

Abstract
Infection and inflammation can disturb immune tolerance at the maternal-fetal interface, resulting in adverse pregnancy outcomes. However, the underlying mechanisms for detrimental immune responses remain ill-defined. Here, we provide evidence for immune programming of fetal loss in response to poly(I:C), a viral mimic and an inducer of inflammatory milieu. IL-10 and uterine NK (uNK) cells expressing the activating receptor NKG2D play a critical role in poly(I:C)-induced fetal demise. In wild type (WT) mice, poly(I:C) treatment induced expansion of NKG2D+ uNK cells and expression of Rae-1 (an NKG2D ligand) on uterine macrophages and led to fetal resorption. In IL-10−/− mice, NKG2D− T cells instead became the source of fetal resorption during the same gestation period. Interestingly, both uterine NK and T cells produced TNF-α as the key cytotoxic factor contributing to fetal loss. Treatment of WT mice with poly(I:C) resulted in excessive trophoblast migration into the decidua and increased TUNEL positive signal. IL-10−/− mice supplemented with recombinant IL-10 induced fetal loss through NKG2D+ uNK cells, similar to the response in WT mice. Blockade of NKG2D in poly(I:C)-treated WT mice led to normal pregnancy outcome. Thus, we demonstrate for the first time that pregnancy disrupting inflammatory events mimicked by poly(I:C) are regulated by IL- 10 and depend on the effector function of uterine NKG2D+ NK cells in WT mice and NKG2D− T cells in IL-10 null mice.
Keywords: IL-10, TLR-3, uterine T cells, uterine NK cells, NKG2D
Introduction
Maintenance of pregnancy is acutely prone to infections and inflammatory triggers. Viral and bacterial infections, clinical or sub-clinical, may disturb immune tolerance at the maternal-fetal interface and predispose to adverse pregnancy outcomes (1-4). However, the mechanisms underlying detrimental immune microenvironment and pregnancy loss remain poorly understood. The immune constitution of the pregnant uterus (decidua) is quite distinct from other organs, consisting of a large number of innate immune cells such as natural killer (NK) cells and macrophages. Uterine NK (uNK) cells comprise 50-60% of the total mononuclear cell population during early to mid-gestation in both humans and mice (5, 6), whereas macrophages make up 20% (7, 8). uNK cells possess alternate phenotype and function when compared to peripheral blood NK cells as they are empowered with angiogenic characteristics and lack killing activity (9-11). In humans, uNK cells are CD56brightCD16dim and play a distinctive role in placental vasculature and decidualization (12, 13). In mice, distinct populations of uNK cells have been identified which mirror the functions of human uNK cells (14, 15). Like uNK cells, macrophages in the decidua also contribute to the balance required for immune protection of the fetus (7, 8). Not surprisingly, conventional T-cells are found in low proportions in human and murine uterine tissue in order to limit reactivity to paternally derived fetal-antigens (16). Moreover, regulatory T cells (Tregs), present at approximately 10% of the uterine immune population in early gestation, may further dampen cytotoxic T cells and guide embryo acceptance in the hormonally receptive uterus (17-19).
Inflammatory events including stress, commensal bacterial activation and excessive local tissue necrosis may also propel an immune tolerant microenvironment toward induction of a detrimental response (20-22). Although the placenta is a very potent immune regulator, it is not known yet whether viral infections alone or in combination with other triggers will elicit fetal ablating immune responses. We recently demonstrated that pregnant IL-10−/− mice when challenged with low doses of lipopolysaccharide (LPS) or CpG, ligands for TLR4 and TLR9 respectively, experienced fetal resorption or preterm birth depending on the gestational age- dependent exposure. These adverse pregnancy outcomes were directly associated with activation and amplification of TNF-α producing uNK cells or macrophages (7, 23, 24). In these models, IL-10 played a critical role as a vascular and anti-inflammatory cytokine for maintaining pregnancy in response to mild or moderate levels of inflammation (4, 25). However, it is not clear which infectious or inflammatory events will overwhelm the IL-10 proficient uterine milieu and cause pregnancy complications. This scenario warrants further analysis of detrimental immune responses activated by diverse inflammatory triggers (26-28). In the TLR cascade, TLR3 can respond to double-stranded nucleic acids emanating from viral infections or necrotic cells (22). In this regard, the viral mimic poly(I:C) has been shown to induce fetal loss (29, 30). However, what detrimental immune responses are elicited at the maternal-fetal interface by poly(I:C) and whether cytokine milieu affects the nature of the response to this viral mimic are poorly understood.
Our study is aimed at addressing the hypothesis that IL-10 regulates the immune response to poly(I:C) that causes fetal demise. Our results support this hypothesis and demonstrate that IL- 10 coupled with TLR3 activation plays a role in inflammatory induction of the activating receptor NKG2D. In particular, NKG2D is best characterized for virus and tumor elimination, and recognizes ligands that are induced by cellular transformation, stress, or infection (31). Our results demonstrate that in response to poly(I:C) treatment of pregnant wild type mice, a significant proportion of uNK cells acquires a fetal damaging, TNF-α producing phenotype marked by IL-10-dependent induction of NKG2D expression. Further, “induced” expression of the cognate NKG2D ligand, Rae-1 (retinoic acid early inducible-1) (32), is predominantly detected on uterine F4/80+ macrophages from poly(I:C)-treated WT mice. In IL-10−/− mice, poly(I:C) treatment amplifies TNF-α producing uterine T cells. Our data suggest that although IL-10 is a pregnancy compatible cytokine (7, 25), TLR3-mediated induction of inflammation at the maternal-fetal interface may alter the anti-inflammatory characteristics of this cytokine.
Materials and Methods
Mice
C57BL/6 WT or IL-10−/− mice were obtained from the Jackson Laboratory and housed in pathogen-free conditions in the Central Research Facility at Rhode Island Hospital. All protocols were approved by the Lifespan Committee for Animal Welfare. Syngeneic matings were used in this study and visualization of a vaginal plug was designated as gestational day (gd) 0.
In vivo treatments
All reagents were injected intraperitoneally (i.p.) in pregnant mice in a time-dependent fashion. Mice were injected on gd6 with saline or varying doses of poly(I:C) (Invivogen) to establish a dose curve for maximum fetal resorption. Neutralization of TNF-α was accomplished by injecting 250 μg/mouse anti-TNF-α antibody (BD Biosciences) on gd5 and gd7 in combination with gd6 injection of poly(I:C) or saline. An isotype matched antibody was used as a control. Asialo-GM1 antibody (Wako) or isotype matched control were injected at a dose of 100 μg/mouse on gd4, 6 and 8 to deplete NK cells. Depletion of T-cells was monitored with dual injection of 200 μg of anti-CD4 and anti-CD8 or isotype control antibodies (BD Bioscience) on gd5 and gd7 in conjunction with saline or poly(I:C) treatment. NK1.1 or NGK2D was blocked with injection of 250 μg/mouse of anti-NK1.1 antibody (BD Bioscience) or anti-NKG2D antibody (eBioscience), respectively, or corresponding isotype controls on gd5 and gd7. Recombinant IL-10 (eBioscience) was injected at a dose of 500 ng/mouse on gd4, 6 and 8 to restore WT phenotype in IL-10−/− mice. When required, uterine horns were visualized and photographed to assess fetal resorption or healthy fetal units.
Tissue sampling
Spleen, implantation units, and serum were harvested from IL-10−/− or WT mice on gd10. Blood was collected and centrifuged at 2000 rpm for 15 min and serum stored at −80°C. Spleen and implantation units were immediately placed in RPMI supplemented with 5% FBS and tissues were manually dissociated between frosted micro-slides. Cell preparations were placed on a Ficoll-Lite (Atlanta Biologicals) gradient and centrifuged at room temperature for 30 min. Lymphocytes and trophoblasts appearing at different densities were collected, washed, counted, and placed in 96 well plates at 1×106 cells per well for further analysis.
Flow cytometry
Analysis was carried out as previously described (7, 23, 24). Surface staining with antibodies for NK1.1, CD45, CD3, CD4, CD8 (BD Bioscience) and NKG2D, F4/80, CD11b, Rae-1 (eBioscience) or isotype matched controls was performed by flow cytometry (FACS Canto, Beckton Dickinson). Intracellular staining was carried out for TNF-α, IFN-γ, IL-12 (eBioscience) as described (7, 23, 24). Briefly, cells were washed with PBS, stained for surface antigens for 30 min, washed, treated with Cytofix/Cytoperm (BD Biosciences), and stained intracellularly in PermWash (BD Bioscience) with antibody or isotype matched control. Cells were washed and analyzed by FACS.
Cytochemistry and Immunocytochemistry
Saline or poly(I:C) treated uterine horns were collected on gd10 and stored in 10% neutral buffered formalin (Protocol) and embedded into paraffin within 48 hours post harvest and section prepared for immunohistochemical analysis as described (7, 23, 24). Serial sections were cut at 5-10 micron intervals. Apoptotic cells were probed by using the ApopTag® Fluorescence In Situ Apoptosis Detection Kit (Millipore) and protocol was performed according to the manufacturer’s instructions. Mouse monoclonal anti-cytokeratin 8 (TROMA-I) (Developmental Hybridoma) and goat anti-mouse Rae-1 (Santa Cruz Biotechnology) were used to stain trophoblasts and Rae-1 positive decidual and placental cells, respectively, as detected with Streptavidin-FITC (Vector Laboratories). For uNK cell identification, Dolichos biflorus (DBA) lectin cytochemistry (14) and perforin (PRF) immunocytochemistry (rabbit-polyclonal anti- perforin antibody, Torrey Pines Biolabs) were performed. Analyses used a Nikon eclipse 80i with SPOT advanced camera (version 4.1.2–Nikon Instruments Inc.) for fluorescence photomicroscopy.
ELISA
Serum samples were analyzed by ELISA to measure TNF-α, IFN-γ, IL-12 (R&D) and IFN-β (Interferon Source). Experiments were performed according to the manufacturer’s instructions.
Statistics
Two groups were compared with two-tailed unpaired Student’s t test. Significance was determined as p<0.05. Time course of multiple groups were compared with two-way ANOVA.
Results
Distinct uterine immune populations amplify in response to poly(I:C) treatment in pregnant WT and IL-10−/− mice
In our previous studies using LPS or CpG to induce adverse pregnancy outcomes, we demonstrated that IL-10−/− mice were highly sensitive to low doses of LPS and CpG for induction of fetal resorption or preterm birth (7, 23, 24). This prompted us to compare responses to viral infections as mimicked by poly(I:C), a TLR3 ligand, when administered i.p. on gd6 of pregnancy. As shown in Figure 1A, poly(I:C) treatment resulted in dose-dependent fetal resorption in both WT and IL-10−/− mice as assessed by evaluation of placental units on gd10. WT and IL-10−/− mice experienced fetal resorption in both uterine horns at the same dose of 100 μg poly(I:C)/mouse with similar kinetics (Fig. 1B), suggesting that IL-10 is not protective against TLR-3 triggered fetal demise.
Figure 1. Fetal resorption and amplification of uterine NK and T cells in WT and IL-10−/− mice in response to poly(I:C) treatment.

(A) poly(I:C) injected i.p. on gd6 was evaluated in a dose dependent manner to induce fetal resorption as assessed by inspection of uterine placental units on gd10. A dose of 100 μg/mouse induced 100% fetal resorption in both IL-10−/− and WT mice. A subset of these mice was allowed to deliver and no pups were born. Data are plotted as mean ± S.E.M (n=6/treatment). (B) Representative gd10 WT and IL-10−/− uterine horns from mice treated with saline or 100 μg/mouse poly(I:C) are depicted. (C) Assessment of splenic and uterine immune cells from WT or IL-10−/− mice treated on gd6 with saline or poly(I:C) (100 μg/mouse) and harvested on gd10. Cellular populations were first gated on CD45+ cells then analyzed for NK1.1 versus CD3. Data from spleen and uterus are representative of 8 mice per condition and numbers are averages of these data. (D) Graphs indicate statistical significance (*, p<0.05) of saline versus poly(I:C) treated cellular populations as indicated.
Since IL-10 has been shown in several viral models to directly suppress T cell function and is concomitantly produced by NK cells (33, 34), we profiled splenic and uterine CD3− NK1.1+ and CD3+ lymphocytes by flow cytometry. Data are presented as representative flow cytometry plots (Fig. 1C) and average numbers from several experiments (Fig. 1D). There were no marked changes in splenic NK1.1+ and CD3+ cells between untreated and poly(I:C)-treated WT or IL-10−/− mice (Fig. 1D). Remarkably, poly(I:C) treatment induced amplification of NK1.1+ cells in WT mice when compared to saline-treated controls (22±4% vs. 10±2%) (Fig. 1C and D). However, in IL-10−/− mice, no significant changes in uNK1.1+ cells were observed in response to poly(I:C) (Fig. 1C and D). In contrast, uterine CD3+NK1.1− cells expanded markedly from 19±2% to 30±3% (Fig. 1C and D). These observations indicate that IL-10 contributes to uNK cell expansion in WT mice in response to poly(I:C) treatment.
TNF-α produced by uNK and T cells is required for fetal resorption in response to poly(I:C)
Since serum cytokines are altered during pregnancy and in response to inflammatory triggers, we first analyzed a panel of inflammatory cytokines that are associated with TLR3 activation (35). We observed no marked changes in serum IFN-γ, IL-12, or IFN-β as measured by ELISA (Fig. 2A). However, TNF-α was significantly increased in WT and IL-10−/− mice exposed to poly(I:C) (Fig. 2A).
Figure 2. Measurement of cytokines and effect of in vivo TNF-α neutralization on fetal resoprtion in WT Mice and IL-10−/− mice.

(A) ELISA was performed using gd10 serum samples from IL-10−/− or WT mice treated on gd6 with saline or poly(I:C) (100 μg/mouse) and the analysis included TNF-α, IFN-β, IFN-γ, and IL-12. Bars represent mean ± S.E.M. (n=8 mice/group) and (*; p<0.05) indicates significance between saline and poly(I:C) treatments. (B) FACS analysis of TNF-α producing uterine NK1.1, CD4+ and CD8+ cells. Uterine mononuclear cells were isolated from gd10 uteroplacental tissue and gated for CD45 positive staining and further analyzed for NK1.1, CD4+CD8+ subpopulations and TNF-α expression. Graphic representation is provided from three independent experiments for intracellular staining of TNF-α produced by the indicated cellular populations in WT or IL-10−/− mice treated with saline or poly(I:C) on gd6. Data are plotted as mean ± S.E.M. (n=9 mice/condition) and (*) indicates statistical significance (p<0.05) between saline and poly(I:C) treated samples. (C) Representative gd10 uterine horns from WT (left) or IL-10−/− (right) mice treated with (from top to bottom of panels) saline + anti-TNF-α antibody, poly(I:C) + anti-TNF- α antibody, or poly(I:C) + isotype antibody (n=4 mice/ condition). (D) Rescue of pregnancy in response to NK cell depletion by asialo-GM1 antibody and T cell depletion by anti-CD4 and anti-CD8 antibodies. a) gd10 uterine horns from WT mice treated with asialo-GM1 antibody + saline, b) asialo-GM1 antibody + poly(I:C), c) gd10 uterine horns from IL-10−/− mice treated with anti-CD4 antibody + anti-CD8 antibody + saline, and d) anti-CD4 antibody + anti-CD8 antibody + poly(I:C). Corresponding dot plots represent data from n=4 independent experiments where TNF-α production was assessed from the cell populations indicated under depletion conditions featured in (b-d).
Next, we identified the uterine cellular source of TNF-α in both WT and IL-10−/− mice as measured by intracellular staining and FACS analysis. Intracellular staining of uNK1.1+, uCD3/CD4+, and uCD3/CD8+ cells showed that uNK cells were the main producer of TNF-α in WT mice in response to poly(I:C) (40±4% vs. 12±2%) (Fig. 2B). No such TNF-α increase occurred in WT mice from CD4+ and CD8+ T-cells. In contrast, IL-10−/− mice showed both CD4+ and CD8+ T cells as the source of marked TNF-α production in response to poly(I:C) as compared to saline treated controls (CD4+: 23±4% vs. 8±2%; CD8+: 50±4% vs. 10±2%) (Fig. 2B).
To ensure that TNF-α was associated with fetal resorption, we neutralized TNF- α in vivo by injecting (i.p.) a neutralizing antibody on gd5 and gd7. As shown in Figure 2C, normal fetal development was observed in poly(I:C)-treated WT and IL-10−/− mice upon TNF-α neutralization. In addition, in cases where mice with TNF-α neutralization were allowed to deliver, they gave birth to healthy litters of normal size at term (data not shown). Next, WT mice treated with NK cell depleting antibody asialo-GM1 and exposed to saline or poly(I:C) exhibited no poly(I:C)- mediated fetal demise (Fig. 2D). Representative FACS data show that TNF- α production is abrogated and uNK cells are depleted in WT mice in response to poly(I:C) upon treatment with asialo-GM1 antibody (Fig. 2D). In contrast, similar NK cell depletion did not rescue pregnancy in IL-10−/− mice and T cell subsets continued to produce TNF- α at high levels (data not shown). In this case, double depletion of both CD4+ and CD8+ T cells as assessed by flow cytometry significantly impaired TNF- α production and rescued pregnancy (Fig. 2D). These data provide evidence for the role of distinct uterine immune populations in inducing poly(I:C)-mediated fetal resorption in WT and IL-10−/− mice, respectively.
NKG2D is induced in NK1.1+/TNF-α+ uNK cells in WT mice in response to poly(I:C)
NKG2D is a molecule that enables NK cell activation and subsequent killing activity (36) and can be further induced in response to TLR3 activation (37). Next, we characterized the expression of NKG2D on splenic and uterine NK1.1+ or CD3+ cells from saline or poly(I:C)- treated WT or IL-10−/− mice. In agreement with the earlier data, we did not observe any changes in numbers of immune cells or NKG2D expression on splenic populations from either WT or IL- 10−/− mice (Fig. 3A). WT mice treated with poly(I:C) exhibited significant upregulation of NKG2D on uNK1.1+ cells as compared to vehicle-treated mice (20±4% vs. 4±2%) (Fig. 3A). Importantly, IL-10−/− mice treated with poly(I:C) did not exhibit increased expression of NKG2D on uNK1.1+ populations, nor was NKG2D induced in uterine CD3+ population in WT or IL-10−/− mice (Fig. 3A and B).
Figure 3. Induction of NKG2D in NK1.1+/TNF- α+ uNK cells in WT mice in response to poly(I:C).

(A) Representative plots of splenic or uterine CD45+NK1.1+NKG2D+ and CD45+CD3+NKG2D+ cells as analyzed by FACS from gd10 WT or IL-10−/− mice treated on gd6 with saline or poly(I:C). Graphs are average of cell populations from 8 mice per condition, (*; p<0.05) indicates significance between saline and poly(I:C) treatment conditions. (B) Representative FACS analysis of intracellular TNF-α in uterine CD45+ cells gated on the NKG2D+ and CD3+ subpopulations from gd10 WT or IL-10−/− mice treated on gd6 with saline or poly(I:C). (C) Summary graphs of TNF-α+ cells gated NK1.1+/NKG2D+ and CD3+/NKG3D− isolated from uterine tissue (n=8 mice/ condition). *; p<0.05 shows significance between saline and poly(I:C) treatment conditions.
To confirm that the expression of NKG2D was specific to the TNF-α+ uNK cell population, we assessed NKG2D+ uNK cells for intracellular TNF-α+ staining by flow cytometry (Fig. 3B). A significant proportion of NK1.1+ cells from WT mice were found to be NKG2D+ in response to poly(I:C) and produced significantly high levels of TNF-α as compared to untreated mice (24±3% vs. 5±2%) (Fig. 3B). In contrast, the NK1.1+/NKG2D+ population did not amplify or exhibit production of TNF- α in IL-10−/− mice. However, uterine CD3+ T cells showed significant TNF- α production in response to poly(I:C) treatment (28±3% vs. 6±2%) (Fig. 3B). A graphical representation of the data from three different experiments is presented in Figure 3C. The activating receptor NKG2D is generally expressed on NK and CD8+ T cells under pathological conditions (38, 39) or in response to TLR3 triggering (37). Our results are suggestive of a critical role of IL-10 in induction of NKG2D on uNK cells when challenged by viral infections.
Direct evidence for IL-10-mediated up-regulation of NKG2D
Our data thus far support the conclusion that the NKG2D receptor was induced in response to poly(I:C) only in WT, not IL-10−/−, mice (Fig. 3). Thus, we aimed to provide direct evidence whether IL-10 contributed to NKG2D induction on uNK cells. We first assessed the outcome of pregnancy in IL-10−/− mice supplemented with recombinant IL-10 (rIL-10) and treated with saline or poly(I:C). Uterine horns on gd10 from IL-10−/− mice supplemented with rIL-10 showed resorbed embryo sites in response to poly(I:C) (Fig. 4A). Allowing a group of these mice to deliver confirmed these results as no pups were born (data not shown).
Figure 4. Recombinant IL-10 (rIL-10) induces NKG2D on uterine NK1.1 cells in IL-10−/− mice.
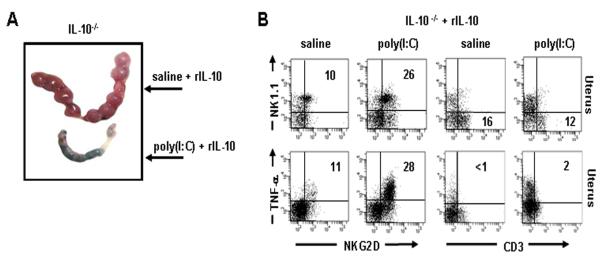
(A) Representative gd10 uterine horns from IL-10−/− mice treated with saline or poly(I:C) + rIL- 10 (n=4 mice/condition). rIL-10 supplementation does not protect against fetal resorption. (B) Uterine populations were analyzed by FACS from IL-10−/− mice supplemented with rIL-10 and treated with saline or poly(I:C). Data represent mean ± S.E.M. (n= 4 mice/condition). IL-10−/− mice supplemented with rIL-10 show NKG2D expression on NK1.1 cells which now produce TNF-α.
In order to determine whether rIL-10 directly induced NKG2D+ uNK cell- expansion in IL-10−/− mice, we harvested uterine lymphocytes from IL-10−/− mice treated with rIL-10 and saline or poly(I:C). Uterine immune cell profile now showed a significant increase in the NK1.1+/NKG2D+ population as compared to saline-treated controls (26±1% vs. 10±1%) (Fig. 4B), and paralleled the uterine immune cell response observed in WT mice when treated with poly(I:C) (see Fig. 3). In contrast, CD3+ uterine cells no longer amplified in response to poly(I:C) in IL-10−/− mice treated with rIL-10 (Fig. 4B). Furthermore, intracellular assessment of TNF-α production proved that NK1.1+/NKG2D+ cells now produced this cytokine (28±3% vs. 11±1%) (Fig. 4B). Analogous to WT mice, uterine CD3+ T cells failed to amplify or produce TNF- α in response to poly(I:C) (Fig. 4B). Taken together, treatment of IL-10 deficient mice with rIL-10 showed similar NKG2D-mediated events as observed in WT mice, suggesting that pregnancy compatible functions of IL-10 are compromised in the context of inflammatory challenges posed by poly(I:C)-like triggers.
NKG2D is necessary for fetal loss induced by poly(I:C)
Based on the data presented above, we claim that “induced” expression of NKG2D on uNK cells is under the control of IL-10 and associated with fetal resorption in response to poly(I:C). Next, we aimed to determine whether NKG2D+ uNK cells were the primary cause of fetal demise. We treated WT mice or IL-10−/− mice supplemented with rIL-10 with an NKG2D blocking antibody and injected either saline or poly(I:C) on gd6 to assess fetal resorption on gd10. Figure 5A shows that examination of uterine horns from WT and rIL-10-treated IL-10−/− mice did not reveal any evidence of fetal resorption, and when allowed, a subset of mice under these conditions gave birth to healthy litters at term (data not shown). Depletion of NKG2D+ NK cells was confirmed by flow cytometry (Fig. 5B). No TNF-α producing NK1.1/NKG2D+ cells from the NKG2D depleted uNK1.1+ population were observed in WT and rIL-10-supplemented IL-10−/− mice in response to poly(I:C) treatment (Fig. 5B).
Figure 5. Depletion of NKG2D+ NK cells rescues pregnancy and leads to loss of TNF-α production in WT and rIL-10 supplemented IL-10−/− mice in response to poly(I:C).

(A) Representative uterine horns from gd10 WT or rIL-10-supplemented IL-10−/− mice treated with anti-NKG2D antibody + saline or anti-NKG2D antibody + poly(I:C). Data represent mean ± S.E.M. (n= 4 mice/condition). NKG2D depletion abrogated fetal resorption as demonstrated by normal fetal units. (B) Assessment of NKG2D depletion and loss of TNF-α production was performed by gating on CD45+NK1.1+NKG2D+ uterine cells from saline or poly(I:C)-treated gd10 WT or rIL-10 + IL-10−/− mice injected i.p. with anti-NKG2D antibody. Dot plots represent data from 4 or more animals per condition.
Poly(I:C) treatment results in induction of NKG2D ligand Rae-1, predominantly in uterine macrophages
The NKG2D activating receptor interacts with the minor histocompatibility antigen ligands, the retinoic acid early inducible-1 (Rae-1) family members and the heat shock 60 (H60), in mice (32, 40). In C57BL/6 mice, Rae-1 is thought to be the key “induced” NKG2D ligand in response to NK cell activation (41). Thus, the NKG2D- Rae-1 axis may further define the mechanism of poly(I:C)-induced fetal loss. Rae-1 has been shown to be expressed at mRNA level in mouse embryonic tissues (32, 42). To assess its induced presence in the mouse placenta from untreated or poly(I:C)-treated WT mice, immunohistochemical analysis was performed using a Rae-1- specific antibody (Fig. 6A). Weak Rae-1 positive staining was observed throughout maternal and placental regions from saline-treated mice (4X and 20X magnification), implying that decidual cells and trophoblasts express baseline levels of this NKG2D ligand. In contrast, Rae-1 immunostaining was notably induced in the decidual region from poly(I:C)-treated mice ( see 20X magnification). To corroborate these data, we characterized decidual immune cells (T cells, NK cells, and macrophages) and trophoblast cells for Rae-1 expression by flow cytometry. Of the uteroplacental cell types (trophoblasts (CD45−CK7+), T cells (CD3+), macrophages (F4/80+), and NK cells (NK1.1+)) examined (data not shown), only macrophages (F4/80+) showed significantly induced expression of Rae-1 in response to poly(I:C) when compared to saline- treated mice (24±4% vs. 5±3%) (Fig. 6B).
Figure 6. Exposure of pregnant WT mice to poly(I:C) results in induced expression of Rae- 1 on macrophages and enhanced trophobalst invasion and cell death.

(A) Representative images from uteroplacental units from gd10 saline or poly(I:C) treated WT mice was assessed by immunohistochemistry for RAE-1 expression. Weak staining for Rae-1 was detected in the decidua and placental regions (4× magnification; Scale Bar: 100 μm and 20× magnification; Scale Bar: 20 μm). (B) FACS analysis of Rae-1 on uterine immune cells. CD45+ and CD45−cytokeratin7+ cells were analyzed. Cells are gated from CD45+ populations obtained from uterine populations. Data represent three independent experiments. (C) Upper panel: immunohistochemical staining with TROMA-I antibody shows unique staining with increased invasion of trophoblast cells into the decidua in response to poly(I:C), but not saline (10× magnification; Scale Bar: 60 μm). Lower panel: TUNEL staining (20× magnification; Scale Bar: 20 μm) demonstrates significantly increased cell death the trophoblastic regions, including invading trophoblasts. Images are representative of a total of 3mice per condition. (D) DBA+ and PRF+ NK cell identification by immunofuorescence staining from uteroplacental unit from gd10. In response to poly(I:C) treatment, DBA+ intensity is diminished but these cells maintain their PRF+ phenotype. However, their distribution appears to be diffused through the placenta (P) region. Images are representative of results obtained from placental samples of 3 mice per condition (10xmagnification; Scale Bar: 60 μm).
It is possible that interaction between NKG2D (uNK cells) and Rae-1 (macrophages and trophoblasts) activates NKG2D+ NK cells to produce TNF-α which in turn causes placental pathology and fetal resorption. We first attempted to assess placental tissue for trophoblast areas by staining with a cytokeratin 8-specific antibody (TROMA-I) to distinguish trophoblastic areas from maternal regions (Fig. 6C). These data demonstrate that trophoblasts in the saline-treated placental units remained flushed throughout the labyrinth and the junctional zone. In stark contrast, poly(I:C)-treated placental units showed excess trophoblast migration into the decidual region and beyond (Fig. 6C). We have previously shown that uNK cell-produced TNF-α caused placental cell death in response to LPS (23, 24). To elucidate whether uNK cells caused apoptosis in migrating trophoblasts as a result of direct interaction with Rae-1 ligand or TNF-α production, we stained uteroplacental tissue from saline or poly(I:C)-treated mice for TUNEL positive regions. Interestingly, TUNEL-positive cells were seen in significant numbers in the placental and decidual regions, particularly in invading trophoblasts from poly(I:C)-treated mice (Fig. 6C). Since TNF-α neutralization and NKG2D blockade abrogate TNF-α production and rescue pregnancy to term, our data in Figure 6 suggest that NKG2D-Rae-1 interactions are critical for poly(I:C)-mediated fetal loss.
It has been suggested that trophoblast migration into the decidua is regulated by uNK cells and their product IFN-γ (43). It is then possible that uNK cells undergo proportional and/or functional changes in response to poly(I:C). Our data suggest that uNK cells, particularly NKG2D+ cells, produce TNF-α and cause apoptosis in trophoblasts. To directly assess all uNK cells and their location in uteroplacental tissue from WT mice treated with poly(I:C), we performed DBA-lectin and PRF staining (14). Analyses of DBA-positive signal revealed greater numbers of DBA+ cells in the mesometrial gland and the decidua basalis regions of uteroplacental tissue from saline-treated WT mice (Fig. 6D). In contrast, DBA staining intensity was poor in tissue from poly(I:C)-treated WT mice. Importantly, DBA+ NK cells appeared to migrate to the placental zone. To rule out that poly(I:C) treatment does not lead to reduction in overall number of uNK cells, we performed PRF staining. As shown in Figure 6D, PRF+ uNK cells were predominant in the decidua basalis region in tissue from saline-treated animals and their presence is similar in intensity and number in tissue from poly(I:C)-treated mice. It is noteworthy that PRF+ uNK cells in poly(I:C)-treated samples are present throughout the tissue and appear to migrate to the placental region, supporting the data for DBA staining. It is possible that NKG2D+ uNK cells may be DBAdim but maintain their PRF+ phenotype. This also supports the TUNEL-positive signal in the placental zone (Fig. 6C).
Discussion
In this report, we identify IL-10 as an integral cytokine that orchestrates TLR3-mediated expansion of effector uterine populations for fetal demise by contributing to induction of cytotoxic NK cell receptor NKG2D on uNK cells. NKG2D+ uNK cells were identified as the source of TNF-α production which led to fetal resorption in WT mice. This was supported by observations that blockade of the NKG2D receptor or neutralization of TNF-α rescued pregnancy to term. The relationship of IL-10 as an inducer of NKG2D was confirmed by direct up-regulation of NKG2D on uNK1.1+TNF-α+ cells in poly(I:C)-treated WT mice, but not IL-10−/− counterparts. Importantly, pregnant IL-10−/− mice supplemented with rIL-10 responded to poly(I:C) in the same manner as WT mice through the expansion of NKG2D+ uNK cells. Although IL-10 is a pregnancy compatible cytokine (4, 7, 25), our results for the first time indicate that TLR3-mediated induction of inflammation at the maternal-fetal interface may alter the anti-inflammatory characteristics of this cytokine. Rather, TLR3 activation and IL-10 together reverse the programming of a uterine immune response from T cell-mediated to NK cell-mediated. However, induced TNF-α production is still a key feature of both cell types in response to poly(I:C). Although not widely accepted a few years back, we proposed and demonstrated that uNK cells could be transformed into foes of pregnancy in response to LPS and this pathway could be triggered in IL-10−/− mice even at a very low dose of LPS (0.5 μg/mouse) (23, 24, 44). In this study, we show that poly(I:C) can utilize IL-10 rich environment to transform uNK cells into pregnancy disrupting and TNF-α producing NKG2D+ NK cells. In the absence of IL-10, uterine T cells become the source of TNF-α and fetal loss. Taken together, our data strongly suggest that different inflammatory triggers are likely to exploit distinct immune cells and cytokine milieu at the maternal-fetal interface to cause pregnancy complications. This study also suggests that intrauterine viral infections alone as mimicked by poly(I:C) or in combination with other inflammatory triggers may transform the uterine immune milieu from tolerant to detrimental resulting in adverse pregnancy outcomes.
Our results provide a mechanistic explanation for poly(I:C)-mediated TLR3 activation at the uterine level and its convergence with IL-10 in regulating innate and adaptive immune responses that lead to fetal loss (Fig. 7). Unscheduled expansion of uterine NKG2D+ inflammatory NK cells and TNF-α production in WT mice support this notion. Importantly, induced expression of the NKG2D ligand Rae-1 by uterine macrophages may trigger over- production of TNF-α by NKG2D+ NK cells. TNF-α alone and/or cell-cell-contact between NKG2D+ uNK cells and invading Rae-1+ trophoblasts may lead to cell death as demonstrated by TUNEL positive staining in these cells. On the other hand, IL-10 deficiency is likely to unleash T cell activation and cytokine storm that may restrain NK cell-mediated responses. Support for this notion comes from the results of Kim et al who suggested that a cytokine storm from adaptive immune cells could temper initial innate immune responses (45).
Figure 7. Schematic representation of the poly(I:C)-induced events in WT and IL-10−/− mice.
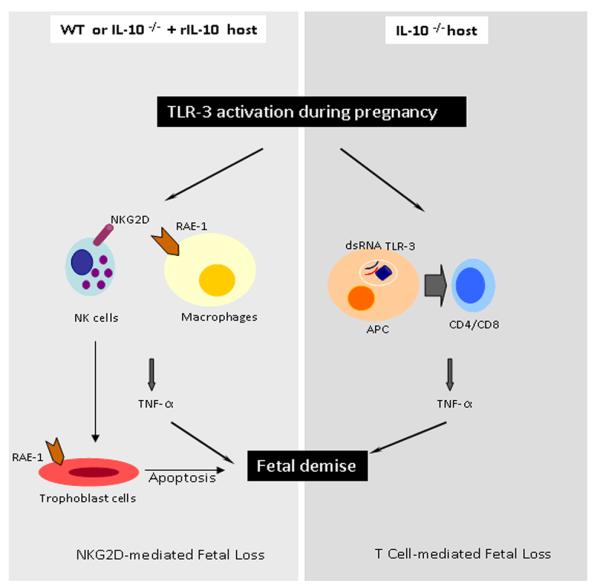
The model recapitulates the pathways leading to uterine NK or T cell-mediated cytotoxicity and fetal resorption in WT and IL-10−/− mice, respectively. Rae-1 positive macrophages or trophoblasts can interact with NKG2D+ NK cells and induce TNF-α production and trophoblast cell death in response to poly(I:C). On the other hand, uterine T cells can be activated by poly(I:C) in the absence of IL-10 to produce TNF-α and to cause fetal demise.
NKG2D is a well characterized lectin-like activating receptor originally detected on NK cells (36). In humans, NKG2D has now been shown to be expressed on CD8+ T cells, γδ T cells (46) and intestinal epithelial cells in pathologic conditions or in response to treatment with poly(I:C) (37). Its blockade in NOD mice has been shown to prevent autoimmune diabetes (47). Surprisingly, we did not observe NKG2D expression on uterine T cells in either WT or IL-10−/− mice treated with poly(I:C) during pregnancy. Since uterine immune cells are specialized in their phenotypic and functional repertoire, it is possible that the response of uterine immune cells to poly(I:C) is equally unique.
Our data support the view that a significant proportion of NK1.1+ NK cells acquires induced expression of NKG2D and produces TNF-α. In this regard, in vivo blockade of NKG2D alone rescued pregnancy in WT mice. It is intriguing that upon blockade of NKG2D, the integrity of the NK1.1+ population was intact, and these cells remained pregnancy compatible. These results agree with a model of transplantation where NKG2D blockade allowed for increased graft survival, but the NK1.1+ population remained unaffected and still migrated into the transplanted organ (38). These findings imply that it is important to identify requirements for molecular cascades that break immune tolerance at the maternal-fetal interface.
Our data in Figure 6 provide important insights into the mechanisms underlying fetal demise in response to inflammatory triggers such as poly(I:C). We show enhanced trophoblast migration into the mesometrial decidual region in poly(I:C)-treated WT mice as demonstrated by TROMA-I positive trophoblast cells. These invading trophoblasts undergo cell death as demonstrated by TUNEL-positive signal in this region. Ain et al have demonstrated that enhanced trophoblast migration occurs on gd14 or thereafter in pregnant rats or mice (48, 49). This timing is linked with reduction in uNK cells and their product IFN-γ in the mesometrial decidua. Since NKG2D+ uNK cells are amplified in response to poly(I:C), it is possible that trophoblast migration occurs not only as a result of reduced uNK cell population but also due to their altered functional characteristics and localization pattern. Our data suggest that NKG2D+ uNK cells now produce TNF-α and maintain their PRF+ phenotype. In the present study, excessive trophoblast invasion into the mesometrial decidua region occurs on gd10 in response to poly(I:C). It is possible that induction of NKG2D on uNK cells alters their regulatory ability which allows trophoblast invasion even on gd10. However, their ability to produce TNF-α and to interact with trophoblasts via NKG2D-Rae-1 coupling also leads to cell death, resulting in defective hemochorial placentation. We reiterate that though uterine NK cells are beneficial in regulating normal pregnancy, they can be transformed into detrimental cells in response to bacterial and viral infections (23, 24, 44). Since NK1.1+ cells represent only a sub-population of uNK cells (15), it is possible that NKG2D+ uNK cells could also belong to non-NK1.1 population and acquire the cytotoxic phenotype as a result of TNF-α production and different regulatory properties. Our data warrant a fresh look at the roles of IL-10, uterine NK cells and T cells in adverse pregnancy outcomes.
Acknowledgements
We would like to thank Paula Weston of the Molecular Pathology Core at Brown University for aiding in initial help with immunohistochemical analysis. We thank Drs. Ann Hill, James Padbury, Sunil Shaw, and Angela Tatum for insightful suggestions and critical reading of the manuscript.
References
- 1.Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N. Engl. J. Med. 2000;342:1500–1507. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- 2.Haun L, Kwan N, Hollier LM. Viral infections in pregnancy. Minerva. Ginecol. 2007;59:159–174. [PubMed] [Google Scholar]
- 3.Romero R, Espinoza J, Chaiworapongsa T, Kalache K. Infection and prematurity and the role of preventive strategies. Semin. Neonatol. 2002;7:259–274. doi: 10.1016/s1084-2756(02)90121-1. [DOI] [PubMed] [Google Scholar]
- 4.Thaxton JE, Nevers TA, Sharma S. TLR-mediated preterm birth in response to pathogenic agents. Infect. Dis. Obstet. Gynecol. 2010;2010 doi: 10.1155/2010/378472. pii.378472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffet A, Loke C. Immunology of placentation in eutharian mammals. Nat. Rev. Immunol. 2006;8:584–594. doi: 10.1038/nri1897. [DOI] [PubMed] [Google Scholar]
- 6.Croy BA, Luross JA, Guimond MJ, Hunt JS. Uterine natural killer cells: insights into lineage relationships and functions from studies of pregnancies in mutant and transgenic mice. Nat. Immun. 1997;15:22–33. [PubMed] [Google Scholar]
- 7.Thaxton JE, Romero R, Sharma S. TLR9 activation coupled to IL-10 deficiency induces adverse pregnancy outcomes. J. Immunol. 2009;183:1144–5114. doi: 10.4049/jimmunol.0900788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houser BL, Tilburgs T, Hill J, Nicrota ML, Strominger JL. Two unique human decidual macrophage populations. J. Immunol. 2011;186:2633–2642. doi: 10.4049/jimmunol.1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kopcow HD, Allan DS, Chen X, Rybalov B, Andzelm MM, Ge B, Strominger JL. Human decidual NK cells form immature activating synapses and are not cytotoxic. Proc. Natl. Acad. Sci., USA. 2005;25:15563–15568. doi: 10.1073/pnas.0507835102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, Gazit R, Yutkin V, Benharroch D, Porgador A, Keshet E, Yagel S, Mandelboim O. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006;12:1065–1074. doi: 10.1038/nm1452. [DOI] [PubMed] [Google Scholar]
- 11.Kalkunte SS, Mselle TF, Norris WE, Wira CR, Sentman CL, Sharma S. Vascular endothelial growth factor C facilitates immune tolerance and endovascular activity of human uterine NK cells at the maternal-fetal interface. J. Immunol. 2009;182:4085–4092. doi: 10.4049/jimmunol.0803769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King A, Balendran N, Wooding P, Carter NP, Loke YW. CD3- leukocytes present in the human uterus during early placentation: phenotypic and morphologic characterization of the CD56+ population. Dev. Immunol. 1991;1:4169–4190. doi: 10.1155/1991/83493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lash GE, Robson SC, Bulmer JN. Functional role of uterine natural killer (uNK) cells in human early pregnancy decidua. Placenta. 2010;31:S87–92. doi: 10.1016/j.placenta.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 14.Paffaro VA, Jr, Bizinotto MC, Joazeiro PP, Yamada AT. Subset classification of mouse uterine natural killer cells by DBA lectin reactivity. Placenta. 2003;24:479–488. doi: 10.1053/plac.2002.0919. [DOI] [PubMed] [Google Scholar]
- 15.Yadi H, Burke S, Madeja Z, Hemberger M, Moffett A, Colucci F. Unique receptor repertoire in mouse uterine NK cells. J. Immunol. 2008;181:6140–6147. doi: 10.4049/jimmunol.181.9.6140. [DOI] [PubMed] [Google Scholar]
- 16.Piccinni MP. T cell tolerance towards the fetal allograft. J. Reprod. Immunol. 2010;85:71–75. doi: 10.1016/j.jri.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 17.Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol. 2004;5:266–271. doi: 10.1038/ni1037. [DOI] [PubMed] [Google Scholar]
- 18.Shima T, Sasaki Y, Itoh M, Nakashima A, Ishii N, Sugamura K, Saito S. Regulatory T cells are necessary for implantation and maintenance of early pregnancy but not late pregnancy in allogeneic mice. J. Reprod. Immunol. 2010;85:121–129. doi: 10.1016/j.jri.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Kahn DA, Baltimore D. Pregnancy induces a fetal antigen-specific maternal T regulatory cell response that contributes to tolerance. Proc. Natl. Acad. Sci., USA. 2010;107:9299–9304. doi: 10.1073/pnas.1003909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nepomnaschy PA, Sheiner E, Mastorakos G, Arck P. Stress, immune function, and women’s reproduction. Ann. N. Y. Acad. Sci. 2007;1113:350–364. doi: 10.1196/annals.1391.028. [DOI] [PubMed] [Google Scholar]
- 21.Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR, Hooper LV, Schmidt RR, von Aulock S, Radek KA, Huang CM, Ryan AF, Gallo RL. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat. Med. 2009;15:1377–1382. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy SP, Fast LD, Hanna NN, Sharma S. Uterine NK cells mediate inflammation-induced fetal demise in IL-10-null mice. J. Immunol. 2005;175:4084–4090. doi: 10.4049/jimmunol.175.6.4084. [DOI] [PubMed] [Google Scholar]
- 24.Murphy SP, Hanna NN, Fast LD, Shaw SK, Berg G, Padbury JF, Romero R, Sharma S. Evidence for participation of uterine natural killer cells in the mechanisms responsible for spontaneous preterm labor and delivery. Am. J. Obstet. Gynecol. 2009;200:1–9. doi: 10.1016/j.ajog.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalkunte S, Nevers T, Norris WE, Sharma S. Vascular IL-10: a protective role in preeclampsia. J. Reprod. Immunol. 2011;88:165–169. doi: 10.1016/j.jri.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galluchi S, Lolkema M, Matzinger P. Natural adjuvents: endogenous activators of dendritic cells. Nat. Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 27.Sauter B, Albert ML, Francisco L, Larsson S, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat.Rev. Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Fougerolles AR, Baines MG. Modulation of the natural killer cell activity in pregnant mice alters the spontaneous abortion rate. J. Reprod. Immunol. 1987;11:147–153. doi: 10.1016/0165-0378(87)90018-0. [DOI] [PubMed] [Google Scholar]
- 30.Koga K, Cardena I, Aldo P, Abrahams VM, Peng B, Fill S, Romero R, Mor G. Activation of TLR3 in the trophoblast is associated with preterm delivery. Am. J. Reprod. Immunol. 2009;61:196–212. doi: 10.1111/j.1600-0897.2008.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 32.Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, Lanier LL. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 33.Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J. Exp. Med. 2009;206:2235–2251. doi: 10.1084/jem.20082387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks DG, Walsh KB, Elsaesser H, Oldstone MB. IL-10 directly suppresses CD4 but not CD8 T cell effector and memory responses following acute viral infection. Proc. Natl. Acad. Sci., USA. 2010;107:3018–3023. doi: 10.1073/pnas.0914500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh TK, Mickelson DJ, Solberg JC, Lipson KE, Inglefield JR, Alkan SS. TLR-TLR cross talk in human PBMC resulting in synergistic and antagonistic regulation of type-1 and 2 interferons, IL-12 and TNF-alpha. Int. Immunopharmacol. 2007;7:1111–1121. doi: 10.1016/j.intimp.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 36.Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 2002;17:19–29. doi: 10.1016/s1074-7613(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 37.Zhou R, Wei H, Sun R, Zhang J, Tian Z. NKG2D recognition mediates Toll-like receptor 3 signaling-induced breakdown of epithelial homeostasis in the small intestines of mice. Proc. Natl. Acad. Sci., USA. 2007;104:7512–7515. doi: 10.1073/pnas.0700822104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Change CK, Hayden T, Feng-Chun L, Benjamin J, Hammerman J, Lanier LL, Kang S. The activating immmunoreceptor NKG2D and its ligands are involved in allograft transplant rejection. J. Immunol. 2007;179:6416–6420. doi: 10.4049/jimmunol.179.10.6416. [DOI] [PubMed] [Google Scholar]
- 39.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 40.Diefenbach A, Jensen ER, Jamiesen AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumor immunity. Nature. 2001;413:165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamerman J, Ogasawara K, Lanier LL. Cutting Edge: Toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J. Immunol. 2004;172:2001–2005. doi: 10.4049/jimmunol.172.4.2001. [DOI] [PubMed] [Google Scholar]
- 42.Xie X, He H, Colonna M, Seya T, Takai T, Croy BA. Pathways participating in activation of mouse uterine natural killer cells during pregnancy. Biol. Reprod. 2005;73:510–518. doi: 10.1095/biolreprod.104.033951. [DOI] [PubMed] [Google Scholar]
- 43.Ain R, Canham LN, Soares MJ. Gestation stage-dependent intrauterine trophoblast cell invasion in the rat and mouse: novel endocrine phenotype and regulation. Dev. Biol. 2003;260:176–190. doi: 10.1016/s0012-1606(03)00210-0. [DOI] [PubMed] [Google Scholar]
- 44.Kalkunte S, Chichester CO, Gotsch F, Sentman CL, Romero R, Sharma S. Evolution of non-cytotoxic uterine natural killer cells. Am. J. Reprod. Immunol. 2008;59:425–432. doi: 10.1111/j.1600-0897.2008.00595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim KD, Zhao J, Auh S, Yang X, Du P, Tang H, Fu YX. Adaptive immune cells temper initial innate responses. Nat. Med. 2007;13:1248–1252. doi: 10.1038/nm1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stern-Ginossar N, Mandelboim O. An integrated view of the regulation of NKG2D ligands. Immunology. 2009;128:1–6. doi: 10.1111/j.1365-2567.2009.03147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, Lanier LL. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–767. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Ain R, Tash JS, Soares MJ. Prolactin-like protein-A is a functional modulator of natural killer cells at the maternal-fetal interface. Mol. Cell. Endocrinol. 2003;204:65–74. doi: 10.1016/s0303-7207(03)00125-4. [DOI] [PubMed] [Google Scholar]
- 49.Ain R, Dai G, Dunmore JH, Godwin AR, Soares MJ. A prolactin family paralog regulates reproductive adaptations to a physiological stressor. Proc. Natl. Acad. Sci. 2004;101:16543–16548. doi: 10.1073/pnas.0406185101. [DOI] [PMC free article] [PubMed] [Google Scholar]