for these diseases often overlaps (2,27–29). However, all susceptibility genes for these diseases have not been yet identified. Thus, association of Pglyrp1 with experimental asthma (9) and changed sensitivity to colitis, psoriasis, atopic dermatitis, and contact dermatitis in Pglyrp-deficient mice raise a possibility of a similar genetic overlap of predisposition to these diseases in Pglyrp deficiencies.
Because of the overlapping predisposing factors and similar immunopathologic mechanisms, asthma is often associated with other allergic diseases, as about half of atopic dermatitis patients develop asthma later in life (1,28,30). Based on this association we hypothesized that Pglyrps have similar effect on asthma as they do on atopic dermatitis. In this report we tested the hypothesis that Pglyrp1 has a proinflammatory effect in experimentally induced asthma, similar to its proinflammatory effect in atopic dermatitis and other inflammatory skin diseases (24,26). Consistent with this hypothesis we show here that Pglyrp1−/− mice develop less severe experimental asthma than wild type (WT) mice following intranasal sensitization with HDM. This less severe asthma in Pglyrp1−/− mice is due to decreased Th2 and Th17 cell responses and increased regulatory T (Treg) cell and plasmacytoid dendritic cell (pDC) responses. Thus, our results suggest that blocking Pglyrp1 activity in the lungs may help to control development of allergic asthma.
Materials and Methods
Mice, HDM asthma model, and lung airway resistance
Female 8–10 week-old Pglyrp1−/− and WT mice, both on BALB/c background, were generated and maintained under conventional pathogen-free conditions as described previously (21,22,24–26). To induce asthma mice were sensitized 5 days/week for 3 to 5 weeks with 10 μl per application of 2.5 mg/ml of purified house dust mite allergen (HDM, from Dermatophagoides pteronyssinus, catalog #XPB82D3A25, lot #145793, endotoxin content 37 EU/mg protein, Greer Laboratories, Lenoir, NC) in PBS instilled into the nose under isoflurane anesthesia (7–9). Three days after the last sensitization mice were evaluated for the extent of asthma and inflammatory response in the lungs, blood, mediastinal lymph nodes (MLN), and spleen. Untreated or PBS-treated mice were used as unsensitized controls. All experiments on mice were approved by the Indiana University School of Medicine–Northwest Institutional Animal Care and Use Committee.
Lung airway resistance was measured in response to aerosol of 1, 2, 4, and 8 mg/ml of methacholine. Mice were anesthetized by subcutaneous injection of 0.2 ml of a mixture of 6.7 mg/ml ketamine hydrochloride and 1.3 mg/ml xylazine, an incision was made to access the trachea, and intratracheal cannula was inserted. Mice were placed on a warming pad in the Buxco Fine Pointe RC System (Buxco Research Systems, Wilmington, NC) and intratracheal cannula was connected to the respirometer, which maintained respirations at 140 respirations/min, delivered methacholine aerosol intratracheally, and measured lung airway resistance and compliance in response to methacholine (7–9). The results are resistance data presented as means ± SEM of 6–12 mice/group. The compliance data are not shown in the Results, because they mirror the resistance with similar statistically significant differences.
To collect bronchoalveolar lavage (BAL) fluid lungs were then lavaged 5 times with 0.9 ml PBS. Total numbers of cells in the BAL fluid were then counted, followed by differential counts of inflammatory cells on hematoxylin/eosin (H&E)- or immunofluorescence-stained cytospin slides and by flow cytometry. Blood was collected for serum IgE and cytokine assays, and lungs, MLN, and spleen were collected for flow cytometry, RNA isolation, and IgE and cytokine assays.
Histology and immunofluorescence
For histological analysis mice were anesthetized with isoflurane, exsanguinated, and the lungs were infused with 10% Buffered Protocol Formalin (Fisher Scientific Co., Kalamazoo, MI) through a tracheal cannula at 20 cm hydrostatic pressure. The trachea was then ligated and the lungs were placed in 10% Buffered Protocol Formalin for 48 hrs and processed for paraffin embedding and periodic acid-Schiff (PAS) or H&E staining of longitudinal sections of the left lung lobe, and evaluated microscopically. PAS+ mucus-producing goblet cells were counted on representative four 20x fields per lung along the bronchial tree. The results are reported as means ± SEM of 6 mice per group.
Cytospin slides of BAL cells were immediately fixed with methanol at 4°C for 5 min, dried, blocked with 5% mouse or rabbit serum in PBS, and incubated for 2 hrs at 20°C with 4 μg/ml of rabbit anti-mouse Pglyrp1 or mouse monoclonal anti-mouse Pglyrp1 Abs (both specific to amino acids 123–182 from Santa Cruz) together with one of the following biotinylated antibodies to mouse cell marker antigens: eosinophil cationic protein (ECP, 10 μg/ml rabbit Ab from Bioss), neutrophil Ly6G (1.25 μg/ml of rat mAb RB6-8C5 from eBioscience), macrophage CD68 (10 μg/ml of rat mAb FA-11 from AbD Serotec), lung epithelial cell podoplanin (2.5 μg/ml hamster mAb 8.1.1 from eBioscience), or pDC CD317 (1.25 μg/ml rat mAb 927 from eBioscience). The slides were washed with PBS, incubated for 1 hr at 20°C with either anti-rabbit IgG-FITC mouse mAb or anti-mouse IgG-FITC rabbit Ab (non-cross-reactive with other species, from Sigma) and with streptavidin-APC (eBioscience), washed with PBS, and observed in Olympus Fluoview FV300 confocal microscope. The excitation and emission spectra were set such that there was no “cross-bleeding” of the signal between FITC and APC. Individual slices were examined and the pictures shown represent merged stacks.
RNA and quantitative real-time reverse transcription PCR (qRT-PCR)
RNA was isolated from right lobes of unsensitized or sensitized lungs using the TRIZOL method (InVitrogen), followed by digestion with RNase-free DNase (Qiagen) and purification on RNeasy spin columns using RNeasy Minikit (Qiagen) (22,24–26). Quantitative reverse transcription real-time PCR (qRT-PCR) was used to quantify the amounts of mRNA in the lungs using custom RT2 Profiler PCR Arrays designed by us and manufactured by Qiagen/SA Biosciences (22,24–26). The arrays typically included 40 assay genes (listed in the figures), 5 housekeeping genes, and reverse transcription efficiency and DNA contamination controls. All primer sets were from Qiagen/SA Biosciences, except for the Pglyrp1 primers (24). cDNA was synthesized from 2 μg of RNA using RT2 PCR Array First Strand Kit (Qiagen/SA Biosciences) and the arrays were performed according to the manufacturer instructions using Qiagen/SA Biosciences Master Mix. The experiments were performed on RNA pooled from 4–5 mice/group and repeated 3 times with new groups of mice.
For each gene, ΔCt was calculated using the same threshold (0.2) for all genes and Ct≥35 considered as no expression, followed by normalization to 5 housekeeping genes (Hsp90ab1, Gusb, Hprt1, Gapdh, and Actb) included in each array, followed by calculation of ΔΔCt for each gene from two arrays: ΔΔCt = ΔCt1 − ΔCt2, where ΔCt1 is the HDM-sensitized mice group and ΔCT2 is the unsensitized mice group, using the program provided by Qiagen/SA Biosciences. This calculation gives the fold increase in expression of each gene in the sensitized mice versus unsensitized mice per μg RNA and does not include in the calculation the increased amount of RNA obtained from the lungs of sensitized compared with unsensitized mice (and thus the increases in gene expression would be greater if calculated per lung, rather than per μg RNA). The genomic DNA contamination controls, reverse transcription controls, and positive PCR controls were included in each array and were all passed. Additional control to assure amplification from RNA, but not from possible contaminating DNA, included parallel reaction sets from which reverse transcriptase was omitted, and which showed no amplification. To compare baseline gene expression in untreated mice, ΔCt1 was from untreated Pglyrp1−/− mice and ΔCt2 was from untreated WT mice.
The results are reported as mean fold increases after HDM sensitization (HDM/PBS) or ratios of fold increases in Pglyrp1−/− mice to WT mice, calculated as follows: [(Pglyrp1−/− HDM)/(Pglyrp1−/− PBS)]/[(WT HDM)/(WT PBS)] and presented as heat maps generated using Java TreeView after converting <1 ratios to negative fold difference using the formula: (−1)/ratio. The fold differences (ratios) of >1 or <1 reflect higher or lower expression levels of the genes (respectively) in Pglyrp1−/− mice than in WT mice. The means ± SEM and the significance of differences in gene activation between groups of mice are shown in Supplemental Table I. The qRT-PCR data have been deposited in NCBI GEO at http://www.ncbi.nlm.nih.gov/geo/, accession number GSE43137.
Isolation of cells and flow cytometry
Mouse lungs were minced into ~1 mm pieces in RPMI-1640 with 5% FBS with 3 mg/ml of collagenase D and 0.15 μg/ml DNAse I, and digested for 2 hrs at 37°C (31). Cells were washed 2 times with RPMI-1640 with 5% FBS, red blood cells were removed with a lysis buffer (BioLegend), and the cells were incubated for 14 hrs in the same medium at 37°C in 5% CO2. Cells were then strained through a 40 μm filter and re-suspended at 2.0 × 107 cells/ml in RPMI-1640 with 5% FBS. Single cells from MLN and spleen were obtained by mincing and passing the tissue through a 40 μm filter; red blood cells were removed from the spleen cells with a lysis buffer (BioLegend), and the cells were suspended at 2.0 × 107 cells/ml in RPMI-1640 with 5% FBS, incubated for 14 hrs in the same medium at 37°C in 5% CO2, and then processed for flow cytometry staining. 14-hr incubation improved recovery of lung cells and did not significantly affect expression of the assayed markers on lung, MLN, and spleen lymphocytes and pDCs.
1 × 106 cells were stained with CD4-APC (clone RM4-5, BioLegend) mAb for 20 min at 4°C. CD4-stained cells were then stained for Foxp3-PE (clone FJK-16s, eBioscience) or for cytokines IFN-γ-PE (clone XMG1.2), IL4-PE (clone 11B11) and IL-17-PE (clone TC11-18H10.1) with mAbs from BioLegend, all used at 2 μg/ml according to BioLegend protocols using BioLegend buffers. Prior to staining for cytokines, CD4-APC stained cells were activated with 12-O-tetradecanoylphorbol 13-acetate (25 ng/ml) and ionomycin (250 ng/ml) in the presence of the Golgi inhibitor, monensin (BioLegend), for 4 hrs at 37°C in 5% CO2. Cells were analyzed by flow cytometry using MACSQuant (Miltenyi Biotec) cytometer. Foxp3, IFN-γ, IL-4 and IL-17 positive cells were measured within the CD4+ gate (shown in the figures) and also within the entire lymphocyte gate (with similar results, not shown). For pDC staining, cells were first treated with Fc blocking reagent (Miltenyi Biotec), followed by one of two protocols. The standard protocol used a cocktail of B220-eFluor450 (clone RA3-6B2, 5 μg/ml, eBioscience), CD11c-APC (clone N418, 2.5 μg/ml, eBioscience), and CD317-PE (clone 927, 2.5 μg/ml, eBioscience) mAbs, where CD11c+CD317+ cells were measured within the B220+ gate. The alternative protocol used a cocktail of MHC class II I-A/I-E-eFluor450 mAb (clone M5/114.15.2, 1.25 μg/ml, eBioscience), CD317-PE mAb (clone 927, 2.5 μg/ml, eBioscience), CD8α-PE-Cy7 mAb (clone 53-6.7, 5 μg/ml, eBioscience), and CD8β-APC mAb (clone H35-17.2, 5 μg/ml, eBioscience), where CD8α+β+, CD8α+β−, and CD8α−β− pDC were measured within the I-A/I-E+CD317+ gate (32). The results are presented as mean ± SEM numbers or percentages of the cell types indicated in the figures; representative dot plots are also shown.
IgE and cytokine ELISA
The amounts of IgE, CCL-11, CCL-24, IL-4, IL-5, IL-17 (IL-17A), and thymic stromal lymphopoietin (TSLP) in the BAL fluid, lungs, and serum were measured by ELISA as previously described (26) using detection kits from BioLegend (IgE, IL-4, IL-5, and IL-17) or R&D Systems (CCL-11, CCL-24, and TSLP). Lungs (left lobes) were homogenized with Polytron in 1 ml of PBS (without Ca/Mg) with 1 mM EDTA, 1 mM PMSF, 1:100 dilution of protease inhibitors (Sigma P1860), and 0.5% Triton X-100, followed by sonication and centrifugation (26).
Depletion of pDC
pDC were depleted in vivo by i.v. injections of anti-CD317 mAb (clone 927) (33–35) or control rat IgG2b (both functional grade from eBioscience), 50 μg twice a week for 5 weeks into Pglyrp1−/− mice, given 1 hr before sensitization with HDM. Significant decreases in the numbers of pDC in the lungs, MLN, and spleen were determined by flow cytometry and are shown in the Results.
Statistical analysis
All quantitative results are presented as means ± SEM, with statistical significance of the differences between groups determined by the two-sample one-tailed Student’s t-test; p≤0.05 was considered significant.
Results
Pglyrp1−/− mice have reduced response in HDM-induced asthma model
To determine the role of Pglyrp1 in allergic asthma we selected the HDM-induced asthma model in mice, because this model most closely mimics human allergic asthma (7–9) and because HDM is one of the most frequent allergens associated with human asthma (6). Repeated intranasal sensitization with HDM for 3 to 5 weeks induced severe asthmatic-like response in WT mice and significantly attenuated response in Pglyrp1−/− mice. Compared with WT mice, after 3 and 5 weeks of HDM sensitization, Pglyrp1−/− mice had significantly reduced lung airway resistance, which is a measure of hyper-responsiveness of airway smooth muscle cells to methacholine, a muscarinic receptor agonist, which induces bronchoconstriction, characteristically more severe in asthma (Fig. 1). There was no significant difference in the response to methacholine in unsensitized WT and Pglyrp1−/− mice. These results indicate that deletion of Pglyrp1 diminishes bronchial hyper-responsiveness in sensitized mice and suggest that in WT mice Pglyrp1 plays a role in the development of allergic asthma upon HDM sensitization.
Figure 1. Reduced lung airway resistance in HDM-sensitized Pglyrp1−/− mice.
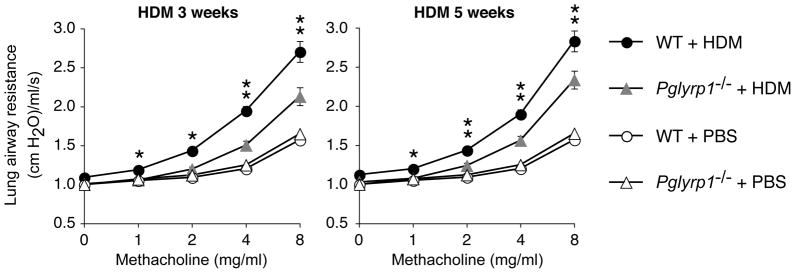
Mice were sensitized with house dust mite (HDM) allergen for 3 or 5 weeks or with PBS (control) and lung airway resistance in response to methacholine was measured. The results are mean ± SEM (SEM were often smaller than the symbols in this and other figures); n = 6 (3 weeks) or 9–12 (5 weeks) mice/group; *, p < 0.05; **, p < 0.005; Pglyrp1−/− versus WT mice.
We then compared the numbers and types of inflammatory cells in BAL fluid and in dissociated lungs in unsensitized and HDM-sensitized WT and Pglyrp1−/− mice, because allergic asthma is characterized by pronounced inflammatory response in the lungs. Inflammatory cells were mostly undetectable in BAL fluid and lungs in unsensitized WT and Pglyrp1−/− mice. HDM sensitization induced high numbers of inflammatory cells in WT mice after 3 weeks of HDM sensitization, and the numbers of these cells further increased after 5 weeks of HDM sensitization (Fig. 2). The inflammatory cells in WT mice in BAL fluid were dominated by eosinophils, with many lymphocytes, neutrophils, and macrophages also present, whereas in dissociated lung cells, the most numerous inflammatory cells were lymphocytes, and the numbers of eosinophils and neutrophils were also high. This prominent eosinophilic response is highly characteristic of allergic asthma. HDM-sensitized Pglyrp1−/− mice, compared with WT mice, had significantly lower total numbers of cells and the numbers of eosinophils in BAL fluid and lungs both at 3 and 5 weeks of sensitization, and the numbers of macrophages at 3 weeks and neutrophils and lymphocytes at 5 weeks of sensitization (Fig. 2). These results demonstrate significantly lower cellular infiltration of HDM-sensitized lungs in Pglyrp1−/− than in WT mice and indicate that in WT mice Pglyrp1 is required for high recruitment of inflammatory cells to the lung during HDM sensitization.
Figure 2. Reduced numbers of inflammatory cells in BAL fluid and lungs in HDM-sensitized Pglyrp1−/− mice.

Mice were sensitized with HDM allergen for 3 or 5 weeks or with PBS (control) and the numbers of the indicated cells in the bronchoalveolar lavage (BAL) fluid or lungs were counted. The results are mean ± SEM, n = 6 (3 weeks) or 9–12 (5 weeks) mice/group; *, p < 0.05; Pglyrp1−/− versus WT mice.
We then compared lung histology of unsensitized and HDM-sensitized WT and Pglyrp1−/− mice to determine the pathologic basis of this lower response to HDM sensitization in Pglyrp1−/− mice. Unsensitized WT and Pglyrp1−/− mice both showed normal histology and no positive cells on PAS staining (Fig. 3A). HDM-sensitized WT mice showed massive inflammatory cell infiltrates mainly around bronchi and also around bronchioles and pulmonary arteries and veins along the bronchial tree (Fig. 3A). The inflammatory infiltrates contained mononuclear cells and numerous eosinophils (Fig. 3B). HDM-sensitized WT mice also showed numerous large PAS+ mucin-producing goblet cells lining the bronchial air spaces, showing pronounced goblet cell hyperplasia and metaplasia, which indicate primarily bronchial involvement and extensive remodeling. There was also smooth muscle hypertrophy and fibrosis, also indicative of remodeling. All of these histopathologic changes are highly characteristic of allergic asthma (1,7–9). HDM-sensitized Pglyrp1−/− mice, compared with WT mice, had substantially smaller cellular infiltrates with fewer eosinophils (consistent with lower inflammatory cell counts in Fig. 2), and smaller and significantly fewer PAS+ mucin-producing goblet cells (Fig. 3A–C). These results demonstrate substantially less severe histopathologic changes characteristic of asthma in HDM-sensitized lungs in Pglyrp1−/− than in WT mice. These results indicate that in WT mice Pglyrp1 is required for the development of severe inflammatory cell infiltrates and airway remodeling with goblet cell hyperplasia and metaplasia following HDM sensitization.
Figure 3. HDM-sensitized Pglyrp1−/− mice have reduced cell infiltration and reduced goblet cell hyperplasia and metaplasia compared with WT mice.

(A) PAS staining of longitudinal lung sections along bronchial tree (Br) of untreated (top) and HDM-sensitized (bottom) WT (left) and Pglyrp1−/− (right) mice with massive inflammatory cell infiltrates (Ci) around bronchi, bronchioles, and pulmonary arteries (Ar) and veins (V) along the bronchial tree, with numerous large PAS+ mucin-producing goblet cells (G) lining the bronchial air spaces in WT, but not in Pglyrp1−/− mice; bar = 200 μm. (B) H&E staining of cellular infiltrates showing higher numbers of eosinophils (E) in WT than in Pglyrp1−/− mice; bar = 50 μm. (C) Numbers of PAS+ mucin-producing goblet cells lining the bronchial air spaces in WT and Pglyrp1−/− mice; means ± SEM of 6 mice/group; *, p < 0.00001; Pglyrp1−/− versus WT mice.
The above results demonstrate development of less severe allergic asthma in Pglyrp1−/− mice compared with WT mice, which indicates that in WT mice Pglyrp1 is required for full manifestation of allergic asthma.
Pglyrp1 is expressed in eosinophils, neutrophils, macrophages, and epithelial cells
We determined the expression of Pglyrp1 in the cells from the BAL fluid from asthmatic lungs using immunofluorescence and confocal microscopy, to identify the cellular sources of Pglyrp1 in HDM-sensitized mice. Pglyrp1 was highly expressed in eosinophils (identified by morphology and expression of eosinophil cationic protein), neutrophils (identified by morphology and expression of Ly-6G), macrophages (identified by morphology and expression of CD68), and lung epithelial cells (identified by morphology and expression of podoplanin) in the lungs of HDM-sensitized WT mice (Fig. 4). Plasmacytoid dendritic cells (pDC, identified by morphology and expression of CD317) expressed low levels of Pglyrp1 (Fig. 4), whereas lymphocytes did not express Pglyrp1 (data not shown), consistent with previous results (14). Eosinophils and neutrophils are known to constitutively express Pglyrp1 (11,14). However, lung epithelial cells in unsensitized mice did not express Pglyrp1 (data not shown). Thus, strong expression of Pglyrp1 in epithelial cells from HDM-sensitized mice indicates that HDM sensitization induces expression of Pglyrp1 in lung epithelial cells. These results demonstrate abundant expression of Pglyrp1 by inflammatory cells in HDM-sensitized lungs.
Figure 4. Expression of Pglyrp1 in inflammatory cells in the lung.

BAL cells from WT mice sensitized with HDM for 5 weeks were stained for Pglyrp1 (green) and the indicated cell marker antigens (red), and visualized in a fluorescent confocal microscope; green, red, overlay of green and red, and overlay of green and transmitted light with representative cells from 5 mice are shown; bar = 5 μm; ECP, eosinophil cationic protein; Pdpn, podoplanin.
Pglyrp1−/− mice have reduced levels of IgE, eotaxins, IL-4, IL-5, and IL-17 in the lungs
We then compared the levels of IgE in the lungs and serum in unsensitized and HDM-sensitized WT and Pglyrp1−/− mice, because atopic allergies, including atopic (allergic) asthma and atopic dermatitis, are characterized by increased local and systemic production of IgE. IgE was undetectable in the BAL fluid, lung homogenates, and serum of unsensitized WT and Pglyrp1−/− mice (Fig. 5). WT mice sensitized with HDM for 3 and 5 weeks had high levels of IgE in BAL fluid, lung homogenates, and serum, whereas similarly sensitized Pglyrp1−/− mice had significantly lower levels of IgE in BAL fluid, lungs, and serum (Fig. 5). These results are consistent with the lower asthmatic responses of Pglyrp1−/− mice and indicate that in WT mice Pglyrp1 is required for high IgE production following HDM sensitization. These results also suggest that Pglyrp1−/− mice have lower Th2 response to HDM, because high IgE production in WT mice is the result of high Th2 polarization of T cell responses.
Figure 5. Reduced BAL fluid, lung, and serum IgE levels and reduced lung CCL-11 (eotaxin-1), CCL-24 (eotaxin-2), IL-4, IL-5, and IL-17 concentrations in HDM-sensitized Pglyrp1−/− mice.

The results are mean ± SEM, n = 6 (3 weeks) or 8–9 (5 weeks) mice/group; *, p < 0.05; **, p < 0.005; Pglyrp1−/− versus WT mice. In unsensitized mice IgE and cytokines were undetectable and are shown at the level of detection.
We also compared the levels of CCL-11 and CCL-24 (eotaxins 1 and 2), IL-4, IL-5, and IL-17 in the lungs and serum in unsensitized and HDM-sensitized WT and Pglyrp1−/− mice, because of high eosinophil and neutrophil infiltrations in the lungs in HDM-sensitized WT, but not Pglyrp1−/− mice (Figs. 2 and 3). CCL-11 and CCL-24 are the main eosinophil-attracting chemokines. IL-4 and IL-5 are the main Th2 cytokines and Th2 polarization promotes IgE responses and type I hypersensitivity. IL-17 promotes Th17 response, which then results in production of neutrophil-attracting chemokines. CCL-11, CCL-24, IL-4, IL-5, and IL-17 were undetectable in the lung homogenates and serum of unsensitized WT and Pglyrp1−/− mice (Fig. 5). WT mice sensitized with HDM for 3 and 5 weeks had high levels of CCL-11, CCL-24, IL-4, IL-5, and IL-17 in the lung homogenates and of IL-4, CCL-11, and CCL-24 also in the serum. However, similarly sensitized Pglyrp1−/− mice had significantly lower levels of CCL-11, CCL-24, IL-4, IL-5, and IL-17 in the lungs at 3 and/or 5 weeks, but similar levels in the serum (except IL-4, which was lower both in the lungs and serum, and IL-5 and IL-17, which were not detectable in the serum) (Fig. 5). These results are consistent with the cell counts and histopathological findings (Figs. 2 and 3) and suggest reduced local (in the lung) eosinophil- and neutrophil-attracting proinflammatory responses in Pglyrp1−/− mice, but no general systemic defect in these responses. These results indicate that in WT mice Pglyrp1 is required for full production of chemokines and cytokines in the lungs.
We also assayed the levels of thymic stromal lymphopoietin (TSLP) in the lungs and serum of HDM-sensitized mice, because TSLP is produced by keratinocytes in allergic individuals, is secreted into the serum, and enhances sensitivity to asthma (36,37). However, TSLP was not detectable in the serum or lungs in WT or Pglyrp1−/− mice sensitized with HDM for 3 or 5 weeks (<5 pg/ml of serum or <100 pg/lung, using ELISA). TSLP mRNA expression in the lungs was also not increased after 3 or 5 weeks of HDM sensitization in WT and Pglyrp1−/− mice (determined by qRT-PCR, data not shown). Thus, TSLP does not play a role the HDM-induced asthma.
Pglyrp1−/− mice have decreased Th2 and Th17 responses in the lungs
We then determined which types of responses and cell subpopulations significantly differed between WT and Pglyrp1−/− mice, to determine the cellular basis for the differences in the inflammatory responses in the lungs of these mice. This was first accomplished by measuring the amounts of mRNA for several marker genes characteristic of the responses generated by various immune and inflammatory cell types in the unsensitized and sensitized lungs. To determine which marker genes are increased or decreased in Pglyrp1−/− mice compared with WT mice (and thus the type of response and the cell type), we calculated how many times higher or lower these genes were induced in Pglyrp1−/− mice than in WT mice (fold induction in Pglyrp1−/− mice/fold induction in WT mice).
Expression of the majority of the genes that we studied was lower in the lungs of unsensitized Pglyrp1−/− mice compared with WT mice (Fig. 6, left panel, and Supplemental Table I). HDM sensitization of WT mice highly induced the expression in the lungs of several marker genes characteristic of Th2 (Il4, Il13, Il33, and Tnfrsf4) and Th17 (Cxcl1, Cxcl2, Cxcl5, Il17a, Il21, IL23a, and Rorgt) responses, and also of alternative macrophage activation (Arg1, Chi3l3, and Fizz1), conventional (myeloid) dendritic cell (cDC) response (Cd273 and Tnfsf4), eosinophil attraction (Ccl11), and goblet cell activation and mucin production (Clca3 and Muc5ac) (Fig. 6, middle panels, and Supplemental Table I). These results are consistent with the high allergic asthmatic response in WT mice. Expression of Pglyrp1 was also increased in the lungs in HDM-sensitized WT mice.
Figure 6. Reduced Th2, Th17, macrophage, cDC, eosinophil, and goblet cell gene expression profiles and increased Th1, Treg, and pDC gene expression profiles in the lungs in HDM-sensitized Pglyrp1−/− mice.

Expression of a panel of cytokines, chemokines, and other marker genes characteristic of the indicated cell types in the lungs of unsensitized mice and mice sensitized with HDM for 3 or 5 weeks measured by qRT-PCR. The results are ratios of the amount of mRNA in unsensitized Pglyrp1−/− to WT mice (No HDM, left panel), or the ratio of the amount of mRNA in HDM-sensitized to unsensitized mice (HDM/PBS, middle panels), or the ratios of fold induction by HDM in Pglyrp1−/− mice to fold induction by HDM in WT mice (which represents the fold difference in the response to HDM in Pglyrp1−/− versus WT mice, right panels). In each panel individual results from 3 experiments with RNA from 4–5 mice/group in each experiment and the averages are shown; the means ± SEM and the significance of differences are shown in Supplemental Table I.
Similarly sensitized Pglyrp1−/− mice had significantly lower expression than WT mice of genes characteristic of Th2, Th17, macrophage (both alternatively and classically activated), cDC, eosinophil, and goblet cell responses at both 3 and 5 weeks of sensitization (Fig. 6, right panel, and Supplemental Table I). These results are consistent with lower allergic asthmatic response in Pglyrp1−/− mice. At 3 weeks of sensitization, Pglyrp1−/− mice had significantly higher expression than WT mice of genes characteristic of Th1 responses (Il2, Ifng, Cxcl9, and Cxcl10), which may reflect Th1 polarization due to decreased Th2 and Th17 responses. These results suggest that Th2, Th17, alternatively activated macrophage, and cDC responses drive high pro-inflammatory allergic asthmatic response in the lungs of HDM-sensitized WT mice and that these responses are attenuated in Pglyrp1−/− mice.
We then used flow cytometry to directly measure Th cell types in the BAL fluid, lungs, draining MLN, and spleen, to further investigate the role and the location of Th cell types in the decreased sensitivity of Pglyrp1−/− mice to HDM-induced asthmatic response. CD4+ T cells were undetectable in the BAL fluid and barely detectable in the lungs of unsensitized mice, but were highly increased in both the BAL fluid and the lungs after 3 and 5 weeks of HDM sensitization in WT mice (Fig. 7A). Similarly sensitized Pglyrp1−/− mice had significantly lower numbers of CD4+ T cells than WT mice in both BAL fluid and the lungs. Th17 (CD4+IL-17+) cells were undetectable in the lungs of unsensitized mice, and they were highly increased at 3 and 5 weeks of sensitization in WT, but not in Pglyrp1−/− mice (Fig. 7A).
Figure 7. Reduced numbers of CD4+, Th2, and Th17 cells, and increased numbers of Treg cells in the lungs in HDM-sensitized Pglyrp1−/− mice.

(A) Numbers of CD4+ and Th17 cells in the BAL fluid and lungs of unsensitized and HDM-sensitized WT and Pglyrp1−/− mice measured by flow cytometry. (B) Percentages of Th1, Th2, Th17, and Treg cells in the BAL fluid, lungs, MLN, and spleen in HDM-sensitized WT and Pglyrp1−/− mice measured by flow cytometry. The results are means ± SEM of 6 mice/group; *, p < 0.05; Pglyrp1−/− versus WT mice. (C) Representative dot plots for Th1, Th2, Th17, and Treg cells in the lungs at 5 weeks of HDM sensitization are shown.
Flow cytometry also revealed that the percentages of Th2 (CD4+IL-4+) and Th17 cells after 3 and 5 weeks of HDM sensitization were significantly higher in the lungs and MLN in WT than in Pglyrp1−/− mice (Fig. 7B, C). In addition, the percentage of Th1 (CD4+IFN-γ+) cells in the lungs was also significantly higher in WT than in Pglyrp1−/− mice at 5 weeks of sensitization. These results confirm and extend the gene expression results (Fig. 6) and show high local Th2 and Th17 responses in the lungs in sensitized WT, but not Pglyrp1−/− mice. Similar percentages of Th1, Th2, and Th17 cells in the spleen in sensitized WT and Pglyrp1−/− mice indicate that Pglyrp1−/− mice do not have systemically defective Th cell responses, and suggest diminished local recruitment and retention of these cells in the lungs and draining lymph nodes of Pglyrp1−/− mice. These results are consistent with lower CCL-11, CCL-24, IL-4, IL-5, and IL-17 levels in the lungs in HDM-sensitized Pglyrp1−/− mice (Fig. 5). Altogether, these results indicate that in WT mice Pglyrp1 is required for high allergic inflammatory cell response to HDM.
Pglyrp1−/− mice have increased Treg and pDC responses in the lungs
Because Pglyrp1−/− mice were able to limit lung inflammation following HDM sensitization more effectively than WT mice, we then tested whether this difference was due to more efficient generation or function of regulatory T cells (Treg) in Pglyrp1−/− mice. Sensitized Pglyrp1−/− mice had higher expression than WT mice of genes characteristic of Treg cell response (Foxp3 and Il10) at both 3 and 5 weeks of HDM sensitization (Fig. 6, right panel, and Supplemental Table I), suggesting higher numbers and/or activation of Treg cells in the sensitized lungs of Pglyrp1−/− than WT mice.
To further verify these findings we then used flow cytometry to directly measure Treg cells (CD4+Foxp3+) in the BAL fluid and lungs in HDM-sensitized WT and Pglyrp1−/− mice. The percentages of Treg cells were significantly higher in the BAL fluid and lungs in Pglyrp1−/− mice compared with WT mice after 3 and 5 weeks of sensitization with HDM (Fig. 7B, C). Two possible reasons for this higher percentage of Treg cells in the lungs of Pglyrp1−/− mice could be either more efficient recruitment of Treg cells to the sensitized lungs or/and higher generation of Treg cells in all lymphoid organs in Pglyrp1−/− mice. Our data support more efficient recruitment of Treg cells to the lungs in Pglyrp1−/− mice for two reasons. First, the lungs of HDM-sensitized Pglyrp1−/− mice had higher expression of Treg-attracting chemokines (Ccl1, Ccl17, and Ccl27a) than WT mice (Fig. 6, right panel, and Supplemental Table I). Second, WT mice had similar percentage of Treg cells in the spleen and higher percentage in the MLN than Pglyrp1−/− mice at 3 weeks of HDM sensitization (Fig, 7B), which indicates sufficient systemic generation of Treg cells in WT mice in lymphoid organs and argues against the possibility of higher systemic capacity for generation of Treg cells in Pglyrp1−/− mice than in WT mice.
We next investigated possible reasons for more efficient recruitment and retention of Treg cells in the lungs of Pglyrp1−/− than WT mice. We first compared the expression of marker genes for various cell types that regulate T cells, including alternatively and classically activated macrophages, cDC, and pDC in WT and Pglyrp1−/− mice. After both 3 and 5 weeks of HDM sensitization Pglyrp1−/− mice had higher expression of pDC signature genes (Aldh1a1, Aldh1a2, and Ido1) than WT mice (Fig. 6, right panel, and Supplemental Table I). These results are consistent with the known capacity of pDC to promote generation of Treg cells (32,34,35,38–40) and suggest pDC-mediated generation of Treg cells as a possible mechanism for the more efficient generation of Treg cells by Pglyrp1−/− mice in the sensitized lungs. By contrast, expression of signature genes for cDC and macrophages, which promote Th2 and Th17 responses, was lower in the lungs of HDM-sensitized Pglyrp1−/− than WT mice (Fig. 6 and Supplemental Table I), which is consistent with the higher Th2 and Th17 responses in WT mice (Figs. 5 and 6).
To verify the above gene expression results, we performed flow cytometry assays, which also revealed significantly higher numbers of pDC (B220+CD11c+CD317+) in the lungs of Pglyrp1−/− than in WT mice after 5 weeks of HDM sensitization (Fig. 8A). We then determined which subpopulations of pDC were increased in HDM-sensitized Pglyrp1−/− mice, because CD8α+β+ and CD8α+β− pDC, but not CD8α−β− pDC, were recently shown to induce Foxp3+ Treg cells and prevent induction of airway hyper-reactivity in ovalbumin-sensitized mice (32). Using flow cytometry, we detected significantly higher percentages of CD8α+β+ and CD8α+β− pDC (I-A/I-E+CD317+) in the lungs and spleens of HDM-sensitized Pglyrp1−/− mice than WT mice (Fig. 8B). These results indicate that in WT mice Pglyrp1 promotes generation or retention of CD8α+β+ and CD8α+β− pDC in asthmatic lungs and suggest that these higher numbers of pDC may be responsible for generation of higher numbers of Treg cells and lower inflammatory responses of Pglyrp1−/− mice.
Figure 8. Increased percentages of pDC and CD8α+β+ and CD8α+β− pDC in the lungs in HDM-sensitized Pglyrp1−/− mice.

WT and Pglyrp1−/− mice were sensitized with HDM for 5 weeks, and the percentages of pDC in their lungs, MLN, and spleens were measured by flow cytometry. (A) B220+CD11c+CD317+ pDC are shown as CD317+ pDC within B220+ gate. (B) CD8α+β+ and CD8α+β− I-A/I-E+CD317+ pDC are shown within I-A/I-E+CD317+ gate. The results are means ± SEM of 6 mice/group, or representative dot plots for pDC in the lungs; *, p < 0.05; Pglyrp1−/− versus WT mice.
Depleting pDC reverses attenuated asthmatic phenotype in Pglyrp1−/− mice
To directly test the role of pDC in the attenuated asthmatic response of Pglyrp1−/− mice, we depleted pDC in vivo with anti-pDC mAb (anti-CD317 clone 927) (33–35) during 5 weeks of sensitization of Pglyrp1−/− mice with HDM. Treatment with anti-pDC mAb significantly reduced the percentages of pDC in the lungs, MLN, and spleen in HDM-sensitized Pglyrp1−/− mice, compared with similarly sensitized mice treated with an isotype control IgG (Fig. 9A). Anti-pDC mAb treatment during HDM sensitization also significantly increased the total numbers of inflammatory cells and the numbers of eosinophils, neutrophils, lymphocytes, and macrophages in the BAL fluid and lungs of Pglyrp1−/− mice (Fig. 9B). Anti-pDC mAb treatment during HDM sensitization also significantly increased the lung airway resistance (Fig. 9C), and resulted in higher numbers of CD4+ cells in the BAL fluid and lungs, higher numbers of Th1, Th2, and Th17 cells in the lungs, and reduced percentages of Treg cells in the BAL fluid and lungs, compared with similarly sensitized Pglyrp1−/− mice treated with isotype control IgG (Fig. 9D). Thus, reducing the numbers of pDC reverses the low responsive phenotype of Pglyrp1−/− mice in the HDM asthma model to resemble the more pro-inflammatory WT phenotype. These results indicate that pDC play a significant role in the generation of Treg cells and low responsiveness of Pglyrp1−/− mice to HDM sensitization.
Figure 9. Depletion of pDC in vivo reverses attenuated asthma phenotype in HDM sensitized Pglyrp1−/− mice.

Pglyrp1−/− mice were treated with anti-pDC (anti-CD317) mAb or isotype control IgG during sensitization with HDM for 5 weeks. Anti-pDC mAb reduced the numbers of pDC in vivo (A). Depletion of pDC increased the numbers of eosinophils, neutrophils, lymphocytes, and macrophages in BAL cells and lungs (B), increased lung airway resistance (C), and increased numbers of CD4+ cells in BAL cells and CD4+, Th1, Th2, and Th17 cells in the lungs, and decreased the percentage of Treg cells in BAL cells and the lungs (D), measured by flow cytometry, with gating as shown in Figs. 7 and 8A; means ± SEM of 6 mice/group; *, p < 0.05; **, p < 0.005; anti-pDC versus IgG.
Altogether, our results indicate that Th2, Th17, alternatively activated macrophage, and cDC responses drive high proinflammatory allergic asthmatic response in the lungs of HDM-sensitized WT mice, and that this response is attenuated in Pglyrp1−/− mice due to their increased pDC responses, which generate increased numbers of Treg cells in the lungs.
Discussion
Allergic asthma is a complex disease, which in addition to the well-known Th2 bias and over-production of IgE, also involves interactions of many other cell types. Thus, the pathogenesis of allergic asthma involves over-activation of Th2 cells, which results in eosinophilic inflammation, and also over-activation of both Th17 cells and Th1 cells, which enhance neutrophilic infiltration (a hallmark of severe asthma) by inducing increased production of neutrophil-attracting chemokines in the lungs (2,41). Th17 cells also indirectly promote eosinophilic infiltration by enhancing Th2 responses and differentiation of B cells (42,43). Moreover, although asthma is classically considered an inflammatory allergic disease, cross-talk between innate and adaptive immune systems is crucial for the initiation and propagation of asthma (2–4), as many allergens, such as HDM or cockroach allergen are also strong activators of innate immunity toll-like receptors (TLR) (44,45). Thus, the presence or absence of TLR activation modulates sensitization in allergic asthma (40).
Regulation of immune and inflammatory responses in asthma is equally complex. cDC promote development and activation of Th2, Th17, and Th1 cells and in general have pro-inflammatory effects, which promote inflammation in asthma (2,46). By contrast, pDC have an opposite anti-inflammatory effect in asthma, because depletion of pDC enhances and adoptive transfer of pDC attenuates allergic asthma (32,34,35,40). pDC oppose the effects of cDC and inhibit generation of T helper and effector cells in lung inflammation by promoting generation and maintenance of Treg cells (32,34,35,38). Treg cells are anti-inflammatory by inhibiting the responses of Th2, Th17, and Th1 cells. Both thymic (natural) Treg cells (generated in the thymus) and peripheral (induced) Treg cells (generated in the peripheral tissues) are recruited to, maintained, and activated at the mucosal sites and prevent and control inflammation (2,41). Thus, genetic predisposition to asthma may be associated with genes that affect any of the above aspects of adaptive and innate immunity.
Here, we reveal a novel innate immunity mechanism that is involved in the development of allergic asthma. We demonstrate that HDM-sensitized Pglyrp1−/− mice develop less severe asthma than WT mice. The mechanism underlying this decreased sensitivity of Pglyrp1−/− mice to allergic asthma is increased generation and activation of CD8α+β+ and CD8α+β− pDC and increased recruitment and activity of Treg cells in the lungs. These cells then decrease production and activation of Th2 and Th17 cells in the lungs and result in attenuated asthmatic response. Thus, our results reveal a previously unknown mechanism of regulation of pDC and Treg cells by Pglyrp1, a member of a family of innate immunity proteins. In WT mice, Pglyrp1 affects the functions of both innate and adaptive immune cells by decreasing the responses of pDC cells in the lungs, which results in reduced generation of Treg cells and increased Th2 and Th17 responses and a more severe asthmatic phenotype. Our results extend recent findings that pDC (especially CD8α+β+ and CD8α+β− pDC) enhance generation, expansion and maintenance of Treg cells in the asthmatic lungs (32,34,35). Pglyrp1 regulates local proinflammatory responses not only in the lungs (reported here), but also in the intestine (22), skin (24,26), and joints (25), with little effect on the systemic inflammatory responses, because the most pronounced changes in the Treg and Th cells in Pglyrp1−/− mice occur in the local inflammatory sites.
How do pDC in Pglyrp1−/− mice induce higher numbers of Treg cells in asthma? Our results suggest three possible mechanisms. First, pDC express a tryptophan-degrading enzyme, indoleamine 2,3-dioxygenase (IDO), which is known to induce Foxp3 expression in naïve CD4+ T cells, to generate Treg cells, and also to activate the existing Treg cells, both in vitro and in vivo (38). Consistent with this model, our HDM-sensitized Pglyrp1−/− mice express higher level of IDO (Ido1, Fig. 6) in their lungs than WT mice, which correlates with the increased numbers of Treg cells in Pglyrp1−/− mice and their attenuated asthma phenotype. Second, generation and maintenance of Treg cells, especially by CD8α+β+ and CD8α+β− pDC, is also promoted by all-trans retinoic acid, which is produced from retinaldehyde by aldehyde dehydrogenases Aldh1a1, Aldh1a2, and Aldh1a3 (32,47). Consistent with this model, both the expression of Aldh1a1 and Aldh1a2 and the percentages of CD8α+β+ and CD8α+β− pDC are increased in the lungs of HDM-sensitized Pglyrp1−/− mice compared with WT mice (Figs. 6 and 8B). Third, in addition to inducing Treg cells, pDC can also suppress inflammation by down-regulating cDC (35), which promote airway inflammation. Indeed, the expression of Cd273, a marker gene for cDC, and the expression of Tnfsf4 (also known as OX40L, which is produced by cDC and amplifies Th2 cell differentiation) are both reduced in HDM-sensitized Pglyrp1−/− mice compared with WT mice (Fig. 6). Consistent with this mechanism, the expression of Tnfrsf4 (Tnfsf4 receptor, also known as OX40, which is produced by Th2 cells) is also reduced in HDM-sensitized Pglyrp1−/− mice compared with WT mice (Fig. 6).
There are also other mechanisms of generating and maintaining Treg cells, which are, however, less likely targets for up-regulation in Pglyrp1−/− mice. Generation of IL-10-secreting Treg cells by pDC can be mediated through the inducible co-stimulator ligand (ICOS-L) (39). This mechanism is less likely in our model, because the expression of Icosl was not up-regulated in the lungs of HDM-sensitized Pglyrp1−/− mice compared with WT mice (data not shown). Another molecule that is required for the induction of Foxp3 expression and that enhances generation and maintenance of Treg cells is programmed death ligand 1 (PD-L1, also known as CD274) (48–50). However, generation of increased numbers of Treg cells in HDM-sensitized Pglyrp1−/− mice through the increased expression of PD-L1 is unlikely, because the expression of PD-L1 (Cd274) is not increased in HDM-sensitized Pglyrp1−/− mice compared with WT mice (Fig. 6).
Classical activation of macrophages and cDC, which both promote inflammation, is decreased in HDM-sensitized Pglyrp1−/− mice compared with WT mice (reduced Nos2, Cd273, and Tnfsf4 expression in Fig. 6), consistent with the attenuated asthma phenotype of Pglyrp1−/− mice. Expression of marker genes for the alternative activation of macrophages (Arg1, Chi3l1, Chi3l3, Fizz1, and Mrc1), which may either promote inflammation or promote healing (51–54) is also mostly decreased in HDM-sensitized Pglyrp1−/− mice compared with WT mice (Fig. 6), consistent with the total decrease in the numbers of macrophages in the lungs of HDM-sensitized Pglyrp1−/− mice compared with WT mice (Fig. 2).
How are pDC recruited into the lungs in HDM-sensitized Pglyrp1−/− mice? Local allergen sensitization recruits pDC into the lungs from MLN as a feedback mechanism to inhibit inflammation (35). The expression of Cxcl9 and Cxcl10 is increased in Pglyrp1−/− mice (Fig. 6) and pDC express CXCR3 (55), which is the receptor for these chemokines. CXCL9 and CXCL10 are considered a part of Th1 response, and the expression of Th1 marker genes (Il2, Ifng, Cxcl9, and Cxcl10) is significantly increased early (at 3 weeks) in HDM-sensitized Pglyrp1−/− mice compared with WT mice (Fig. 6). Thus, CXCL9 and CXCL10 are likely candidates for preferential recruitment of pDC to the sensitized lungs in Pglyrp1−/− mice. As significantly more pDC accumulate in the lungs, more Treg cells are generated, which results in greatly diminished numbers of Th2, Th17, and Th1 cells in Pglyrp1−/− mice than in WT mice (Fig. 7, and also lower expression of marker genes for all Th cells at 5 weeks, Fig. 6).
Pglyrp1−/− mice are not only more resistant than WT mice to asthma, but also to experimentally induced atopic dermatitis, contact dermatitis, and psoriasis-like skin inflammation (24,26). These results are consistent with our current asthma data showing increased numbers of Treg cells at the sites of inflammation in Pglyrp1−/− mice and decreased numbers of Th2, Th17, and Th1 cells. All these Th cells are subject of negative regulation by Treg cells, because ablation of Treg cells results in increased numbers and activity of all Th cell types (56). Lower sensitivity to asthma in HDM-sensitized Pglyrp1−/− mice is determined by lower local response in the lungs and not by lower systemic response in lymphoid organs or in other tissues, such as skin. This conclusion is based on the most pronounced changes in chemokines, cytokines, and other inflammatory mediators and cells in the lungs, but not systemically in Pglyrp1−/− mice, compared with WT mice.
Thus, our results indicate that in WT mice Pglyrp1 promotes cDC, macrophage, Th2, and Th17 pro-inflammatory responses. Such responses to allergens are not desirable, but the same responses to infectious agents could be beneficial. Note that Pglyrp1 is delivered to the sites of inflammation by eosinophils, neutrophils, and macrophages (Fig. 4) (11,14,20,21), which are usually recruited early to fight infections or parasitic infestations, and thus enhancement of inflammatory responses in early stages of infection is beneficial for host defense. Pglyrp1 expression is also induced in lung epithelial cells by allergen sensitization (Fig. 4). However, enhancement of responses to allergens is undesirable and can be targeted for treatment of allergic diseases.
What is the mechanism responsible for the increased numbers and activity of pDC in Pglyrp1−/− mice? Pglyrp1 is a secreted bactericidal protein (10–13) involved not only in defense against infections (10,21), but also in maintaining healthy beneficial microbiome (22). Pglyrp1−/− mice have significant changes in the intestinal microflora compared with WT mice, and these changes increase the sensitivity of these mice to colitis (22). It is therefore possible that the effect of Pglyrp1 on the sensitivity to lung or skin inflammatory diseases is indirect through Pglyrp1-dependent effects on the intestinal or local microflora. This possibility is consistent with the prominent role of microflora in the generation of Treg cells and pDC (56–58). Especially relevant to our results are recent findings showing that germ-free mice have decreased numbers of pDC and enhanced sensitivity to asthma, which can be reversed by colonization with certain bacteria (59), and that some commensal bacteria induce Treg cells through their effect on pDC (60). Moreover, metabolites from commensal bacteria, such as fatty acids (61) or ATP (62), greatly affect not only the metabolism of the host, but also its immune system. For example, generation of Treg cells is promoted by fatty acids, whereas generation of Th cells is promoted by glucose (63). Thus, changes in commensal bacteria (such as those in Pglyrp1−/− mice), could influence the development of pDC or Treg cells. Another possibility is that induction of Pglyrp1 expression in lung epithelial cells or delivery of Pglyrp1 by eosinophils, neutrophils, and macrophages (Fig. 4) (11, 14,20,21) have direct effects on the secretion of chemokines and cytokines by epithelial cells in the lungs, creating a proinflammatory microenvironment. These possibilities will be investigated in future studies.
Peptidoglycan is the ligand for several pattern recognition molecules and a potent stimulant of innate immunity (15,17). Pglyrp1 can inhibit some of these immunostimulatory activities by binding to peptidoglycan and preventing its recognition by innate immunity receptors (14,18). Pglyrp2 can also hydrolyze peptidoglycan and eliminate some of its immunostimulatory activities (15,19). However, these effects of Pglyrp1 and other Pglyrps on peptidoglycan are probably unrelated to their ability to modulate inflammation, because Pglyrps modulate inflammatory responses induced not only by peptidoglycan (14,18,25), but also by unrelated allergens and antigens (22–26). Therefore, as discussed above, modulation of inflammation by Pglyrps is most likely indirect, through the effects of Pglyrps on the microbiome and local cytokine-secreting cells.
Our results on the HDM mouse model are relevant to human asthma for several reasons. First, HDM is a frequent allergen in human asthma (6). Second, asthma is associated with atopic dermatitis in 50% of patients (30), and our previous (26) and current results reveal a role for Pglyrp1 in both atopic dermatitis and asthma. These results suggest Pglyrp1 as a possible candidate for susceptibility to both lung and skin inflammatory diseases (24,26). Third, increased numbers of cDC (which are pro-inflammatory and oppose anti-inflammatory effects of pDC) are present in BAL fluid in patients with asthma (64), whereas these patients have low numbers of pDC (65) or impaired functions of pDC (66,67). And fourth, deficiency of circulating pDC during infancy is a risk factor for more frequent and more severe respiratory tract infections, wheezing, and diagnosis of asthma, whereas infants with higher numbers of pDC are protected against these outcomes (68). Thus, our results are consistent with these clinical observations that increasing numbers or activity of pDC may be beneficial for prevention and treatment of asthma and other allergic diseases. Our results also open a new possibility that blocking the pro-inflammatory activity of Pglyrp1 or decreasing its expression in the lungs may be a new approach to prevention and treatment of asthma.
Supplementary Material
Acknowledgments
This work was supported by USPHS Grants AI028797 and AI073290 from NIH.
We are grateful to Stewart Levine (NHLBI, NIH) for sharing with us his unpublished data on Pglyrp1 and asthma, to Chang H. Kim (Purdue University) for advice on T cell subpopulations, and Julie Cook for maintaining and breeding our mice.
Abbreviations
- BAL
bronchoalveolar lavage
- cDC
conventional (myeloid) dendritic cells
- ECP
eosinophil cationic protein
- HDM
house dust mite (allergen)
- MLN
mediastinal lymph nodes
- PAS
periodic acid-Schiff
- pDC
plasmacytoid dendritic cells
- Pglyrp
peptidoglycan recognition protein
- Treg
regulatory T (cells)
- TSLP
thymic stromal lymphopoietin
- WT
wild type
Footnotes
The qRT-PCR data have been deposited in NCBI GEO at http://www.ncbi.nlm.nih.gov/geo/, accession number GSE43137.
References
- 1.Fanta CH. Asthma. N Engl J Med. 2009;360:1002–1014. doi: 10.1056/NEJMra0804579. [DOI] [PubMed] [Google Scholar]
- 2.Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18:673–683. doi: 10.1038/nm.2731. [DOI] [PubMed] [Google Scholar]
- 3.Minnicozzi M, Sawyer RT, Fenton MJ. Innate immunity in allergic disease. Immunol Rev. 2011;242:106–127. doi: 10.1111/j.1600-065X.2011.01025.x. [DOI] [PubMed] [Google Scholar]
- 4.Sly PD, Holt PG. Role of innate immunity in the development of allergy and asthma. Curr Opin Allergy Clin Immunol. 2011;11:127–131. doi: 10.1097/ACI.0b013e32834487c6. [DOI] [PubMed] [Google Scholar]
- 5.Hammad H, Lambrecht BN. Dendritic cells and airway epithelial cells at the interface between innate and adaptive immune responses. Allergy. 66:579–587. doi: 10.1111/j.1398-9995.2010.02528.x. [DOI] [PubMed] [Google Scholar]
- 6.Bessot JC, Pauli G. Mite allergens: an overview. Eur Ann Allergy Clin Immunol. 2011;43:141–56. [PubMed] [Google Scholar]
- 7.Johnson JR, Wiley RE, Fattouh R, Swirski FK, Gajewska BU, Coyle AJ, Gutierrez-Ramos JC, Ellis R, Inman MD, Jordana M. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am J Respir Crit Care Med. 2004;169:378–385. doi: 10.1164/rccm.200308-1094OC. [DOI] [PubMed] [Google Scholar]
- 8.Yao X, Dai C, Fredriksson K, Dagur PK, McCoy JP, Qu X, Yu ZX, Keeran KJ, Zywicke GJ, Amar MJ, Remaley AT, Levine SJ. 5A, an apolipoprotein A-I mimetic peptide, attenuates the induction of house dust mite-induced asthma. J Immunol. 2011;186:576–83. doi: 10.4049/jimmunol.1001534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao X, Fredriksson K, Yu ZX, Xu X, Raghavachari N, Keeran KJ, Zywicke GJ, Kwak M, Amar MJ, Remaley AT, Levine SJ. Apolipoprotein E negatively regulates house dust mite-induced asthma via a low-density lipoprotein receptor-mediated pathway. Am J Respir Crit Care Med. 2010;182:1228–1238. doi: 10.1164/rccm.201002-0308OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu X, Wang M, Qi J, Wang H, Li X, Gupta D, Dziarski R. Peptidoglycan recognition proteins are a new class of human bactericidal proteins. J Biol Chem. 2006;281:5895–5907. doi: 10.1074/jbc.M511631200. [DOI] [PubMed] [Google Scholar]
- 11.Tydell CC, Yuan J, Tran P, Selsted ME. Bovine peptidoglycan recognition protein-S: antimicrobial activity, localization, secretion, and binding properties. J Immunol. 2006;176:1154–1162. doi: 10.4049/jimmunol.176.2.1154. [DOI] [PubMed] [Google Scholar]
- 12.Wang M, Liu LH, Wang S, Li X, Lu X, Gupta D, Dziarski R. Human peptidoglycan recognition proteins require zinc to kill both Gram-positive and Gram-negative bacteria and are synergistic with antibacterial peptides. J Immunol. 2007;178:3116–3125. doi: 10.4049/jimmunol.178.5.3116. [DOI] [PubMed] [Google Scholar]
- 13.Kashyap DR, Wang M, Liu LH, Boons GJ, Gupta D, Dziarski R. Peptidoglycan recognition proteins kill bacteria by activating protein-sensing two-component systems. Nature Med. 2011;17:676–683. doi: 10.1038/nm.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu C, Gelius E, Liu G, Steiner H, Dziarski R. Mammalian peptidoglycan recognition protein binds peptidoglycan with high affinity, is expressed in neutrophils, and inhibits bacterial growth. J Biol Chem. 2000;275:24490–24499. doi: 10.1074/jbc.M001239200. [DOI] [PubMed] [Google Scholar]
- 15.Royet J, Dziarski R. Peptidoglycan Recognition Proteins: pleiotropic sensors and effectors of antimicrobial defenses. Nature Rev Microbiol. 2007;5:264–277. doi: 10.1038/nrmicro1620. [DOI] [PubMed] [Google Scholar]
- 16.Royet J, Gupta D, Dziarski R. Peptidoglycan recognition proteins: modulators of the microbiome and inflammation. Nature Rev Immunol. 2011;11:837–851. doi: 10.1038/nri3089. [DOI] [PubMed] [Google Scholar]
- 17.Sorbara MT, Philpott DJ. Peptidoglycan: a critical activator of the mammalian immune system during infection and homeostasis. Immunol Rev. 2011;243:40–60. doi: 10.1111/j.1600-065X.2011.01047.x. [DOI] [PubMed] [Google Scholar]
- 18.Sharma P, Dube D, Singh A, Mishra B, Singh N, Sinha M, Dey S, Kaur P, Mitra DK, Sharma S, Singh TP. Structural basis of recognition of pathogen-associated molecular patterns and inhibition of proinflammatory cytokines by camel peptidoglycan recognition protein. J Biol Chem. 2011;286:16208–16217. doi: 10.1074/jbc.M111.228163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang ZM, Li X, Cocklin RR, Wang M, Wang M, Fukase K, Inamura S, Kusumoto S, Gupta D, Dziarski R. Human peptidoglycan recognition protein-L is an N-acetylmuramoyl-L-alanine amidase. J Biol Chem. 2003;278:49044–49052. doi: 10.1074/jbc.M307758200. [DOI] [PubMed] [Google Scholar]
- 20.Liu C, Xu Z, Gupta D, Dziarski R. Peptidoglycan recognition proteins: a novel family of four human innate immunity pattern recognition molecules. J Biol Chem. 2001;276:34686–34694. doi: 10.1074/jbc.M105566200. [DOI] [PubMed] [Google Scholar]
- 21.Dziarski R, Platt KA, Gelius E, Steiner H, Gupta D. Defect in neutrophil killing and increased susceptibility to infection with non-pathogenic Gram-positive bacteria in peptidoglycan recognition protein-S (PGRP-S)-deficient mice. Blood. 2003;102:689–697. doi: 10.1182/blood-2002-12-3853. [DOI] [PubMed] [Google Scholar]
- 22.Saha S, Jing X, Park SY, Wang S, Li X, Gupta D, Dziarski R. Peptidoglycan recognition proteins protect mice from experimental colitis by promoting normal gut flora and preventing induction of interferon-γ. Cell Host Microbe. 2010;8:147–162. doi: 10.1016/j.chom.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zenhom M, Hyder A, de Vrese M, Heller KJ, Roeder T, Schrezenmeir J. Peptidoglycan recognition protein 3 (PglyRP3) has an anti-inflammatory role in intestinal epithelial cells. Immunobiology. 2012;217:412–419. doi: 10.1016/j.imbio.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Park SY, Gupta D, Hurwich R, Kim CH, Dziarski R. Peptidoglycan recognition protein Pglyrp2 protects mice from psoriasis-like skin inflammation by promoting regulatory T cells and limiting Th17 responses. J Immunol. 2011;187:5813–5823. doi: 10.4049/jimmunol.1101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saha S, Qi J, Wang S, Wang M, Li X, Kim Y-G, Núñez G, Gupta D, Dziarski R. PGLYRP-2 and Nod2 are both required for peptidoglycan-induced arthritis and local inflammation. Cell Host Microbe. 2009;5:137–150. doi: 10.1016/j.chom.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park SY, Gupta D, Kim CH, Dziarski R. Differential effects of peptidoglycan recognition proteins on experimental atopic and contact dermatitis mediated by Treg and Th17 cells. PLoS One. 2011;6:e24961. doi: 10.1371/journal.pone.0024961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 28.Martinez FD. Genes, environments, development and asthma: a reappraisal. Eur Respir J. 2007;29:179–184. doi: 10.1183/09031936.00087906. [DOI] [PubMed] [Google Scholar]
- 29.Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut. 2011;60:1739–1753. doi: 10.1136/gut.2009.199679. [DOI] [PubMed] [Google Scholar]
- 30.Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233–246. doi: 10.1111/j.1600-065X.2011.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolfi DV, Duttagupta PA, Boesteanu AC, Mueller YM, Oliai CH, Borowski AB, Katsikis PD. Dendritic cells and CD28 costimulation are required to sustain virus-specific CD8+ T cell responses during the effector phase in vivo. J Immunol. 2011;186:4599–4608. doi: 10.4049/jimmunol.1001972. [DOI] [PubMed] [Google Scholar]
- 32.Lombardi V, Speak AO, Kerzerho J, Szely N, Akbari O. CD8α+β+ and CD8α+β− plasmacytoid dendritic cells induce Foxp3+ regulatory T cells and prevent the induction of airway hyper-reactivity. Mucosal Immunol. 2012;5:432–443. doi: 10.1038/mi.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asselin-Paturel C, Brizard G, Pin JJ, Brière F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. 2003;171:6466–6477. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- 34.de Heer HJ, Hammad H, Soullié T, Hijdra D, Vos N, Willart MA, Hoogsteden HC, Lambrecht BN. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med. 2004;200:89–98. doi: 10.1084/jem.20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kool M, van Nimwegen M, Willart MA, Muskens F, Boon L, Smit JJ, Coyle A, Clausen BE, Hoogsteden HC, Lambrecht BN, Hammad H. An anti-inflammatory role for plasmacytoid dendritic cells in allergic airway inflammation. J Immunol. 2009;183:1074–1082. doi: 10.4049/jimmunol.0900471. [DOI] [PubMed] [Google Scholar]
- 36.Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, de Waal-Malefyt R, Bazan F, Kastelein RA, Liu YJ. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 37.Demehri S, Morimoto M, Holtzman MJ, Kopan R. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. 2009;7:e1000067. doi: 10.1371/journal.pbio.1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito T, Yang M, Wang YH, Lande R, Gregorio J, Perng OA, Qin XF, Liu YJ, Gilliet M. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med. 2007;204:105–115. doi: 10.1084/jem.20061660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crother TR, Schröder NW, Karlin J, Chen S, Shimada K, Slepenkin A, Alsabeh R, Peterson E, Arditi M. Chlamydia pneumoniae infection induced allergic airway sensitization is controlled by regulatory T-cells and plasmacytoid dendritic cells. PLoS One. 2011;6:e20784. doi: 10.1371/journal.pone.0020784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, Saito Y, Hatano M, Tokuhisa T, Iwakura Y, Puccetti P, Iwamoto I, Nakajima H. IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008;178:1023–1032. doi: 10.1164/rccm.200801-086OC. [DOI] [PubMed] [Google Scholar]
- 43.Mitsdoerffer M, Lee Y, Jäger A, Kim HJ, Korn T, Kolls JK, Cantor H, Bettelli E, Kuchroo VK. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci USA. 2010;107:14292–14297. doi: 10.1073/pnas.1009234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, Thorne PS, Wills-Karp M, Gioannini TL, Weiss JP, Karp CL. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Page K, Ledford JR, Zhou P, Wills-Karp M. A TLR2 agonist in German cockroach frass activates MMP-9 release and is protective against allergic inflammation in mice. J Immunol. 2009;183:3400–3408. doi: 10.4049/jimmunol.0900838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lambrecht BN, Hammad H. Lung dendritic cells in respiratory viral infection and asthma: from protection to immunopathology. Annu Rev Immunol. 2012;30:243–270. doi: 10.1146/annurev-immunol-020711-075021. [DOI] [PubMed] [Google Scholar]
- 47.Goswami S, Angkasekwinai P, Shan M, Greenlee KJ, Barranco WT, Polikepahad S, Seryshev A, Song LZ, Redding D, Singh B, Sur S, Woodruff P, Dong C, Corry DB, Kheradmand F. Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat Immunol. 2009;10:496–503. doi: 10.1038/ni.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci USA. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Francisco LM, V, Salinas H, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–2309. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGee HS, Yagita H, Shao Z, Agrawal DK. Programmed Death-1 antibody blocks therapeutic effects of T-regulatory cells in cockroach antigen-induced allergic asthma. Am J Respir Cell Mol Biol. 2010;43:432–442. doi: 10.1165/rcmb.2009-0258OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, Van Rooijen N, Urban JF, Jr, Wynn TA, Gause WC. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. 2012;18:260–266. doi: 10.1038/nm.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, ElKasmi KC, Smith AM, Thompson RW, Cheever AW, Murray PJ, Wynn TA. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5:e1000371. doi: 10.1371/journal.ppat.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nair MG, Du Y, Perrigoue JG, Zaph C, Taylor JJ, Goldschmidt M, Swain GP, Yancopoulos GD, Valenzuela DM, Murphy A, Karow M, Stevens S, Pearce EJ, Artis D. Alternatively activated macrophage-derived RELM-α is a negative regulator of type 2 inflammation in the lung. J Exp Med. 2009;206:937–952. doi: 10.1084/jem.20082048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cook PC, Jones LH, Jenkins SJ, Wynn TA, Allen JE, MacDonald AS. Alternatively activated dendritic cells regulate CD4+ T-cell polarization in vitro and in vivo. Proc Natl Acad Sci USA. 2012;109:9977–9982. doi: 10.1073/pnas.1121231109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Furuhashi K, Suda T, Hasegawa H, Suzuki Y, Hashimoto D, Enomoto N, Fujisawa T, Nakamura Y, Inui N, Shibata K, Nakamura H, Chida K. Mouse lung CD103+ and CD11bhigh dendritic cells preferentially induce distinct CD4+ T-cell responses. Am J Respir Cell Mol Biol. 2012;46:165–172. doi: 10.1165/rcmb.2011-0070OC. [DOI] [PubMed] [Google Scholar]
- 56.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chinen T, Rudensky AY. The effects of commensal microbiota on immune cell subsets and inflammatory responses. Immunol Rev. 2012;245:45–55. doi: 10.1111/j.1600-065X.2011.01083.x. [DOI] [PubMed] [Google Scholar]
- 59.Herbst T, Sichelstiel A, Schär C, Yadava K, Bürki K, Cahenzli J, McCoy K, Marsland BJ, Harris NL. Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Respir Crit Care Med. 2011;184:198–205. doi: 10.1164/rccm.201010-1574OC. [DOI] [PubMed] [Google Scholar]
- 60.Konieczna P, Groeger D, Ziegler M, Frei R, Ferstl R, Shanahan F, Quigley EM, Kiely B, Akdis CA, O’Mahony L. Bifidobacterium infantis 35624 administration induces Foxp3 T regulatory cells in human peripheral blood: potential role for myeloid and plasmacytoid dendritic cells. Gut. 2012;61:354–366. doi: 10.1136/gutjnl-2011-300936. [DOI] [PubMed] [Google Scholar]
- 61.Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K. ATP drives lamina propria TH17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 63.Michalek RD, V, Gerriets A, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kayserova J, Zentsova-Jaresova I, Budinsky V, Rozkova D, Kopecka J, Vernerova E, Pohunek P, Skalicka V, Spisek R, Sediva A. Selective increase in blood dendritic cell antigen-3-positive dendritic cells in bronchoalveolar lavage fluid in allergic patients. Scand J Immunol. 2012;75:305–313. doi: 10.1111/j.1365-3083.2011.02649.x. [DOI] [PubMed] [Google Scholar]
- 65.Silver E, Yin-DeClue H, Schechtman KB, Grayson MH, Bacharier LB, Castro M. Lower levels of plasmacytoid dendritic cells in peripheral blood are associated with a diagnosis of asthma 6 yr after severe respiratory syncytial virus bronchiolitis. Pediatr Allergy Immunol. 2009;20:471–476. doi: 10.1111/j.1399-3038.2008.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gill MA, Bajwa G, George TA, Dong CC, Dougherty II, Jiang N, Gan VN, Gruchalla RS. Counterregulation between the FcεRI pathway and antiviral responses in human plasmacytoid dendritic cells. J Immunol. 2010;184:5999–6006. doi: 10.4049/jimmunol.0901194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tversky JR, Le TV, Bieneman AP, Chichester KL, Hamilton RG, Schroeder JT. Human blood dendritic cells from allergic subjects have impaired capacity to produce interferon-alpha via Toll-like receptor 9. Clin Exp Allergy. 2008;38:781–788. doi: 10.1111/j.1365-2222.2008.02954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Upham JW, Zhang G, Rate A, Yerkovich ST, Kusel M, Sly PD, Holt PG. Plasmacytoid dendritic cells during infancy are inversely associated with childhood respiratory tract infections and wheezing. J Allergy Clin Immunol. 2009;124:707–173. doi: 10.1016/j.jaci.2009.07.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.