

Abstract
Autophagy is an evolutionarily conserved cytoplasmic process regulated by the energy rheostats mTOR and AMPK that recycles damaged or unused proteins and organelles. It has been described as an important effector arm of immune cells. We have shown that the cytoplasmically oriented calcium/calmodulin-dependent protein kinase I α (CaMKIα) regulates the inflammatory phenotype of the macrophage (Mφ). Here, we hypothesize that CaMKIα mediates Mφ autophagy. LPS induced autophagy in RAW 264.7 cells and murine peritoneal Mφ that was attenuated with biochemical CaMK inhibition or CaMKIα siRNA. Inhibition of CaMKIα reduced LPS-induced p-Thr172AMPK and TORC1 activity, and expression of a constitutively active CaMKIα but not a kinase-deficient mutant induced p-Thr172AMPK and autophagy that was attenuated by the AMPK inhibitor Compound C. Co-immunoprecipitation and in vitro kinase assays demonstrated that CaMKIα activates AMPK, thereby inducing ATG7, which also localizes to this CaMKIα-AMPK complex. During LPS-induced lung inflammation, C57Bl/6 mice receiving CaMKIαsiRNA displayed reduced lung and bronchoalveolar immune cell autophagy that correlated with reduced neutrophil recruitment, myeloperoxidase activity, and air space cytokine concentration. Independently inhibiting autophagy, using siRNA targeting the PI3 kinase VPS34, yielded similar reductions in lung autophagy and neutrophil recruitment. Thus, a novel CaMKIα-AMPK pathway is rapidly activated in Mφ exposed to LPS and regulates an early autophagic response, independent of TORC1 inhibition. These mechanisms appear to be operant in vivo in orchestrating LPS-induced lung neutrophil recruitment and inflammation.
Introduction
Acute lung injury (ALI) and respiratory distress syndrome (ARDS) remain significant causes of ICU morbidity, with an estimated case mortality of 27–43%.(1–3) The most common etiology is sepsis, in particular pneumonia.(4) Experimental evidence has shown a strong cause-and-effect relationship between persistent inflammation and progression to ALI.(5) However, the efficacy of current anti-inflammatory agents and the development of effective immunomodulatory therapies have been modest. This slow progress may stem, in part, from an incomplete understanding of the cellular and molecular mechanisms deterministic of disease progression or recovery. Specifically, we have yet to distinguish the causal pathogenic mechanisms necessitating targeted therapy from those adaptive and fundamental for disease resolution, which should be protected.
Autophagy is an ancient cytoplasmic homeostatic process regulating cellular biomass quantity, quality, and distribution. It endows cells with the ability to sacrifice and reuse portions of their cytoplasm to support vital functions during periods of stress. In the setting of nutrient deprivation, the mechanisms orchestrating this dynamic process involve the main growth rheostat mammalian target of Rapamycin (mTOR) and AMP kinase, a sensor of cellular energy status.(6–12) Fundamental for embryological development and survival, it integrates extensively with apoptosis and may itself function as a form of programmed (type II) cell death.(12–14) Hence, it is not surprising that it is preserved in all eukaryotic organisms, from yeast to humans.
Though the quintessential function of autophagy is to autodigest and to recycle portions of the cytoplasm to sustain vital cellular functions during starvation, recent data support a fundamental role in innate immunity as an anti-microbial effector of Toll-like receptors.(14–18) In Mφ, LPS induces autophagy through mechanisms dependent on TLR4, TRIF, RIP1 and p38 MAPK.(17, 19) Biologically, it has been shown to be important in the elimination of intracellular microbes, to contribute to major histocompatibility complex class II presentation, and to assist pattern recognition receptors by delivering cytosolic pathogen-associated molecular patterns (PAMP) to endosomal Toll-like receptors.(18, 20, 21) However, a complete understanding of the signaling mechanisms regulating autophagy during sepsis and the functional role of autophagy in cell survival and recovery of organ function has yet to be developed.
The calcium/calmodulin-dependent protein kinases (CaMK), a family of serine/threonine kinases responsive to intracellular Ca2+ concentration [Ca2+], are also integral to the immune response, mediating Ca2+-dependent Mφ function and regulating septic inflammation.(22–24) CaMKIV, upon its phosphorylation and activation by an upstream CaMKK, translocates into the nucleus and regulates the nucleocytoplasmic shuttling of HMGB1 in Mφ exposed to LPS.(23) As a component of a signaling cascade initiated by LPS activation of TLR4, CaMKIV also facilitates survival of dendritic cells by phosphorylating CREB and regulating expression of the Bcl-2 gene.(25) By contrast, CaMKIα regulates the cytoplasmic release of HMGB1 through a mechanism involving secretory lysosomes.(26)
Recent data are defining regulatory roles for members of this multifunctional family in autophagy. CaMKKβ may function as an upstream kinase for AMP kinase and regulate autophagy in response to elevations in cytosolic calcium through Bcl-2.(27) More recent data suggest indirect mechanisms by which CaMKKβ regulates autophagy, and that CaMK downstream in this cascade (i.e. CaMKI, CaMKIV) may be involved.(28) In this study we reveal that CaMKIα regulates autophagy in Mφ responding to LPS. Furthermore, these mechanisms are operant in vivo during LPS-induced lung inflammation and may underlie the subsequent progression of lung dysfunction.
Materials and Methods
Reagents
Ultra Pure LPS (Escherichia coli 0111:B4) was obtained from List Biologicals (Campbell, CA). KN62 ((S)-5-Isoquinolinesulfonic acid 4-[2-[(5 isoquinolinylsulfonyl) methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl]phenyl ester, 1-[N,O-bis(5-Isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine), obtained from Calbiochem (San Diego, CA), was dissolved in sterile dimethyl sulfoxide (DMSO) and used at a concentration of 20μM. STO690 (7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid - acetic acid) was obtained from Calbiochem (San Diego, CA) and used at concentrations of 1–20μM. STO609 is highly specific for CaMKK: it has an in vitro IC50 of 0.13–0.38 μM for CaMKK and 32 μM for CaMK II with little or no inhibition of CaMKI, CaMKIV, PKA, PKC, ERK, or myosin light chain kinase.(29) Monoclonal antibody (mAb) for Thr177/180 phosphorylated CaMKI (pCaMKI) was the generous gift of Dr. Naohito Nozaki (Kanagawa, Japan).(30) This mAb has been shown to recognize the active, threonine phosphorylated species of CaMKIα (Thr177), CaMKIβ (Thr180), and CaMKIδ (Thr178).(26, 30) Antibodies for p-Thr172AMPK, p-Ser2448mTOR, p-Thr389p70 S6 kinase and p-Ser235/236S6 were obtained from Abcam (Cambridge, MA). Antibodies for immunoblot of CaMKIα, ATG7, ATG5–12, LC3B, AMPK, mTOR, VPS34, and actin were obtained from Abcam (Cambridge, MA). For immunofluorescent microscopy rabbit anti-ATG7 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), mouse anti-LC3b (Novus Biologicals, Littleton, CO), goat anti-rabbit-Alex Fluoro488 (Invitrogen, Grand Island, NY), and goat anti-mouse-Cy3 (Invitrogen) were used.
Animal experimentation
We performed all animal experiments in accordance with the National Institutes of Health guidelines under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pittsburgh. We randomly grouped 6–8 week old male C57BL/6J mice and assigned them to a specific experiment. Investigators who treated animals knew the treatment groups and collected samples, which were then analyzed by other investigators blinded to the specific treatment.
In vivo RNAi
We performed in vivo RNAi to selectively inhibit CaMKIα or VPS34 as previously performed.(26) C57BL/6 mice (The Jackson Laboratory) were administered CaMKIα or scrambled, non-target (NT) siRNA (6mg/kg) by hydrodynamic tail vein injection delivered in (animal mass/10) mL lactated ringers. Others and we have shown that this technique efficiently and specifically inhibits the expression of CaMKIα.(26) For all experiments, confirmation of successful knockdown was assessed by immunoblot.
In vivo LPS-induced acute lung inflammation
Mice were anesthetized using a mixture of isoflurane and oxygen bled in at 3.5L/min. LPS (List Biologicals, Inc., Campbell, CA) dissolved in DNase/RNase free distilled water (1mg/ml) was instilled intratracheally at a concentration of 1.5 mg/kg by direct laryngoscopy. Animals were sacrificed 6 and 12 hours after LPS, and blood was isolated by intracardiac puncture. Whole blood was stored overnight at 4°C and centrifuged at 17,000g for 10min. The thorax was then opened, and the left hilum was ligated with a 4-0 silk. The left lung was then excised, snap frozen, and stored at −80°C for further studies. We cannulated the trachea with an 18-guage angiocatheter, and 5 sequential lavages of 0.5mL of bronchoalveolar lavage (BAL) fluid (0.9% saline with 0.6mmol/L ethylenediaminetetraacetic acid at room temperature (RT)) were instilled into the right lung and pooled for analysis. Cytospin slides were created using 200μl aliquots of BAL fluid and the Shandon Cytospin 3 centrifuge (Thermo Fisher Scientific, Inc., Waltham, MA). Manual cell counts and differentials were performed.
Cell isolation and treatment
Murine macrophage cell line RAW 264.7 (American Type Culture Collection, Rockville, MD) was grown in DMEM (BioWhittaker, Walkersville, MD) supplemented with 10% fetal calf serum (Sigma, San Diego, CA), 50U/mL penicillin, and 50μg/mL streptomycin (Cellgro Mediatech Inc., Kansas City, MO). Peritoneal Mφ were isolated from C57BL/6 mice by lavaging the peritoneal cavity with 5-2mL aliquots of 4°C PBS. The lavage was centrifuged at 300g for 5 minutes, and the cells were resuspended in RPMI supplemented with 10% fetal calf serum, 50U/mL penicillin, and 50μg/mL streptomycin. Selected cells were pretreated with KN62 (20 μM) or STO609 (1–20 μM) for 30 min or transfected with siRNA targeting CaMKIα. DMSO, at concentrations similar to comparative experiments, was used as an appropriate control and never exceeded 0.2%. Selected cells were then treated with LPS (1–100ng/mL).
siRNA
RAW 264.7 Mφ (2 × 104) or murine peritoneal Mφ (1 × 105) were plated in 0.5 ml of growth medium (without antibiotics) in each well of a 24-well plate, resulting in 30% or 80% confluence, respectively. Non-target and CaMKIα siRNA obtained from Dharmacon (Lafayette, Colorado) was added to 50 μl serum-free DMEM in a final concentration of 25 nM. We utilized the Smartpool siRNA from Dharmacon that incorporates 4 separate siRNA sequences for each CaMK. The following CaMKIα sequences were used:
| Target | Sequence |
|---|---|
|
| |
| CaMKIα: | sense GAACGAGAUUGCCGUCUUAUU antisense UAAGACGGCAAUCUCGUUCUU sense CGGAAGACAUUAGGGAUAUUU antisense AUAUCCCUAAUGUCUUCCGUU sense GGAGAGCUGUUUGACCAGGUU antisense UUCGGUCAAACAGCUCUCCUU sense AUACAGCUCUGGAUAAGAAUU antisense UUCUUAUCCAGAGCUGUAUUU |
In a separate tube, 3 μl Hiperfect were diluted in 50 μl serum-free DMEM and incubated at RT for 5 min. These two solutions were combined, and the final transfection mixture was incubated for 20 min at RT. This transfection mixture was applied to each well and incubated for 6 h, after which it was replaced by 500 μl of cell medium and incubated for 72 h. Inhibition of each targeted protein was determined by immunoblot. All experiments and cell number determinations were performed in triplicate.
Plasmid construction and transfection
Plasmids encoding a constitutively active CaMKI (CaMKI293) or a kinase-inactive CaMKI293K49A mutant were the generous gifts of Dr. Marinna Picciotto (Yale University, New Haven, CT). CaMKI293 contains a C-terminally truncated version of the CaMKI-encoding gene, truncated to 293. CaMKI293K49A was constructed by changing lysine 49 to alanine in CaMKI293, which negatively affects ATP binding at the catalytic site.(31, 32) For transient transfection, RAW 264.7 cells were seeded in a 24-well plate at 3 × 105 cells/well. After 2 h of adhesion, Mφ were transfected with 1 μg of plasmid CaMKI293 or CaMKI293K49A using the Lipofectamine 2000 reagent accordingto the instructions specified by the manufacturer (Life Technologies, Carlsbad, CA). Following transfection, cells were handled as detailed in the figure legends.
Cellular protein extraction
Total cellular lysate was extracted at 4°C in 500 μL of lysis buffer (20 mM Tris, 137 mM NaCl, 2 mM EDTA, 10% glycerol, 1% Triton X-100, 1 μM sodium orthovanadate, 100 μM dithiothreitol [DTT], 200 μM phenylmethylsulfonyl fluoride [PMSF], 10 μg/mL leupeptin, 0.15 U/mL aprotinin, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 2.5 μg/mL pepstatin A, 1 mM benzamidine, and 40 mM [alpha]-glycerophosphate). Protein concentration was determined using a bicinchoninic acid protein assay (Pierce, Rockford, IL).
Western blots
Total cellular lysate was electrophoresed in a 6%, 10%, 15%, or gradient SDS-PAGE gel (based on protein of interest) and was transferred to a Hybond-enhanced chemiluminescence nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway, NJ). The membrane was blocked for 1 h at room temperature with 5% milk and incubated with primary antibody for 12 h at 4°C. Blots were then incubated in a horseradish peroxidase-conjugated secondary antibody against the primary antibody at room temperature for 1 h. The blot was developed using the SuperSignal chemiluminescent substrate (Pierce, Rockford, IL) and was exposed on KAR-5 film (Eastman Kodak, Rochester, NY). Densitometry was performed by the NIH.image program (National Institutes of Health, Bethesda, MD) to quantitate optical density. All gels were reblotted for total protein to confirm equal loading or to assess knockdown after RNAi.
Cytokine production
IL-10, TNFα, IL-6, CXCL/MIP2 and CXCL1/KC concentrations were quantified using an enzyme immunoassay kit (R&D Systems, Minneapolis, MN) that is based on a coated-well, sandwich enzyme immunoassay.
Immunofluorescence labeling of pulmonary lavage cells
BAL fluid was centrifuged at 300g for 10 minutes and resuspended in RPMI-1640 (BioWhittaker Inc., Walkersville, MD). Cells were then plated on sterilized glass slides and allowed to adhere for 2 hours at 37°C. Cells underwent 3 washes with PBS and were then fixed overnight in 2% paraformaldehyde at 4°C. Following fixation, cells were permeabilized in 0.1% Triton X (Sigma-Aldrich, St. Louis, MO) at 4°C and rehydrated with 3 washes of PBS. Cells were then washed 3 times with 0.5% bovine serum albumin (BSA) in PBS and blocked in 2% BSA for 45 minutes at RT. Cells were then washed 3 times with PBS+0.5% BSA (PBB) and then incubated with mouse anti-LC3B and rabbit anti-ATG7 in PBB for 60 minutes at RT. Cells were then washed 3 times with PBB and incubated with secondary antibody (goat anti-mouse Cy3 (red) or goat anti-rabbit Alexa488 (green) and resuspended in PBB for 60 minutes at RT. Following 3 washes of PBB, a single wash of PBS was used to remove trace amounts of BSA prior to use of Hoescht stain. Cell monolyaers were stained with Hoescht for 30 seconds, cover slips were adhered with gelvatol and slide were placed at 4°C overnight before imaging.
Statistical analysis
Statistical analyses were performed using Stata 11SE software (College Station, TX). Values are expressed as means ± SEM. Groups are compared by ANOVA. A p<0.05 was considered statistically significant.
Results
LPS-induced Autophagy is CaMKIαdependent
Prior studies have reported that LPS induces autophagy in macrophages.(17, 19) A CaMKKβ-dependent mechanism regulating autophagy in response to elevations in cytosolic calcium has been described, and we explored whether the CaMK regulated autophagy in Mφ responding to LPS using the broad CaMK inhibitor KN62.(24, 27) As shown in Figure 1, KN62 inhibited LPS induction of ATG7, LC3B, and ATG5–12 in RAW 264.7 cells. Rapamycin, which induces autophagy through mTOR inhibition, served as a positive control (data not shown).(33) Thus, CaMK inhibition by KN62 suppressed LPS-induced autophagy in Mφ.
Figure 1. CaMK cascade mediates LPS-induced autophagy in macrophages.

RAW 264.7 cells were incubated with the broad CaMK inhibitor KN62 (20uM) for 30 minutes and then exposed to LPS (100ng/ml) for 30 minutes. Total cellular lysate (20μg per lane) was isolated and the induction of ATG7, LC3B, and ATG5–12 assessed by immunoblot (n=4 independent experiments).
These observations prompted us to explore whether CaMKIα, a downstream effector of CaMKKα and CaMKKβ, is involved.(24) As we have previously shown, LPS increased p-Thr177/200CaMKIα that was inhibited by STO609, a specific CaMKK inhibitor, supporting that CaMKIα activation is dependent upon upstream CaMKKα/β activity (Figure 2A).(26) To study CaMKIα we utilized CaMKIα siRNA (CaMKIαsiRNA), which we have previously shown is specific to CaMKIα, and notably, does not alter CaMKKα/β expression.(23) By comparison to Mφ treated with a scrambled, non-target siRNA (NTsiRNA), CaMKIαsiRNA Mφ exhibited attenuated ATG7, LC3B, and ATG5–12 after exposure to LPS (Figure 2B).
Figure 2. CaMKI regulates LPS-induced autophagy in macrophages.

A, Primary peritoneal macrophages were incubated with either media or the highly selective CaMKK inhibitor STO609 (10μM) for 60 minutes and then exposed to LPS (100ng/ml) for the durations indicated. Total cellular lysate (20μg per lane) was isolated and the induction of active p-Thr177/200-CaMKI assessed by immunoblot (n=3 independent experiments). B, RAW 264.7 cells and primary peritoneal macrophages were subjected to RNAi using Non-target (NT) or CaMKIα siRNA. After 72 h, cells were exposed to LPS (100ng/mL), total cellular lysate (20ug per lane) was isolated and the induction of ATG7, LC3B, and ATG5–12 assessed by immunoblot (n=4 independent experiments).
These data suggest that CaMKIα is integral to LPS-induced autophagy in Mφ. Whether CaMKIα is sufficient for LPS-induced autophagy was tested utilizing a constitutively active CaMKIα (CaMKI293) or its kinase defective mutant CaMKI293K49A. As shown in Figure 3, CaMKI293 induced ATG7, LC3B, and ATG5–12 that at 6 hours was 14-, 2-, and 1.7 fold relatively greater than the respective timepoints observed with CaMKI293K49A, suggesting that active CaMKIα is sufficient to induce autophagy.
Figure 3. CaMKIα is sufficient to induce autophagy in macrophages.

RAW 264.7 cells were transfected with either a constitutively active CaMKIα(CaMKI293) or a kinase-deficient mutant (CaMKI293K49A) for the durations indicated. Total cellular lysate (20μg per lane) was isolated and analyzed for the induction of ATG7, LC3B, ATG5–12, CaMKIα, and actin by immunblot (representative blot of 3 independent experiments). Lipo=lipofectamine.
CaMKIα is an AMP kinase kinase and regulates mTOR (TORC1) activity
Substantial evidence supports that the mTOR (mTORC1) pathway functions as a negative regulator of autophagy, and thus inhibiting mTORC1 induces autophagy.(7, 33) AMP kinase (AMPK), by inducing Tuberous Sclerosis Complex (TSC) 1/2-Rheb inhibition of mTOR, activates autophagy.(7, 33) Thus, we explored whether CaMKIα regulates autophagy through AMPK-dependent inhibition of mTORC1. LPS increased p-Thr172AMPK expression in NTsiRNA treated Mφ, and this effect was attenuated in CaMKIαsiRNA treated Mφ, indicating that CaMKIα is upstream of AMPK and regulates its activity following LPS (Figure 4). However, LPS induction of p-Thr172AMPK correlated with increased rather than decreased mTORC1 activity as evidenced by increased p-Ser2448mTOR, p-Thr421Ser424 p70 S6 kinase and p-Ser235/236S6 as shown in NTsiRNA treated Mφ. These events were also attenuated in CaMKIαsiRNA Mφ (Figure 4). Although CaMKIα induced p-Thr172AMPK following LPS stimulation, AMPK phosphorylation correlated with increased, rather than decreased, mTORC1 activity, suggesting that CaMKIα induction of autophagy utilizes an mTORC1-independent mechanism.
Figure 4. CaMKIα is an AMPK kinase.

RAW 264.7 cells were subjected to RNAi using Non-target (NT) or CaMKIα siRNA. After 72 h, cells were exposed to LPS (100ng/mL) for 30 minutes. Total cellular lysate (20μg per lane) was isolated and analyzed for p-AMPK, AMPK, p-mTOR, mTOR, p-p70 S6 kinase, p-S6, and CaMKIα by immunblot (representative blot of 3 independent experiments).
We further explored a CaMKIα-AMPK dependent mechanism regulating autophagy by transfecting Mφ with either active CaMKI293 or inactive CaMKI293K49A in the presence or absence of the AMPK inhibitor, Compound C. By comparison to CaMKI293K49A, CaMKI293 induced p-Thr172AMPK, ATG7, LC3B, and ATG5–12, which were significantly inhibited by Compound C (Figure 5). CaMKI293 also induced mTORC1 signaling, as evidenced by increased p-Thr421Ser424p70S6K and p-Ser235/236S6, which were not notably affected by Compound C (Figure 5). These data suggest that CaMKIα regulates autophagy through an AMPK-dependent mechanism, but mTORC1 suppression does not appear to be involved in the induction of autophagy.
Figure 5. CaMKIα induces autophagy through an AMPK-dependent mechanism.

RAW 264.7 cells were incubated with the AMPK inhibitor Compound C for 30 minutes and then transfected with either a constitutively active CaMKIα (CaMKI293) or a kinase-deficient mutant (CaMKI293K49A) for 8 hours. Total cellular lysate (20μg per lane) was then isolated and analyzed for the expression of ATG7, LC3B, ATG5–12, p-AMPK, AMPK, p-mTOR, mTOR, p-p70 S6 kinase, p-S6, CaMKIα, and actin by immunblot (representative blot of 3 independent experiments).
We explored the possibility that in Mφ responding to LPS, p-CaMKIα mediates autophagy through direct threonine-172 phosphorylation and activation of AMPK. As shown in Figure 6A, CaMKIα and AMPK coimmunoprecipitated after LPS stimulation, suggesting that LPS induces a CaMKIα-AMPK complex, and that CaMKIα phosphorylates AMPK. Indeed, in vitro kinase assay demonstrated that active CaMKIα directly phosphorylates AMPK on threonine 172 (Figure 6B). ATG7, in addition to being dependent upon CaMKIα and AMPK activity, colocalized to this CaMKIα-AMPK complex (Figure 6A). One mechanism by which CaMKIα might regulate autophagy is through ATG7, which is proximal and integral to the induction of LC3B. As shown in Figure 7, RNAi of ATG7 attenuated LPS-induced LC3B (Figure 7). Thus CaMKIα-dependent autophagy, specifically LC3B, appears to involve a CaMKIα-AMPK pathway inducing ATG7.
Figure 6. LPS induces a CaMKIα-AMPK-ATG7 complex.

A, RAW 264.7 macrophages were exposed to LPS (100ng/ml) for the durations indicated at which point total cell lysate was harvested, immunoprecipitated for p-AMPK, subjected to immunoblot analysis (20μg per lane), and probed for CaMKIα, AMPK, or ATG7 (n=3 independent experiments). B, AMPK was isolated by immunoprecipitation from RAW 264.7 cells and then incubated in the presence or absence of active p-CaMKI (25ng) for 10 min at 30°C with the following additions: 10 mM MgCl2, 0.2 mM ATP, 1 mM CaCl2, and 1 μM CaM. Reactions were terminated by boiling in SDS-2-ME dissociation solution, subjected to immunoblot, and probed with anti-p-Thr172 AMPK or AMPK antibody (representative blot of 3 independent experiments).
Figure 7. ATG7 siRNA inhibits LPS-induced LC3B.
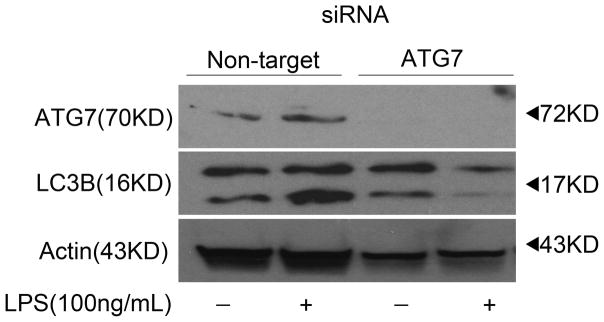
RAW 264.7 macrophages were subject to RNAi of ATG7 or LC3. After 72 hours cells were exposed to LPS (100ng/ml) for 30 minutes, at which point total cell lysate was harvested and subjected to immunoblot analysis (20μg per lane) and probed for ATG7, LC3B, and actin (n=3 independent experiments).
CaMKIα regulates autophagy in vivo during LPS-induced lung inflammation
We explored whether these mechanisms are operant in vivo during sepsis using a murine model of LPS-induced acute lung inflammation. There is difficulty in generating a constitutive CaMKI knockout mouse, potentially because of embryonic lethality (Marina Picciotto, personal communication). Hence, we utilized in vivo RNAi to selectively inhibit CaMKIα as previously demonstrated.(26) As shown in Figure 8, mice receiving CaMKIα siRNA, by comparison to NT siRNA, demonstrated a reduced CaMKIα expression in the lung. Densitometry showed that CaMKIα siRNA reduced CaMKIα siRNA expression by 68% (91 vs. 29; p=0.03). These CaMKIαsiRNA mice exhibited reduced induction of ATG7 and LC3B in the lung tissue, by comparison to NTsiRNA animals, at both 6 hours (data not shown) and 12 hours (Figure 8) after intratracheal administration of LPS. The degree of inhibition of autophagy correlated with the efficacy of siRNA knockdown of CaMKIα. CaMKIαsiRNA mice also exhibited reduced induction of ATG7 (green, Alexa488) and LC3B (red, Cy3) in bronchoalveolar immune cells (Figure 9).
Figure 8. CaMKIα siRNA inhibits pulmonary CaMKIα expression and autophagy in vivo.

C57Bl/6 mice were administered either CaMKIα (CaMKIαsiRNA) or Non-target (NTsiRNA) siRNA (6mg/kg) by hydrodynamic tail vein injection. After 72 hours, CaMKIαsiRNA and NTsiRNA mice were subjected to intratracheal (IT) administration of LPS (1.5mg/kg). After 12 hours, mice were euthanized, the lungs were harvested, and pulmonary CaMKIα, ATG7, LC3B and actin expression assessed by immunoblot (20μg per lane) (representative blot of 3 independent experiments where 5 mice per experimental condition is shown; N=12 mice total per experimental condition for all 3 experiments combined). Densitometry of CaMKIα was performed, adjusted for densitometry of actin control, and then tested by Mann-Whitney.
Figure 9. CaMKIα regulates autophagy in bronchoalveolar immune cells.

CaMKIαsiRNA and NTsiRNA mice were subjected to intratracheal (IT) administration of LPS (1.5mg/kg). After 6 and12 hours, mice were euthanized, bronchoalveolar lavage (BAL) was performed, and BAL immune cells were plated by cytospin. The expression of ATG7 (green, Alexa 488), LC3B (red, Cy3), and Hoechst (blue) staining was determined by immunofluorescence (n=3 independent experiments). 40X magnification.
CaMKIα RNAi alters pulmonary chemokine/cytokine concentration and reduces neutrophil influx during LPS-induced lung inflammation
Our prior study identified a CaMKIα-dependent mechanism mediating systemic TNFα, IL-6, IL-10 and HMGB1 release during intraabdominal CLP sepsis.(26) We explored the biological relevance of these mechanisms in the lungs following LPS-induced inflammation. By comparison to NTsiRNA mice, CaMKIαsiRNA mice exhibited significantly reduced bronchoalveolar lavage (BAL) concentrations of TNFα and IL-6 concentrations 6 hours after LPS (Figure 10). CaMKIαsiRNA mice also exhibited reduced pulmonary concentrations of CXCL1/KC at 6 hours after LPS instillation, though there were no significant reductions in IL-10 or MIP-2 (Figure 10). There were no significant reductions in BAL cytokine or chemokine concentrations at 12 hours (data not shown). There were no significant reductions in systemic concentrations of TNFα, IL-6 or IL-10 at any of the timepoints evaluated (data not shown).
Figure 10. CaMKIα regulates bronchoalveolar cytokine and chemokine concentrations during LPS-induced lung inflammation.

CaMKIαsiRNA and NTsiRNA mice were subjected to intratracheal (IT) administration of LPS (1.5mg/kg). After 6 hours, mice were euthanized, the lungs were lavaged (BAL), and BAL fluid was analyzed for cytokine and chemokines by ELISA. Data represent mean ± SEM for each group. Statistical significance was determined by Mann-Whitney non-parametric analysis (3 independent experiments with 4 mice per experimental condition; N=12 mice total per experimental condition for all 3 experiments combined).
In the context of these alterations in pulmonary cytokine/chemokine concentrations, we assessed the influence of CaMKIα inhibition on neutrophil recruitment. As shown in Figure 11, CaMKIαsiRNA mice exhibited reduced neutrophil recruitment after LPS (Figure 11A), and myeloperoxidase activity was attenuated throughout the entire period of observation (Figure 11B). This correlated with a trend toward decreased lung injury as demonstrated by attenuated alveolar protein concentration at 6 h but not at 12 h (Figure 11C).
Figure 11. CaMKIα regulates neutrophil recruitment in LPS-induced lung inflammation.

CaMKIαsiRNA and NTsiRNA mice were subjected to intratracheal (IT) administration of LPS (1.5mg/kg). After 6 and12 hours, mice were euthanized, the lungs were lavaged (BAL), and BAL fluid was analyzed for neutrophil count, myeloperoxidase (MPO) activity, and protein concentration (3 independent experiments with 4 mice per experimental condition; N=12 mice total per experimental condition for all 3 experiments combined).
VPS34 RNAi recapitulates the results of CaMKIαRNAi
We hypothesized that the effect of CaMKIα siRNA in reducing lung neutrophil accumulation was mediated by inhibition of autophagy. To corroborate these findings, we utilized a second, independent method of inhibiting autophagy by inhibiting the class III PI3kinase VPS34, which has been shown to regulate autophagy.(5) Immunoblotting revealed a reduction in both LC3B and ATG7 in whole lung lysate of VPS34siRNA mice by comparison to NTsiRNA mice (Figure 12A). The degree of inhibition of autophagy correlated with the efficacy of siRNA knockdown of VPS34. Although there were no significant differences in BAL cytokine concentrations 12h following LPS (data not shown), VPS34siRNA mice exhibited reduced bronchoalveolar recruitment of neutrophils and MPO activity (Figure 12B) similar to the finding of CaMKIαsiRNA mice.
Figure 12. Inhibition of VPS34 attenuates autophagy and neutrophil recruitment in LPS-induced lung inflammation.

A, VPS34siRNA and NTsiRNA mice were subjected to intratracheal (IT) administration of LPS (1.5mg/kg). After 12 hours, mice were euthanized, the lungs were harvested, total cell lysate was isolated, subjected to immunoblot (20μg per lane), and probed for ATG7, LC3B, VPS34, and actin (representative blot of 2 independent experiments where 6 mice per experimental condition is shown; N=11 mice total per experimental condition for all 2 experiments combined). B, VPS34siRNA and NTsiRNA mice were subjected to intratracheal (IT) administration of LPS (1.5mg/kg). After 12 hours mice were euthanized, the lungs were lavaged (BAL), and BAL fluid was analyzed for neutrophil count, myeloperoxidase (MPO) activity, and protein concentration. Data represent mean ± SEM for each group. Statistical significance was determined by Mann-Whitney non-parametric analysis (2 independent experiments with 4 mice per experimental conditions; N=8 mice total per experimental condition for all 2 experiments combined).
Discussion
Autophagy is an evolutionarily conserved lysosomal mechanism that enables cells to conserve and maintain cellular biomass quality and quantity by targeting damaged or unused proteins and even organelles for autophagosomal degradation.(9) Though essential for tissue homeostasis, the mechanisms regulating this complex process, and the ramifications of any defects, are just being elucidated. First described in the context of nutrient deprivation and under the regulation of the energy rheostats mTOR and AMPK, autophagy has recently been described as an important effector arm of immune cells responding to exogenous (i.e. PAMP: LPS)(19, 34) and endogenous (DAMP: HMGB1)(35) stimuli.(7, 15, 33, 34, 36–38) Here we describe a novel CaMKIα-AMP kinase pathway rapidly activated in Mφ exposed to LPS and regulating an early autophagic response. CaMKIα concomitantly and positively regulates AMPK and TORC1, suggesting that CaMKIα invokes an AMPK-dependent, yet mTOR-independent, mechanism in the regulation of autophagy. The mechanisms identified appear to be operant in vivo during LPS-induced lung inflammation.
These data complement prior investigations that describe mechanisms by which members of the family of multifunctional CaMK regulate autophagy. HØyer-Hansen and colleagues, in in vitro experiments using carcinoma cell lines and NIH3T3 fibroblasts, observed that calcium-mobilizing agents (e.g. vitamin D, thapsigargin) inhibited the activity of mTOR, a negative regulator of macroautophagy, and induced the accumulation of autophagosomes in a Beclin-1 and ATG-7 manner.(27) This process was mediated by CaMKKβ and AMP kinase.(27) Other studies have identified a CaMKKα/β-AMPK pathway in which CaMKKα/β functions as an indirect AMPK kinase.(7, 39) Collectively, these data create a compelling paradigm in which a CaMKKα/β-AMPK pathway mediates autophagy in cells responding to calcium mobilization. In Mφ, LPS induces endoplasmic reticulum stress and calcium mobilization that we have shown leads to the activation of all members of the CaMK cascade.(23, 26, 40, 41) Thus, we hypothesized that CaMKIα, a downstream and predominantly cytoplasmic effector of CaMKKα/β, may also be involved in Mφ autophagy. We observed that in Mφ, LPS activates CaMKIα as an essential event for the subsequent induction of autophagy. Similar to CaMKKβ, CaMKIα also regulates AMPK, and a direct interaction (i.e. co-immunoprecipitation) and Thr172 phosphorylation of AMPK by CaMKIα was observed. Importantly, our methods do not alter CaMKK expression(23), suggesting that our observations are specific to CaMKIα. We have previously shown that LPS-induced CaMKIα activity in Mφ is CaMKKα/β-dependent; thus, the most likely mechanism is a CaMKKα/β-CaMKIα-AMPK pathway.
AMPK-mediated inhibition of mTOR has been fairly well established, and direct evidence is accumulating that this mechanism can induce autophagy in a variety of cells and experimental models.(7, 33, 42, 43) Our data suggest, however, that CaMKIα-AMPK functions through mechanisms other than mTOR inhibition, at least in Mφ responding to LPS. Though RNAi of CaMKIα correlated with reduced p-AMPK and autophagy, we also observed reduced phosphorylation of ribosomal protein S6 kinase (p-Thr412Ser424p70S6K) and ribosomal S6 (p-Ser235/236S6), the downstream targets of TORC1. These observations contrast with the results of previous studies noting that active AMPK, through phosphorylation and increased GTPase activity of Rheb, reduced TORC1 activity, and thereby increased autophagy.(7, 33, 42, 43) LPS has been shown to induce both the mTOR/p70S6K/S6 (TORC1) pathway(44, 45) and autophagy(16, 19, 34) in Mφ, which raises doubt as to whether the link between AMPK-dependent mTOR inhibition and autophagy is preserved in innate immune cells responding to septic insult. Though active CaMKI293 induced AMPK-dependent LC3B, and CaMKIα siRNA inhibited LPS-induced autophagy and p-AMPK, additional CaMKIα mechanisms may be induced during LPS exposure that regulate autophagy. In Mφ, LPS induction of mTOR is mediated in part by PI3 kinases and AKT.(44, 45) Class III PI3 kinases (i.e. hVPS34) initiate autophagy through a complex involving hVPS34/Beclin1 that initiates ATG7.(9) Recent data suggest hVPS34 can also regulate TORC1/p70S6K signaling(9, 13, 46) Hence, a general regulation of PI3 kinases by CaMKIα could account for all of our observations. The mechanisms by which the CaMKIα-AMPK pathway regulates autophagy independent of TORC1 in Mφ exposed to LPS remains to be determined, though our data suggest that it may directly regulate the induction of ATG7. Indeed, inhibition of ATG7 reduced LPS-induced ATG5–12 and LC3B conjugation.
In addition to being operant in vitro, our studies suggest that these mechanisms are similarly induced in vivo in the early response to sepsis, and regulate bronchoalveolar cytokine/chemokine production and neutrophil accumulation. Interestingly, LC3B induction indirectly correlated with the degree of CaMKIα inhibition, further supporting a causal relationship between CaMKIα and autophagy. We observed similar attenuation in airspace neutrophil accumulation when autophagy was inhibited with siRNA targeting VPS34. These data corroborate CaMKIα-dependent autophagy as the mechanism by which CaMKIα regulates pulmonary inflammation. What awaits further investigation is the role this autophagic response serves at the level of the cell, organ and host in appropriately responding to and recovering from the septic insult.
In summary, CaMKIα mediates LPS-induced autophagy in the Mφ through mechanisms involving the regulation of AMPK, independent of TORC1 activity. These CaMKIα-dependent processes appear to be operant in vivo during the inflammatory response to sepsis, lending support to the hypothesis that CaMKIα signaling mediates the autophagic response to sepsis. The observation that CaMKIα regulates autophagy in the lungs, underscores the need to study these mechanisms in the context of organ-specific injury and dysfunction in response to septic insult. Furthermore, additional studies are needed to translate these cellular events to organ physiology and the host response, in attempts to identify the clinical utility of CaMK and autophagic modulation during inflammatory states.
Acknowledgments
We would like to thank Dr. Marina Picciotto for the generous gift of the CaMKI plasmids.
Footnotes
This work supported by National Institutes of Health grants R01 GM082852 (MRR) and R01 HL086884 (JSL).
References
- 1.Blank R, Napolitano LM. Epidemiology of ARDS and ALI. Crit Care Clin. 2011;27:439–458. doi: 10.1016/j.ccc.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Phua J, Badia JR, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, Scales DC, Stather DR, Li A, Jones A, Gattas DJ, Hallett D, Tomlinson G, Stewart TE, Ferguson ND. Has mortality from acute respiratory distress syndrome decreased over time?: A systematic review. American journal of respiratory and critical care medicine. 2009;179:220–227. doi: 10.1164/rccm.200805-722OC. [DOI] [PubMed] [Google Scholar]
- 3.Frutos-Vivar F, Ferguson ND, Esteban A. Epidemiology of acute lung injury and acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27:327–336. doi: 10.1055/s-2006-948287. [DOI] [PubMed] [Google Scholar]
- 4.Luce JM. Acute lung injury and the acute respiratory distress syndrome. Critical care medicine. 1998;26:369–376. doi: 10.1097/00003246-199802000-00043. [DOI] [PubMed] [Google Scholar]
- 5.Lang JD, McArdle PJ, O’Reilly PJ, Matalon S. Oxidant-antioxidant balance in acute lung injury. Chest. 2002;122:314S–320S. doi: 10.1378/chest.122.6_suppl.314s. [DOI] [PubMed] [Google Scholar]
- 6.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoyer-Hansen M, Jaattela M. AMP-activated protein kinase: a universal regulator of autophagy? Autophagy. 2007;3:381–383. doi: 10.4161/auto.4240. [DOI] [PubMed] [Google Scholar]
- 8.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self- eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9:65–76. [PMC free article] [PubMed] [Google Scholar]
- 12.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swanson MS, Molofsky AB. Autophagy and inflammatory cell death, partners of innate immunity. Autophagy. 2005;1:174–176. doi: 10.4161/auto.1.3.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delgado M, Singh S, De Haro S, Master S, Ponpuak M, Dinkins C, Ornatowski W, Vergne I, Deretic V. Autophagy and pattern recognition receptors in innate immunity. Immunol Rev. 2009;227:189–202. doi: 10.1111/j.1600-065X.2008.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ. 2009;16:976–983. doi: 10.1038/cdd.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waltz P, Carchman EH, Young AC, Rao J, Rosengart MR, Kaczorowski D, Zuckerbraun BS. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy. 2011;7:315–320. doi: 10.4161/auto.7.3.14044. [DOI] [PubMed] [Google Scholar]
- 18.Deretic V. Autophagy in innate and adaptive immunity. Trends in immunology. 2005;26:523–528. doi: 10.1016/j.it.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 21.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 22.Rosengart MR, Arbabi S, Garcia I, Maier RV. Interactions of calcium/calmodulin-dependent protein kinases (CaMK) and extracellular-regulated kinase (ERK) in monocyte adherence and TNFalpha production. Shock. 2000;13:183–189. doi: 10.1097/00024382-200003000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Wheeler D, Tang Y, Guo L, Shapiro RA, Ribar TJ, Means AR, Billiar TR, Angus DC, Rosengart MR. Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J Immunol. 2008;181:5015–5023. doi: 10.4049/jimmunol.181.7.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Means AR. The Year in Basic Science: calmodulin kinase cascades. Mol Endocrinol. 2008;22:2759–2765. doi: 10.1210/me.2008-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Illario M, Giardino-Torchia ML, Sankar U, Ribar TJ, Galgani M, Vitiello L, Masci AM, Bertani FR, Ciaglia E, Astone D, Maulucci G, Cavallo A, Vitale M, Cimini V, Pastore L, Means AR, Rossi G, Racioppi L. Calmodulin-dependent kinase IV links Toll-like receptor 4 signaling with survival pathway of activated dendritic cells. Blood. 2008;111:723–731. doi: 10.1182/blood-2007-05-091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Guo L, Collage RD, Stripay JL, Tsung A, Lee JS, Rosengart MR. Calcium/calmodulin-dependent protein kinase (CaMK) I{alpha} mediates the macrophage inflammatory response to sepsis. J Leukoc Biol. 2011;90:249–61. doi: 10.1189/jlb.0510286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Fogarty S, Hawley SA, Green KA, Saner N, Mustard KJ, Hardie DG. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem J. 2010;426:109–118. doi: 10.1042/BJ20091372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- 30.Schmitt JM, Wayman GA, Nozaki N, Soderling TR. Calcium activation of ERK mediated by calmodulin kinase I. J Biol Chem. 2004;279:24064–24072. doi: 10.1074/jbc.M401501200. [DOI] [PubMed] [Google Scholar]
- 31.Xie J, Black DL. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature. 2001;410:936–939. doi: 10.1038/35073593. [DOI] [PubMed] [Google Scholar]
- 32.Chatila T, Anderson KA, Ho N, Means AR. A unique phosphorylation-dependent mechanism for the activation of Ca2+/calmodulin-dependent protein kinase type IV/GR. J Biol Chem. 1996;271:21542–21548. doi: 10.1074/jbc.271.35.21542. [DOI] [PubMed] [Google Scholar]
- 33.Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–865. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- 34.Xu Y, Liu XD, Gong X, Eissa NT. Signaling pathway of autophagy associated with innate immunity. Autophagy. 2008;4:110–112. doi: 10.4161/auto.5225. [DOI] [PubMed] [Google Scholar]
- 35.Ghavami S, Eshragi M, Ande SR, Chazin WJ, Klonisch T, Halayko AJ, McNeill KD, Hashemi M, Kerkhoff C, Los M. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 2010;20:314–331. doi: 10.1038/cr.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klionsky DJ. Autophagy. Curr Biol. 2005;15:R282–283. doi: 10.1016/j.cub.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 37.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, Eissa NT. Autophagy in innate and adaptive immunity. Proc Am Thorac Soc. 2010;7:22–28. doi: 10.1513/pats.200909-103JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 40.Gotoh T, Oyadomari S, Mori K, Mori M. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J Biol Chem. 2002;277:12343–12350. doi: 10.1074/jbc.M107988200. [DOI] [PubMed] [Google Scholar]
- 41.Nakayama Y, Endo M, Tsukano H, Mori M, Oike Y, Gotoh T. Molecular Mechanisms of the LPS-induced Non-apoptotic ER Stress-CHOP Pathway. J Biochem. 2010;147:471–83. doi: 10.1093/jb/mvp189. [DOI] [PubMed] [Google Scholar]
- 42.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 43.Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P. AMPK signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285:9100–13. doi: 10.1074/jbc.M109.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinstein SL, Finn AJ, Dave SH, Meng F, Lowell CA, Sanghera JS, DeFranco AL. Phosphatidylinositol 3-kinase and mTOR mediate lipopolysaccharide-stimulated nitric oxide production in macrophages via interferon-beta. J Leukoc Biol. 2000;67:405–414. doi: 10.1002/jlb.67.3.405. [DOI] [PubMed] [Google Scholar]
- 45.Salh B, Wagey R, Marotta A, Tao JS, Pelech S. Activation of phosphatidylinositol 3-kinase, protein kinase B, and p70 S6 kinases in lipopolysaccharide-stimulated Raw 264.7 cells: differential effects of rapamycin, Ly294002, and wortmannin on nitric oxide production. J Immunol. 1998;161:6947–6954. [PubMed] [Google Scholar]
- 46.Nobukuni T, Kozma SC, Thomas G. hvps34, an ancient player, enters a growing game: mTOR Complex1/S6K1 signaling. Curr Opin Cell Biol. 2007;19:135–141. doi: 10.1016/j.ceb.2007.02.019. [DOI] [PubMed] [Google Scholar]