

Abstract
The major human pathogen Staphylococcus aureus has very efficient strategies to subvert the human immune system. Virulence of the emerging community-associated methicillin-resistant S. aureus (CA-MRSA) depends on phenol-soluble modulin (PSM) peptide toxins, which are known to attract and lyse neutrophils. However, their influences on other immune cells remain elusive. Here, we analyzed the impact of PSMs on dendritic cells (DCs) playing an essential role in linking innate and adaptive immunity. In human neutrophils, PSMs exert their function by binding to the formyl peptide receptor (FPR) 2. We show that mouse DCs express the FPR2 homologue mFPR2 as well as its paralog mFPR1 and that PSMs are chemoattractants for DCs at non-cytotoxic concentrations. PSMs reduced clathrin-mediated endocytosis and inhibited TLR2 ligand-induced secretion of the proinflammatory cytokines TNF, IL-12 and IL-6 while inducing IL-10 secretion by DCs. As a consequence, treatment with PSMs impaired the capacity of DCs to induce activation and proliferation of CD4+ T cells, characterized by reduced Th1 but increased frequency of FOXP3+ regulatory T cells (Tregs). These Tregs secreted high amounts of IL-10 and their suppression capacity was dependent on IL-10 and TGF-β. Interestingly, the induction of tolerogenic DCs by PSMs appeared to be independent of mFPRs as shown by experiments with mice lacking mFPR2 (mFPR2−/−) and the cognate G protein (p110γ−/−). Thus, PSMs from highly virulent pathogens affect DC functions thereby modulating the adaptive immune response and probably increasing the tolerance towards the pathogen.
Introduction
Staphylococcus aureus is a major cause of invasive infectious diseases ranging from skin and soft tissue infections to severe systemic infections such as endocarditis or sepsis (1). The increasing prevalence of methicillin-resistant S. aureus (MRSA) strains, being highly resistant to antibiotic treatment, has become a significant public health threat (2). While MRSA strains used to be restricted to hospital settings, there was a dramatic spread in the last decade of new community-associated (CA-) MRSA strains such as USA300, causing severe infections also in healthy individuals (3). Several virulence factors contribute to the pathogenicity of CA-MRSA including α-hemolysin, Panton-Valentine leukocidin, and phenol-soluble modulin (PSM) peptides (4-7). PSMs play essential roles in the virulence of CA-MRSA as they are strongly expressed in CA-MRSA strains and knockout of these peptides leads to loss of CA-MRSA virulence (6). This group of virulence factors consists of four PSMα peptides (PSMα1-4), two PSMβ peptides (PSMβ1-2), and the long-known δ-toxin, which all share an α-helical, amphipathic structure but limited sequence similarity (6). PSMs act as chemoattractants for neutrophils at nanomolar concentrations leading to a massive influx of these cells to the infection site. However, at micromolar concentrations these peptides cause potent lysis of neutrophils through the destruction of the cell membrane, which abrogates the neutrophil capacity to clear the infection and provides the bacterium with nutrients (5, 6). While cell lysis seems to be a receptor-independent mechanism, chemotaxis of neutrophils is induced by binding to the human formyl peptide receptor (FPR) 2 (5). In the mouse FPR-related receptors are expressed on neutrophils, dendritic cells (DCs) and microglial cells (8). The mouse FPR family consists of more members than the human family: mFPR1 is the direct homologue of human FPR1 whereas mFPR2 and fpr-rs1 seem to be orthologs of human FPR2. In addition, there are several additional mouse receptors of this family without direct human counterparts.
The extraordinary virulence of S. aureus depends on its multiple ways of compromising host defense mechanisms. S. aureus is known to secrete several immune-modulatory proteins interfering with the innate immune system such as inhibitors of the complement cascade (9), of leukocyte chemotaxis and extravasation (10), and of the bactericidal activity of defensin (11) to name just a few. However, while S. aureus modulation of innate immunity has been explored to some extent it has remained largely unclear how these bacteria interfere with human adaptive immunity. Recent studies strongly suggest that such mechanisms contribute to S. aureus virulence thereby limiting the efficacy of antibodies, T cells, and vaccines (12, 13). If and how S. aureus modulates the function of antigen-presenting cells has hardly been investigated.
DCs are unique antigen-presenting cells linking the innate and adaptive immunity. In their immature state DCs are characterized by efficient antigen uptake in the periphery. After stimulation of pattern recognition receptors (PRRs) e.g. toll-like receptors (TLRs) they undergo maturation, characterized by antigen processing and presentation on MHCII molecules, up-regulation of co-stimulatory molecules as well as cytokine secretion. Mature DCs enter the lymphatic organs where they efficiently activate T cells and thereby induce antigen-specific T-cell responses (14, 15).
Various T helper cell subsets play important roles in the immune response against S. aureus infections (16-20). In a mouse infection model it has been shown that IFN-γ is important for survival of S. aureus-induced sepsis by activating macrophages and neutrophils leading to enhanced elimination of the pathogen (20). Furthermore, IFN-γ produced by CD4+ T cells induces the secretion of several CXC chemokines, leading to the recruitment of neutrophils to the site of infection (18). In addition, IL-17 secreted by Th17- and γδ-T cells further activates the recruitment of neutrophils and induces the secretion of antimicrobial peptides by keratinocytes upon S. aureus infection of the skin (16, 19). Vaccination with the recombinant N terminus of the candidal Als3p adhesin with aluminum hydroxide led to the induction of Th1 and Th17 cells and thereby protected against systemic S. aureus infection (17).
While the influence of PSMs on neutrophils has been well described, their impact on DC functions and the potential consequences for T-cell activation are completely unknown. In the present study, we show that PSMs modulate the capacity of DCs to respond to TLR2 ligands leading to a tolerogenic phenotype. This is characterized by inhibited pro-inflammatory cytokine but increased IL-10 secretion and reduced antigen uptake. As a consequence, PSM-treated DCs had a reduced capability to activate T cells and specifically induced Tregs while inhibiting Th1 differentiation. Although PSMs bound to mFPR2-expressing mouse DCs mFPR2 did not seem to play an important role in the DC-modulatory capacity of PSMs. Thus we propose that in addition to their effects on neutrophils PSM peptides subvert the adaptive immunity by modulating DCs leading to immune evasion.
Materials and Methods
Mice
Female C57BL/6JolaHsd mice were purchased from Janvier (St BerthevinCedex, France). OT-II (21), TLR2−/−(22), , FPR2−/− (23) and p110γ−/− mice (24) with a genetic C57BL/6 background were bred in the animal facilities of the University Clinic of Tübingen and the University of Tübingen. FOXP3-eGFP mice (25) were generously provided by G.J. Hämmerling (DKFZ, Heidelberg, Germany). All mice were held under specific pathogen-free conditions, were provided food and water ad libitum, and used for experiments were between 6–10 weeks of age Animal experiments were performed in strict accordance with the German regulations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). The protocol was approved by the Regierungspräsidium Tübingen (Anzeige 1.12.2010 and 12.9.2011).
Reagents and bacteria
Formylated PSM peptides (PSMα1, PSMα2, PSMα3, PSMα4, and δ-toxin) with the recently published sequences (6), a formylated control peptide with the reversed sequence of amino acids from PSMα4 (sequence: KAFIDIIAKIIKIITGVIAM), M M K 1 ( sequence: LESIFRSLLFRVM-NH2) and OVA323–339 peptide (sequence: ISQAVHAAHAEINEAGR) were synthesized at the Interfaculty Institute of Cell Biology, Department of Immunology, Tübingen.
BM-DCs were treated with S. aureus cell lysates containing lipopeptides, the major staphylococcal TLR2 ligands. S. aureus cell lysates were prepared from a protein A-deficient SA113 (26) mutant strain as the S. aureus protein A turned out to disturb subsequent ELISA assays (data not shown).
The S. aureus USA300 strain and the mutant strains deficient for PSMs (δα, δβ, δδ, and δαβδ) have recently been described in detail (6, 27). Bacterial culture supernatants were obtained by centrifugation of either overnight cultures in tryptic soy broth (TSB) or culture for 9 h in DMEM medium (cytokine assay) and filtered through 0.22 μm pore size filters.
Generation of bone marrow-derived DCs (BM-DCs)
RPMI-1640 medium (Biochrom) supplemented with 10% fetal calf serum (FBS; Sigma-Adrich), 2 mM glutamine (Biochrom), 100 U/ml penicillin/streptomycin (Biochrom), 50 μM 2-mercaptoethanol (Roth), 1mM sodium pyruvate (Biochrom) and 1× non essential amino acids (Biochrom) was used in all cell culture experiments. BM-DCs were prepared using granulocyte–macrophage colony-stimulating factor (GM-CSF) as previously described (28, 29). Briefly, 2 × 106 bone marrow cells, flushed from the femurs and tibias of C57BL/6, TLR2−/−, FPR2−/− and p110γ−/− mice, were seeded in 100-mm dishes in 10 ml medium containing 200 U/ml GM-CSF. After 3 days, an additional 10 ml of fresh medium containing 200 U/ml GM-CSF was added to the cultures. On day 6 and day 8, half of the culture supernatant was collected and centrifuged, and the resultant cell pellet was resuspended in 10 ml of fresh medium containing 200 U/ml GM-CSF and given back to the original plate. At day 7-9, the slightly attached cells were used for the experiments described in this report.
Chemotaxis of BM-DCs
Chemotaxis of BM-DCs towards PSMα3 was determined by using fluorescently labeled BM-DCs that migrated through a membrane fitted into an insert of a 24-well microtiter plate transwell system (Costar) containing a prewetted 5-μm-pore-size polycarbonate filter as described recently (30, 31). Briefly, 1 × 106 BM-DCs per ml were labeled with 1.1 μM BCECF-AM (acetoxymeth y l e s t e r o f b i s-2-carboxyethyl-5-[and-6]-carboxyfluorescein; Molecular Probes, Invitrogen) for 20 min at room temperature, washed, and resuspended in HBSS containing 0.05% HSA (HBSS-HSA). The upper compartment of the transwell system was filled with 100 μl of labeled BM-DCs and placed into a well containing 600 μl HBSS-HSA with different concentrations of PSMα3 or diluted S. aureus culture filtrates as indicated. After incubation at 37°C under 5% CO2 for 100 min, the inserts were removed and the fluorescence of the wells was read in a fluorescence-reader with excitation and emission filters of 485 nm and 530 nm respectively. The fluorescence measured was used to monitor migration after subtracting the background migration observed when only buffer was added to the lower compartment.
Preparation of splenocytes
Splenocytes were prepared as previously described (32). Briefly, the spleen was aseptically removed, cut into small pieces and then digested for 30 min at 37 °C in 2 ml modified RPMI 1640 + 2% FCS medium containing collagenase (1 mg/ml; type IV; Sigma-Aldrich) and DNase I (150 mg/ml, Roche). To disrupt DC-T cell complexes, EDTA (0.1 M (pH 7.2)) was added, and mixing continued for 5 min. Single cell suspensions were made by pipetting the digested organs. Undigested fibrous material was removed by filtration and erythrocytes were lysed with lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 2 mM NaEDTA).
To enrich DCs and neutrophils prior to cell sorting CD19-expressing cells were depleted from splenic single cell suspensions by MACS technology using CD19 magnetic beads (Miltenyi Biotec) following the manufacturer’s protocol. DCs (CD11chiMHC II+) and neutrophils (Gr-1+CD11bhi) were sorted on a FACS Aria cell sorter (BD Biosciences) and reanalyzed on a Canto-II flow cytometer.
RNA isolation, reverse transcription-PCR and quantitative PCR
Total RNA was isolated from BM-DCs or sorted splenic DCs and neutrophils using the High Pure RNA Tissue Kit (Roche).
Quantitative PCR was performed with the kit PowerSYBR Green RNA-to-Ct 1-Step Kit (Applied Biosystems). QuantiTect Primer Assay (Qiagen) was used for mouse FPR1, FPR2 und GAPDH. qPCR was performed on AbiPrism 7500 Fast Real-Time with the following settings: Reverse transcription at 48 °C for 30 min, polymerase activation at 98 °C for 10 min followed by 40 cycles of primer annealing and DNA extension at 60 °C for 1 min and denaturation at 95 °C for 15 s. The samples were normalized to GAPDH.
Flow cytometry
FACS buffer (PBS containing 1% FBS (Sigma-Aldrich) and 0.09% NaN3 (Sigma-Aldrich)) was used for all incubations and washing steps. Cells were stained with FITC, PE, APC, PE-Cy7, APC-efluor780 or efluor450 conjugates of antibodies against CD11c (N418), Gr-1 (RB6-8C5), CD4 (RM4-5), CD25 (PC61), CD80 (16-10A1), CD40 (HM40-3), MHC II (M5/114.15.2), CD11b (M1/70) or TLR4/MD-2 (MTS510) for 20 min at 4°C. To exclude dead cells, 7-aminoactinomycin D (7-AAD; Sigma-Aldrich) or aqua life dead (Invitrogen) was used.
To detect intracellular production of IFN-γ a n d I L-17 cells were fixed with 1% paraformaldehyde (Sigma-Aldrich) in PBS, permeabilized with 0.1% saponin (Sigma-Aldrich) and 0.5% BSA (Sigma-Aldrich) in PBS, and stained for intracellular cytokines with anti-IFN-γ (XMG1.2) and anti-IL-17 (TC11-18H10) for 15 min at 4°C. For staining with anti-FOXP3 antibody (FJK-16s) the FOXP3 Staining Buffer Set (eBioscience) was used. Samples were acquired for analysis using a Canto-II flow cytometer (BD Biosciences) with DIVA software (BD Biosciences) and were further analyzed using FlowJo 7.6 software (TreeStarInc).
For BM-DC surface marker analysis, BM-DCs (5 × 105) were seeded in 48-well plates and stimulated with 3 μg/ml S. aureus cell lysate and PSMα3 (10 μM) for 6h. For PSMα3 binding analysis 5 × 105 BM-DCs or 2 × 106 splenocytes were incubated with TAMRA-marked PSMα3 (10 μM) for 30 min at 37°C in complete medium. The cells were then stained with fluorescently labeled antibodies as described above and analyzed by flow cytometry.
Measurement of antigen uptake by flow cytometry
BM-DCs (5 × 105) prestimulated for 24h with 3 μg/ml S. aureus cell lysate and PSMα3 as indicated were incubated in medium with OVA-AlexaFluor647 (10 μg/ml) or lucifer yellow (2 mg/ml; both from Molecular Probes, Invitrogen) for 30 min or 10 min at 37°C, respectively. Unspecific binding was assessed by incubation of the DCs with the markers on ice. For phagocytosis BM-DCs were incubated with GFP-expressing Yersinia enterocolitica WA-314 (serotype 0:8) (33), kindly provided by Jürgen Heesemann, at a ratio of one bacteria per DC for 30 min at 37°C. Cells were washed three times in ice-cold phosphate-buffered saline containing 2% FCS. Subsequently, the BM-DCs were labeled with fluorescent antibodies and analyzed by flow cytometry.
For measurement of LPS-induced uptake of TLR4, BM-DCs were incubated with or without PSMα3 1 h prior to stimulation with LPS (100 ng/ml) for 30 min at 37°C. The TLR4 surface expression was determined by flow cytometry as described above.
Cytokine production by BM-DCs
BM-DCs (5 × 105) were seeded in 48-well plates and stimulated with 3 μg/ml S. aureus cell lysate. PSM peptides or culture supernatants of USA300 and its knockout mutant δαβδ were added in different concentrations as indicated and incubated for 6 h (TNF analysis) or 24 h (IL-6, IL-12 and IL-10 analysis). Cytokines in the supernatants were analyzed by sandwich ELISA according to the manufacturer’s instructions (TNF, IL-6 and IL-10 ELISAs from BD Bioscience, IL-12p70 ELISA from Biolegend).
In vitro T-cell stimulation
BM-DCs (5 × 104) were seeded in 96-well U-bottom plates and stimulated with 3 μg/ml S. aureus cell lysate. Simultaneously, PSM peptides were added in different concentrations as indicated and incubated for 24 h. CD4+ T cells (2 × 105) from the spleen of C57BL/6 mice were purified by CD4 negative selection using MACS beads (Miltenyi Biotech) and added to the BM-DCs. After 72 h T cells were analyzed for CD25 and FOXP3 expression by flow cytometry. T-cell proliferation was determined by incorporation of 3[H]-thymidine during the last 18 h of the culture.
For T-cell priming analysis of Th1 cells 1 × 104 BM-DCs were stimulated with S. aureus cell lysate in the presence or absence of PSMα3 for 24 h, then splenic CD4+ T cells from OT-II mice, purified as described above, and OVA peptide (aa 323-339; 25 ng/ml) were added. For analysis in a Th17 favoring condition 1 × 104 BM-DCs were co-cultured with 5 × 105 CD4+ T cells from OT-II mice in the presence of IL-6 (20 ng/ml; eBioscience), TGF-β (2 ng/ml; eBioscience), anti-IFN-γ (2 ng/ml; XMG1.1; e-Bioscience), anti-IL-4 (2 ng/ml; 11B11; eBioscience), OVA peptide (0,3 μg/ml) with or without PSMα3. In both settings the cells were co-cultured for 4 days. During the last 5 h of co-culture T cells were re-stimulated with OVA peptide (1 μg/ml) and anti-CD3 antibody (1 μg/ml; 145-2C12; eBioscience) together with Monensin (eBioscience). The cells were analyzed for CD25 expression, IFN-γ, and IL-17 expression by flow cytometry.
Treg suppression assay
BM-DCs (5 × 104) were seeded in 96-well U-bottom plates and stimulated with 3 μg/ml S. aureus cell lysate with or without PSMα3 (6 μM) for 24 h. 2 × 105 CD4+ T cells from the spleen of FOXP3-eGFP mice were purified as described above and added to the BM-DCs. 72 h after co-culture cells were stained with anti-CD4 (RM4-5; eBioscience) antibody and the CD4+FOXP3+ Tregs were purified by FACS sorting according to the expression of CD3, CD4 and eGFP. In addition, CD3+CD4+FOXP3− from the spleen of FOXP3-eGFP mice were purified by FACS sorting and used as Teff cells. The CD3+CD4+FOXP3+ from a naive mouse were used as control Treg cells. The Teff cells were labeled with 10 μM efluor670 Cell Proliferation Dye (eBioscience) for 10 min at 37°C. Teff cells (5 × 104) were cultured for 72 h in U-bottom 96 well plates with irradiated splenocytes from C57BL/6 mice, anti-CD3 antibody (1 μg/ml; 145-2C12; eBioscience) and the indicated numbers of the sorted Treg cells. Proliferation of T cells was measured by flow cytometry and analyzed with the proliferation platform of flowjo software 7.6. The division index is depicted in the histograms and is defined as the average number of cell divisions that a cell in the original population has undergone. Neutralizing antibodies against IL-10 (10 μg/ml; JES5-16E3; eBioscience) and TGF-β (10 μg/ml; 1D11; R&D) were added as indicated. TGF-β and IL-10 secretion by Treg cells after BM-DC coculture (1 × 105 per well in 250 μl medium) was assessed by ELISA (eBioscience) after further culture for 72h in an anti-CD3-coated 96-well flat-bottom plate (0.5 μg/ml anti-CD3).
Statistical analysis
Statistical analysis was performed with the GraphPadPrism 5.0 software (GraphPad, San Diego, CA) using one-way ANOVA with Bonferroni post-test or the unpaired two-tailed Student’s t-test as indicated. The differences were considered as statistical significant if p<0,05 (*), p<0,005 (**) or p<0,001 (***).
Results
mFPR expression by mouse DCs
As PSM peptides exert their activity toward human neutrophils by binding with high affinity to FPR2 and a FPR2-specific inhibitor blocked their capacity to activate mouse neutrophils (5), the members of the FPR family are also likely targets for PSM peptides on mouse DCs. Analysis of the expression of receptors of the FPR family on mouse BM-DCs revealed that both mFPR1 and mFPR2 were expressed on RNA level (Figure 1A). Quantitative PCR revealed a 500-fold or 260-fold higher expression of mFPR1 and mFPR2 on neutrophils compared to splenic DCs, respectively (Figure 1A left). Expression of mFPR1 and mFPR2 by BM-DCs was slightly higher than that of splenic DCs. To determine if the expression of these receptors changes during maturation, quantitative PCR from immature and mature BM-DCs was performed (Figure 1A right). Both receptors were slightly up-regulated 24 h after DC maturation with S. aureus cell lysate. Furthermore, BM-DCs and splenic DCs bound fluorescently labeled PSMα3 as determined by flow cytometry (Figure 1C). Splenic neutrophils served as control and bound PSMα3 with similar affinity as DCs, whereas CD4+ T cells, which do not express members of the mFPR receptor family (8) revealed no binding of PSMα3. These data show that mouse DCs express mFPR1 and mFPR2 and are able to bind PSMs.
Figure 1. FPR expression by mouse DCs.

A, Expression of mFPR1 and mFPR2 by the indicated cells (left, DCs (CD11c+MHCIIhi) and neutrophils (GR1+CD11bhi) purified by cell sorting from splenocytes) or BM-DCs (right) was determined by quantitative RT-PCR and normalized to splenic DCs or medium, respectively. B, BM-DCs or splenocytes were incubated for 30 min with TAMRA-marked PSMα3 and the binding was analyzed by flow cytometry. The data show one representative out of three independent experiments with similar results (A-B left graph) or represent means of three independent experiments ± SD (B right graph and C). * indicates statistically significant differences (one-way ANOVA with Bonferroni post-test or unpaired students t-test, B left and right, respectively).
PSMs elicit chemotaxis of DCs
S. aureus PSMs have been shown to attract neutrophils by binding to FPR2 (5). Since we could show that mFPR2 is expressed by DCs we hypothesized that PSMs might also be chemoattractants for DCs and might alter DC functions. PSMα3, one of the most effective PSM peptides regarding receptor-mediated effects on human neutrophils, also induced chemotaxis in mouse BM-DCs at nanomolar concentrations (Figure 2A). The chemotaxis was characterized by a typical bell-shaped dose response curve with a decrease at concentrations higher than 0.5 μM. No cytotoxicity toward BM-DCs was detectable using these PSM concentrations (Supplementary Figure 1). Chemotaxis was measurable at concentrations above 100 nM, indicating that slightly higher concentrations are necessary to attract DCs compared to human neutrophils (5). With PSMα2 at least 0.5 μM were needed to induce chemotaxis. Furthermore, culture filtrates from S. aureus wild-type strain USA300 induced chemotaxis of BM-DCs whereas chemotaxis towards culture filtrates from a USA300 mutant strain deficient in PSMs was abrogated (Figure 2B). These data suggest that S. aureus strains secreting high amounts of PSMs also attract DCs to the site of infection.
Figure 2. PSMs elicit chemotaxis of DCs.
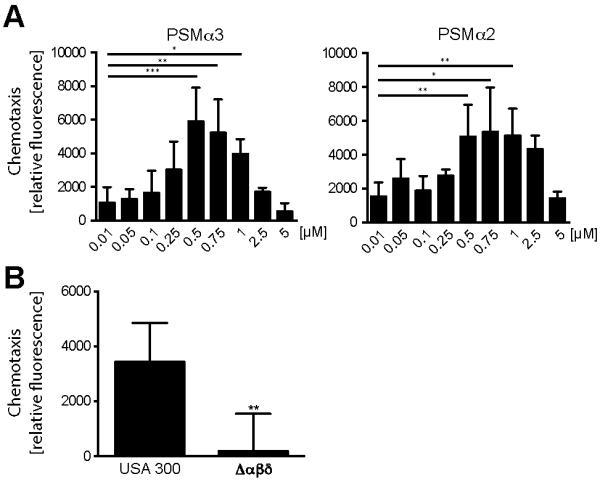
Chemotaxis of BM-DCs after treatment with the indicated concentrations of A PSMα3 (left graph) and PSMα2 (right graph) or B culture supernatants of USA300 and its deletion-mutant strain δαβδ for 100 min. Data represent means ± SD of three to four independent experiments done in duplicates. * indicates statistically significant differences (one-way ANOVA with Bonferroni post-test (A) or unpaired students t-test (B)).
PSMs modulate the cytokine profile of TLR2-stimulated DCs
Encounter of bacterial products by PRRs leads to the up-regulation of MHCII and co-stimulatory molecules as well as the production of pro-inflammatory cytokines by DCs. To analyze if PSMs modulate TLR2-mediated DC activation, BM-DCs were treated with S. aureus cell lysates containing lipopeptides, the major staphylococcal TLR2 ligands. This treatment led to up-regulation of MHCII as well as the co-stimulatory molecules CD40 and CD80. Treatment of DCs with S. aureus cell lysates in combination with PSMα3 had no influence on the phenotypic DC activation induced by S. aureus cell lysates (Figure 3A).
Figure 3. PSMα3 modulates cytokine production of TLR2-stimulated DCs.

A, BM-DCs were incubated with S. aureus cell lysate with or without PSMα3 (10 μM) for 6 h. The cells were stained with antibodies against CD11c, MHCII, CD80 and CD40 and analyzed by flow cytometry. B, BM-DCs were incubated with S. aureus cell lysate and PSMα3 or randomly synthesized control peptide, with the indicated concentrations for 6 h (TNF) or 24 h (IL-12, IL-10, IL-6) and cell culture supernatants were analyzed by ELISA. The data represent means of triplicates ± SD. C, BM-DCs were incubated with S. aureus cell lysate and PSMα3 in the presence or absence of neutralizing anti-IL10 antibody or isotype control. D, BM-DCs were incubated with S. aureus cell lysate and culture supernatants of USA300 and its deletion-mutant strain δαβδ for 6 h and analyzed as described in A. The data show one representative out of three to four independent experiments with similar results. * indicates statistically significant differences (one-way ANOVA with Bonferroni post-test).
The production of the pro-inflammatory cytokines TNF, IL-6, and IL-12 by DCs was also induced by S. aureus cell lysate (Figure 3B). This induction was TLR2-dependent, as DCs from TLR2−/− mice did not secrete any of these cytokines (Supplementary Figure 1A) upon stimulation with S. aureus cell lysate. Simultaneous treatment of DCs with S. aureus cell lysate and PSMα3 inhibited the secretion of TNF, IL-6 and IL-12, whereas the secretion of the anti-inflammatory cytokine IL-10 was increased in a dose-dependent manner (Figure 3B). Interestingly, PSMα3 alone did not induce secretion of any of the measured cytokines. PSMα3 was not cytotoxic toward DCs at concentrations that modulated cytokine secretion and had only little or no cytokine-modulating effects at concentrations lower than 100 nM (Figure 3B). A reversed scrambled alpha-PSM congener lacking the α-helical structure of the PSM peptides served as control peptide and had no effects on TLR2 ligand-induced cytokine secretion by DCs (Figure 3B). Similar results as for PSMα3 regarding cytokine production by DCs were obtained using the other PSMα peptides and δ-toxin, with only PSMα4 having weaker effects (Supplementary Figure 2).
Autocrine IL-10 has been shown to modulate IL-12 production in macrophages and DCs (34, 35). Addition of an IL-10-blocking antibody reversed the inhibition of IL-12 and partly of TNF expression by PSMα3 in DCs (Figure 3C), indicating that the induction of IL-10 expression by PSMα3 in DCs leads to an autocrine inhibition of IL-12 and TNF production. To further strengthen the results obtained with synthetic PSMs culture filtrates from S. aureus strain USA300 and a mutant strain deficient in PSMs were used to elucidate the effects of secreted PSMs on DC cytokine production (Figure 3D). The culture filtrate of the USA300 wild type strain, harboring the α- and β-PSMs and δ-toxin, inhibited TNF secretion by DCs as shown above for the synthetic PSMs, whereas the mutant strain deficient in all PSMs did not. These data demonstrate that PSM peptides are responsible for modulation of DC cytokine secretion by S. aureus.
PSMs inhibit clathrin-mediated endocytosis of DCs
To elucidate possible impacts of PSMs on antigen uptake, a hallmark of DCs and prerequisite for T-cell activation, we used fluorescently labeled OVA (OVA-AlexaFluor647) as a model antigen and measured OVA uptake by flow cytometry. DCs were cultured for 24 h with PSMα3 in the presence or absence of S. aureus cell lysate and subsequently incubated with OVA-AlexaFluor647 for 30 min. Ovalbumin is mainly taken up by clathrin-mediated endocytosis via the macrophage mannose receptor and to a lesser extent by macropinocytosis (36-38). Accordingly, OVA uptake by S. aureus cell lysate-treated and thereby matured DCs (downregulated macropinocytosis) was reduced by 32% compared to immature DCs. Increasing amounts of PSMα3 reduced the uptake of OVA both by DCs incubated with S. aureus cell lysate or without (Figure 4A) to the same level, indicating that PSMs inhibit OVA uptake by clathrin-mediated endocytosis. This is supported by unchanged macropinocytosis of lucifer yellow by DCs in the presence or absence of PSMα3 (Figure 4B) and reduced LPS-induced endocytosis of TLR4 by PSMα3 (Figure 4C), which is accomplished by clathrin-mediated endocytosis (39). Furthermore, PSMα3 did not affect phagocytosis of bacteria by DCs as shown for GFP-expressing Yersinia enterocolitica (Figure 4D).
Figure 4. PSMα3 inhibits clathrin-mediated endocytosis of DCs.

BM-DCs were incubated with 10 μM PSMα3 or with the indicated concentrations in the presence or absence of S. aureus cell lysate for 24 h and subsequently incubated with OVA-AlexaFluor647 (10 μg/ml) for 30 min (A), Lucifer yellow (2 mg/ml) for 10 min (B) or GFP-expressing Yersinia enterocolitica for 30 min (D) at 37 °C and analyzed by flow cytometry. C, Internalization of TLR4 was induced by LPS treatment (100 ng/ml) for 30 min in the presence or absence of PSMα3. TLR4 surface expression by BM-DCs was analyzed by flow cytometry. Representative histograms from three independent experiments are shown (A - C). Graphical summary of OVA- (A) or bacterial uptake (D); mean ± SD from three independent experiments. * indicates statistically significant differences compared to OVA-treated BM-DCs without PSMα3 (two-way ANOVA with Bonferroni post-test).
Impaired T-cell proliferation and Th1 priming by PSM-modulated DCs
Th1 and Th17 cells play important roles in the immune response against S. aureus (16-20). To examine, whether the aforementioned effects of PSMs on DC functions affect the ability of DCs to activate and prime CD4+ T cells, BM-DCs were treated with S. aureus cell lysate and PSMα3 overnight and subsequently co-cultured with splenic CD4+ T cells from congenic mice for 72 h. Incubation with S. aureus cell lysate led to strongly increased T-cell proliferation as analyzed by [3H]-thymidine incorporation (Figure 5A), which is in agreement with the activating capacity of TLR2 ligands (40). However, T-cell proliferation was reduced in a dose-dependent manner when DCs were co-incubated with PSMα3 (Figure 5A). In accord with this finding, the number of activated CD4+CD25+ T cells was reduced after stimulation with PSMα3-treated DCs (Figure 5B).
Figure 5. Impaired T-cell proliferation and Th1-priming by PSM-modulated DCs.

A BM-DCs were incubated with S. aureus cell lysate and the indicated concentrations of PSMα3 for 24h. Splenic CD4+ T cells from C57BL/6 mice were added to the culture and T-cell proliferation was analyzed by [3H]-thymidine incorporation 72 h later. B, The frequency of CD4+CD25+ T cells after DC-T-cell co-culture with a PSMα3 concentration of 10 μM as described in A. C, BM-DCs treated as in A with a PSMα3 concentration of 6 μM and co-cultured with OVA-specific splenic CD4+ T cells from OT-II mice for four days in the presence of OVA323-339 peptide. IFN-γ production by OVA-specific CD4+ T cells was assessed by flow cytometry. D, PSM-treated BM-DCs were co-cultured as in C in the presence of IL-6, TGF-β, anti-IFN-γ, anti-IL-4 for 4 days. IL-17 production by OVA-specific CD4+ T cells was assessed by flow cytometry. The data show one representative out of three independent experiments with similar results (A, C and D) or means of four independent experiments (B). The graphs represent means ± SD. * indicates statistically significant differences (one-way ANOVA with Bonferroni post-test (A) or unpaired students t-test (B-D)).
To analyze the influence of PSMs on T-cell priming DCs were incubated with S. aureus cell lysate in the presence or absence of PSMα3, loaded with OVA-peptide and subsequently co-cultured with CD4+ T cells from OVA-specific T-cell receptor transgenic mice. Flow cytometry analysis revealed a significantly reduced number of IFN-γ-secreting CD4+CD25+ T cells after co-culture of DCs incubated with S. aureus cell lysate in the presence of PSMα3 compared with S. aureus cell lysate-treated DCs (Figure 5C). No significant differences could be observed in the percentage of IL-17 secreting CD4+CD25+ T cells (Figure 5D). These data demonstrate that PSMs do not only inhibit T-cell activation but also T-cell priming towards Th1 cells.
Induction of Tregs by PSM-treated DCs
The reduced T-cell proliferation observed with PSM-modulated DCs could be due to a reduced stimulatory capacity of DCs or to the induction of regulatory T cells (Tregs). The frequency of CD4+CD25+FOXP3+Tregs was significantly increased after 72 h co-culture with DCs preincubated with S. aureus cell lysate in the presence of PSMα3 compared to DCs pretreated with S. aureus cell lysate alone (Figure 6A). These data suggest that incubation of DCs with PSMα3 induces tolerogenic DCs characterized by IL-10 production and priming of Tregs leading to the inhibition effector T cells. Next we tested whether there are differences in the suppression capacity of Tregs induced by DCs preincubated with or without PSMα3. Therefore, CD4+ T cells from FOXP3-eGFP mice were co-cultured with DCs as described above. 72 h later the FOXP3+ T cells were purified by cell sorting according to their expression of eGFP and their capacity to suppress T effector cell (Teff) proliferation was analyzed. No differences in the suppressive capacity of Tregs, generated by DCs prestimulated either in the presence or absence of PSMα3, could be observed (Figure 6B). Interestingly, Tregs induced by PSM-treated DCs produced higher amounts of IL-10 compared to control Tregs, whereas no difference in their TGF-β secretion could be observed (Figure 6C). Thus, PSMα3 modulates DCs to induce IL-10 producing Tregs. To address the mechanism by which the generated Tregs act on Teff we performed suppression assays in the presence of function-blocking antibodies for IL-10 and TGF-β. Both, blocking of IL-10 or TGF-β in co-culture experiments increased Teff proliferation (Figure 6D). Blocking of both cytokines further slightly reduced the suppression capacity of Tregs, but T-cell proliferation was still reduced by 27%, indicating that in addition to TGF-β and IL-10 other yet unknown factors lead to suppression capacity of Tregs in this assay. In summary, PSM-modulated DCs show impaired Th1 priming most likely due to the induction of IL-10 producing CD4+CD25+FOXP3+ Tregs.
Figure 6. Induction of IL-10 secreting Tregs by PSM-treated DCs.

A, BM-DCs were incubated with S. aureus cell lysate and the indicated concentrations of PSMα3 for 24 h and then co-cultured with CD4+ T cells for 72 h. The frequency of CD4+CD25+FOXP3+T cells was analyzed by flow cytometry. B, Experiment as in A with a PSMα3 concentration of 6 μM with CD4+ T cells from FOXP3-eGFP mice. Splenic CD3+CD4+FOXP3− Teff cells from the FOXP3-eGFP mice and the CD4+FOXP3+ Tregs from the co-culture were purified by cell sorting. Proliferation of cell proliferation dye efluor670 labeled Teff cells cultured with or without CD4+FOXP3-eGFP+ Tregs was analyzed by flow cytometry. The histogram overlays show proliferation of Teff cells without (gray dotted line) or with (black line) Tregs. The numbers depicted in the histogram overlays correspond to division indices (the average number of cell divisions that a cell in the original population has undergone) as analyzed by FlowJo software. The graph shows division indices at a Teff:Tref ratio of 2:1. C, Experiments as in B. Sorted CD4+FOXP3-eGFP+ Tregs were stimulated with anti-CD3 antibody for 72 h and cell culture supernatants were analyzed for IL-10 and TGF-β by ELISA. D, Experiment as in B (Teff:Treg 2:1) with neutralizing antibodies against IL-10 and TGF-β. Division indices are depicted in the histograms. The data represent means of three independent experiments ± SD (A and C) or show one representative out of three independent experiments performed in triplicates with similar results (B and D). * indicates statistically significant differences (A, one-way ANOVA with Bonferroni post-test. ,C unpaired students t-test).
PSM-modulated cytokine pattern by DCs is independent of FPRs
PSMs bind with high affinity to FPR2 on neutrophils (5). To determine, whether the induction of tolerogenic DCs by PSMs is mediated via mFPR2, BM-DCs from wild type and FPR2−/− mice were analyzed for cytokine production. TLR2 activation led to slightly higher production of TNF and IL-12 by DCs from FPR2−/− mice compared to wild type mice, whereas IL-10 production was reduced (Figure 7A). Similar to wild type DCs, PSMα3 inhibited the production of TNF and IL-12, while inducing IL-10 secretion by DCs from mFPR2−/− mice (Figure 7A), indicating that PSMα3 modulates DC cytokine production independently of mFPR2.
Figure 7. PSMα3 modulates cytokine production independently of mFPR2.
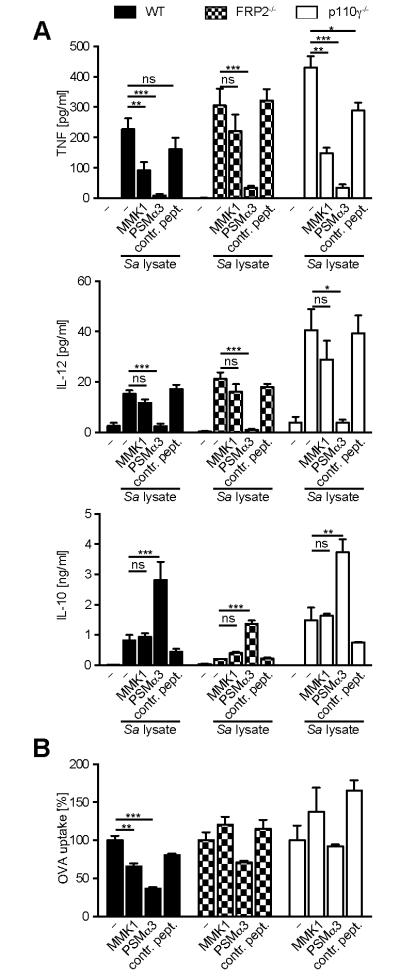
BM-DCs from wild type, FPR2−/−, and p110γ−/− mice were incubated with S. aureus cell lysate and 10 μM PSMα3 or MMK1 for 6 h (TNF) or 24 h (IL-12, IL-10) and cell culture supernatants were analyzed by ELISA (A) or were incubated with OVA-AlexaFluor647 for 30 min and OVA uptake was analyzed by flow cytometry (B). The data represent means of three independent experiments ± SD (A) or means of triplicates ± SD from one representative out of three independent experiments with similar results (B). * indicates statistically significant differences compared to S. aureus cell lysate treated BM-DCs or differences compared to the respective condition without inhibitor (one-way ANOVA with Bonferroni post-test).
To analyze whether the changes in cytokine profile by DCs are specific for PSMs or a general DC response to FPR2 ligands we simultaneously treated DCs with S. aureus cell lysates and MMK1, a non-cytotoxic FPR2 ligand. MMK1 inhibited TNF production by DCs, but not as efficient as PSMα3 (Figure 7A). This inhibition was abrogated using mFPR2−/− DCs, showing that MMK1-mediated inhibition of TNF was FPR2-dependent. Moreover, in contrast to PSMα3 MMK1 did not affect IL-12 and IL-10 secretion, further indicating that DC modulation by PSMα3 is not a general response to FPR2 ligands. Furthermore, by using BM-DCs from p110γ−/− mice, in which the downstream PI3K class Ib signaling of GPCRs is blocked (41) we ruled out the involvement of GPCRs in the induction of tolerogenic DCs by PSMs, as p110γ−/− DCs revealed similar cytokine secretion patterns as wild type mice (Fig. 7A). In contrast to the modulated cytokine secretion by DCs, the reduction of OVA uptake mediated by PSMα3 and MMK1 was dependent on FPR2 and downstream signaling of GPCRs (Fig. 7B). Taken together, these data point to an FPR2-independent induction of tolerogenic DCs by PSMs produced by CA-MRSA strains.
Discussion
PSMs are a group of virulence factors contributing to the virulence of emerging CA-MRSA strains such as USA300 (6). Previous studies have shown the modulatory effects of PSMs on innate immune cells e.g. recruitment and activation of neutrophils and induction of cell lysis (5, 6). However, the impact of PSMs on the adaptive immune response has not yet been addressed. We demonstrate that PSMs produced by CA-MRSA strains modulate the adaptive immune response because they effectively prevent the development of a Th1 response induced by the TLR2 ligands of S. aureus, at least in vitro, via multiple mechanisms, such as inhibited pro-but increased anti-inflammatory cytokine secretion as well as reduced antigen uptake by DCs thereby inducing Treg differentiation.
PSM peptides bind to FPR2 on human neutrophils thereby inducing chemotaxis (5). Herein we show that the mouse mFPR2 as well as its paralog mFRP1 binding formylated peptides are expressed by DCs. PSMs elicited chemotaxis of DCs even though at slightly higher concentrations as for neutrophils. This can be explained by reduced expression of mFPR2 mRNA by DCs compared to neutrophils. It should be further addressed whether the slight differences in mFPR1 and mFPR2 expression by BM-DCs and splenic DCs have relevance for chemotaxis. Our data suggest that DCs are actively attracted to the infection site where they may take up antigen, e.g. lysed neutrophils, undergo maturation and initiate adaptive immunity. We found that almost all of these DC functions are counteracted by PSMs, at least in in vitro studies. Analysis of antigen uptake revealed reduced clathrin-mediated endocytosis (OVA (36-38) and TLR4 endocytosis (39)), while there were no influences on macropinocytosis and phagocytosis. The inhibited clathrin-mediated endocytosis could result in less efficient antigen presentation thereby contributing to the reduced T-cell activation observed upon PSM treatment. PSMs had no influence on S. aureus cell lysate-induced BM-DC maturation, characterized by unaltered up-regulation of MHC class II and co-stimulatory molecules upon stimulation with S. aureus lysates. In contrast, LPS-induced up-regulation of co-stimulatory molecules by human monocyte-derived DCs is reduced by a synthetic FPR2 ligand (42).
PSMs dramatically changed the cytokine secretion pattern of DCs upon TLR2 ligand stimulation, namely they inhibited the production of the pro-inflammatory cytokines TNF, IL-6, and IL-12 while inducing the secretion of anti-inflammatory IL-10. Furthermore, we could show that, similar to mycobacterial infection (34), IL-10 inhibits IL-12 and also TNF production by DCs in an autocrine manner. It is long known that systemic S. aureus infections are associated with the endogenous production of TNF, IL-6, and IFN-γ (43). This pro-inflammatory response plays a significant role in the clearance of the bacterium as TNF/Lymphotoxin-α double-mutant mice show increased mortality upon S. aureus i.v infection (44).
An effective evasion strategy by a pathogen would be to target and instruct DCs to become tolerogenic and prime regulatory IL-10 responses blocking inflammation and allowing uncontrolled spread of the pathogen (45). Several groups have described the induction of tolerogenic DC by several pathogens (46-49). Thus, we speculate that the anti-inflammatory effects of PSMs on DCs may be beneficial for the spread of the bacterium.
PSM-treated DCs showed a reduced capability to activate T cells and more specifically blocked Th1 differentiation while Th17 differentiation was unaffected. Such an impaired T cell response might lead to reduced bacterial clearance during infection, as e.g. patients with HIV disease are highly susceptible to S. aureus colonization and skin infection due to reduced CD4+ T-cell counts (50). Protective CD4+ T cell responses have been described for Th1 and Th17 cells due to their secretion of IFN-γ and IL-17 (16-19, 51). Accordingly, IFN-γ plays a protective role during systemic infection, by activation of macrophages and neutrophils enhancing phagocytosis of the bacteria (51), and wound infection by CXC chemokine-induced neutrophil recruitment (18), while it is dispensable in skin infections (16). The findings in the present study point to a reduced capability of the host to clear systemic and invasive infections caused by PSM-producing S. aureus infections.
IL-17 produced by Th17 and γδ T cells is important for immunity against skin and other epithelial infections due to neutrophil recruitment and activation of keratinocytes leading to production of antimicrobial peptides (19). PSMs did not affect priming of Th17 cells, although secretion of the Th17-inducing cytokine IL-6 by DCs was reduced. This is in contrast to data from a non-eosinophilic asthma mouse model showing that activation of FPRs by a synthetic W-peptide inhibits both Th17 and Th1 responses by modulating airway DCs (52). This difference might be explained by the different TLR stimuli used or that our in vitro system lacks certain characteristics of an in vivo system, which might influence priming of Th17 cells.
During infection a fine regulation between regulatory and effector T cells is needed to control the immune response and contain the infection without inducing immune pathologies (53). Thus, induction of Tregs by the pathogen itself can lead to evasion of protective T cell responses. Inducible populations of Tregs are characterized by high secretion of IL-10, which has been shown to be the key regulator for inhibiting the immune responses against several pathogens (46, 54). Here, PSMα3-treated DCs induced the differentiation of naïve CD4+ T cells into IL-10-producing Tregs. However, their suppression capacity was only partly dependent on IL-10 and TGF-β, indicating that other mechanisms such as cell contact-mediated suppression may be involved in Treg-mediated suppression of T-cell differentiation. Similar to our findings filamentous hemagglutinin (FHA) from Bordetella pertussis stimulates IL-10 production thereby inhibiting IL-12 release by DCs (46). This leads to the induction of IL-10 producing regulatory T cells and the inhibition of a Th1 response. FHA functions as an adhesin and mediates this effect by binding and thereby activating CR3 or CD47 on DC. It is possible that PSMs induce via modulating DCs mixed populations of activated T cells thereby probably influencing the outcome of an infection as we observed less Th1 cells, more Tregs and unchanged numbers of Th17 cells upon pretreatment of DCs with PSMs. A direct effect of PSMs on T cells can be excluded as T cells do not express mFPR2 (8) and do not bind PSMα3 (Figure 1).
We show that PSMs bind to FPRs on mouse DCs and human neutrophils (5). However, the induction of tolerogenic DCs appears to be independent of binding to FPRs as DCs from mFPR2−/− mice as well as combined inhibition of mFPR1 and mFPR2 did not affect the induction of tolerogenic DCs. Recently, another pore-forming toxin, β hemolysin/cytolysin from Group B Streptococcus, has been described to induce IL-10 while inhibiting IL-12 and NOS2 production in macrophages (35). The authors of this study tested other pore forming toxins such as S. aureus α toxin that showed similar abilities to inhibit IL-12 expression but not to increase expression of IL-10. Furthermore, they ruled out the responsibility of membrane damage for the induction of tolerogenic DCs. Together with the fact that non-cytotoxic PSM concentrations were used in our study suggest that other mechanisms than pore formation or binding to FPRs mediate induction of tolerogenic DCs by PSMs.
In summary the net effect of PSMs on the adaptive immune system appears to be the inhibition of Th1-cell activation by the induction of tolerogenic DCs that direct the induction of Tregs. This may represent a novel immune subversion strategy employed e.g. by CA-MRSA strains producing high amounts of PSMs. However, additional experiments are needed to examine whether the findings presented herein are relevant in in vivo infection models reflecting different diseases of S. aureus.
Supplementary Material
Acknowledgements
The authors would like to thank Nele Nikola, Manina Günter, Daniela Gunst, and Patricia Hrstić for excellent technical support, Sabrina Grimm for operating the cell sorter, and Hans-Georg Rammensee for critical discussion.
Footnotes
This work was financed by the German Research Foundation grants SFB685 to S.A. and A.P. and TRR34 to A.P., the Fortüne program of the University of Tübingen Medical Faculty to D.K, and the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), NIH, to M.O..
Abbreviations used in this article: CA-MRSA, community-associated methicillin-resistant S. aureus; PSM, phenol-soluble modulin; DC, dendritic cells; BM-DCs, bone marrow-derived DCs; FPR, formyl peptide receptor; Tregs, regulatory T cells; Teff, T effector cells.
References
- 1.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Fridkin SK, Hageman JC, Morrison M, Sanza LT, Como-Sabetti K, Jernigan JA, Harriman K, Harrison LH, Lynfield R, Farley MM. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352:1436–1444. doi: 10.1056/NEJMoa043252. [DOI] [PubMed] [Google Scholar]
- 3.Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu Rev Microbiol. 2010;64:143–162. doi: 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- 4.Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13:1405–1406. doi: 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- 5.Kretschmer D, Gleske AK, Rautenberg M, Wang R, Koberle M, Bohn E, Schoneberg T, Rabiet MJ, Boulay F, Klebanoff SJ, van Kessel KA, van Strijp JA, Otto M, Peschel A. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell host & microbe. 2010;7:463–473. doi: 10.1016/j.chom.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 7.Diep BA, Chan L, Tattevin P, Kajikawa O, Martin TR, Basuino L, Mai TT, Marbach H, Braughton KR, Whitney AR, Gardner DJ, Fan X, Tseng CW, Liu GY, Badiou C, Etienne J, Lina G, Matthay MA, DeLeo FR, Chambers HF. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5587–5592. doi: 10.1073/pnas.0912403107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Laarman A, Milder F, van Strijp J, Rooijakkers S. Complement inhibition by gram-positive pathogens: molecular mechanisms and therapeutic implications. Journal of molecular medicine (Berlin, Germany) 2010;88:115–120. doi: 10.1007/s00109-009-0572-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bestebroer J, De Haas CJ, Van Strijp JA. How microorganisms avoid phagocyte attraction. FEMS microbiology reviews. 2010;34:395–414. doi: 10.1111/j.1574-6976.2009.00202.x. [DOI] [PubMed] [Google Scholar]
- 11.Peschel A, Sahl HG. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nature reviews. Microbiology. 2006;4:529–536. doi: 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- 12.Broker BM, van Belkum A. Immune proteomics of Staphylococcus aureus. Proteomics. 2011;11:3221–3231. doi: 10.1002/pmic.201100010. [DOI] [PubMed] [Google Scholar]
- 13.Kim HK, Thammavongsa V, Schneewind O, Missiakas D. Recurrent infections and immune evasion strategies of Staphylococcus aureus. Current opinion in microbiology. 2012;15:92–99. doi: 10.1016/j.mib.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 15.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nature immunology. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 16.Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, Cheng G, Modlin RL, Miller LS. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. The Journal of clinical investigation. 2010;120:1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, Fu Y, French SW, Edwards JE, Jr., Spellberg B. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 2009;5:e1000703. doi: 10.1371/journal.ppat.1000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLoughlin RM, Lee JC, Kasper DL, Tzianabos AO. IFN-gamma regulated chemokine production determines the outcome of Staphylococcus aureus infection. J Immunol. 2008;181:1323–1332. doi: 10.4049/jimmunol.181.2.1323. [DOI] [PubMed] [Google Scholar]
- 19.Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol. 2011;11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao YX, Nilsson IM, Tarkowski A. The dual role of interferon-gamma in experimental Staphylococcus aureus septicaemia versus arthritis. Immunology. 1998;93:80–85. doi: 10.1046/j.1365-2567.1998.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 23.Chen K, Le Y, Liu Y, Gong W, Ying G, Huang J, Yoshimura T, Tessarollo L, Wang JM. A critical role for the g protein-coupled receptor mFPR2 in airway inflammation and immune responses. J Immunol. 2010;184:3331–3335. doi: 10.4049/jimmunol.0903022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konrad S, Ali SR, Wiege K, Syed SN, Engling L, Piekorz RP, Hirsch E, Nurnberg B, Schmidt RE, Gessner JE. Phosphoinositide 3-kinases gamma and delta, linkers of coordinate C5a receptor-Fcgamma receptor activation and immune complex-induced inflammation. The Journal of biological chemistry. 2008;283:33296–33303. doi: 10.1074/jbc.M804617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Kissenpfennig A, Mingueneau M, Richelme S, Perrin P, Chevrier S, Genton C, Lucas B, DiSanto JP, Acha-Orbea H, Malissen B, Malissen M. Th2 lymphoproliferative disorder of LatY136F mutant mice unfolds independently of TCR-MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J Immunol. 2008;180:1565–1575. doi: 10.4049/jimmunol.180.3.1565. [DOI] [PubMed] [Google Scholar]
- 26.Schlag M, Biswas R, Krismer B, Kohler T, Zoll S, Yu W, Schwarz H, Peschel A, Gotz F. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Molecular microbiology. 2010;75:864–873. doi: 10.1111/j.1365-2958.2009.07007.x. [DOI] [PubMed] [Google Scholar]
- 27.Joo HS, Cheung GY, Otto M. Antimicrobial activity of community-associated methicillin-resistant Staphylococcus aureus is caused by phenol-soluble modulin derivatives. The Journal of biological chemistry. 2011;286:8933–8940. doi: 10.1074/jbc.M111.221382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erfurth SE, Grobner S, Kramer U, Gunst DS, Soldanova I, Schaller M, Autenrieth IB, Borgmann S. Yersinia enterocolitica induces apoptosis and inhibits surface molecule expression and cytokine production in murine dendritic cells. Infect Immun. 2004;72:7045–7054. doi: 10.1128/IAI.72.12.7045-7054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 30.de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, Poppelier MJ, Van Kessel KP, van Strijp JA. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. The Journal of experimental medicine. 2004;199:687–695. doi: 10.1084/jem.20031636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Somerville GA, Cockayne A, Durr M, Peschel A, Otto M, Musser JM. Synthesis and deformylation of Staphylococcus aureus delta-toxin are linked to tricarboxylic acid cycle activity. J Bacteriol. 2003;185:6686–6694. doi: 10.1128/JB.185.22.6686-6694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Autenrieth SE, Warnke P, Wabnitz GH, Estrada C. Lucero, Pasquevich KA, Drechsler D, Gunter M, Hochweller K, Novakovic A, Beer-Hammer S, Samstag Y, Hammerling GJ, Garbi N, Autenrieth IB. Depletion of Dendritic Cells Enhances Innate Anti-Bacterial Host Defense through Modulation of Phagocyte Homeostasis. PLoS Pathog. 2012;8:e1002552. doi: 10.1371/journal.ppat.1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oellerich MF, Jacobi CA, Freund S, Niedung K, Bach A, Heesemann J, Trulzsch K. Yersinia enterocolitica infection of mice reveals clonal invasion and abscess formation. Infect Immun. 2007;75:3802–3811. doi: 10.1128/IAI.00419-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demangel C, Bertolino P, Britton WJ. Autocrine IL-10 impairs dendritic cell (DC)-derived immune responses to mycobacterial infection by suppressing DC trafficking to draining lymph nodes and local IL-12 production. European journal of immunology. 2002;32:994–1002. doi: 10.1002/1521-4141(200204)32:4<994::AID-IMMU994>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 35.Bebien M, Hensler ME, Davanture S, Hsu LC, Karin M, Park JM, Alexopoulou L, Liu GY, Nizet V, Lawrence T. The Pore-Forming Toxin beta hemolysin/cytolysin Triggers p38 MAPK-Dependent IL-10 Production in Macrophages and Inhibits Innate Immunity. PLoS Pathog. 2012;8:e1002812. doi: 10.1371/journal.ppat.1002812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Autenrieth SE, Soldanova I, Rosemann R, Gunst D, Zahir N, Kracht M, Ruckdeschel K, Wagner H, Borgmann S, Autenrieth IB. Yersinia enterocolitica YopP inhibits MAP kinase-mediated antigen uptake in dendritic cells. Cellular microbiology. 2007;9:425–437. doi: 10.1111/j.1462-5822.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 37.Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316:612–616. doi: 10.1126/science.1137971. [DOI] [PubMed] [Google Scholar]
- 38.Burgdorf S, Lukacs-Kornek V, Kurts C. The mannose receptor mediates uptake of soluble but not of cell-associated antigen for cross-presentation. J Immunol. 2006;176:6770–6776. doi: 10.4049/jimmunol.176.11.6770. [DOI] [PubMed] [Google Scholar]
- 39.Husebye H, Halaas O, Stenmark H, Tunheim G, Sandanger O, Bogen B, Brech A, Latz E, Espevik T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. The EMBO journal. 2006;25:683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volz T, Nega M, Buschmann J, Kaesler S, Guenova E, Peschel A, Rocken M, Gotz F, Biedermann T. Natural Staphylococcus aureus-derived peptidoglycan fragments activate NOD2 and act as potent costimulators of the innate immune system exclusively in the presence of TLR signals. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:4089–4102. doi: 10.1096/fj.09-151001. [DOI] [PubMed] [Google Scholar]
- 41.Koyasu S. The role of PI3K in immune cells. Nature immunology. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- 42.Kang HK, Lee HY, Kim MK, Park KS, Park YM, Kwak JY, Bae YS. The synthetic peptide Trp-Lys-Tyr-Met-Val-D-Met inhibits human monocyte-derived dendritic cell maturation via formyl peptide receptor and formyl peptide receptor-like 2. J Immunol. 2005;175:685–692. doi: 10.4049/jimmunol.175.2.685. [DOI] [PubMed] [Google Scholar]
- 43.Nakane A, Okamoto M, Asano M, Kohanawa M, Minagawa T. Endogenous gamma interferon, tumor necrosis factor, and interleukin-6 in Staphylococcus aureus infection in mice. Infect Immun. 1995;63:1165–1172. doi: 10.1128/iai.63.4.1165-1172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hultgren O, Eugster HP, Sedgwick JD, Korner H, Tarkowski A. TNF/lymphotoxin-alpha double-mutant mice resist septic arthritis but display increased mortality in response to Staphylococcus aureus. J Immunol. 1998;161:5937–5942. [PubMed] [Google Scholar]
- 45.Manicassamy S, Pulendran B. Dendritic cell control of tolerogenic responses. Immunological reviews. 2011;241:206–227. doi: 10.1111/j.1600-065X.2011.01015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGuirk P, McCann C, Mills KH. Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin 10 production by dendritic cells: a novel strategy for evasion of protective T helper type 1 responses by Bordetella pertussis. The Journal of experimental medicine. 2002;195:221–231. doi: 10.1084/jem.20011288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Depaolo RW, Tang F, Kim I, Han M, Levin N, Ciletti N, Lin A, Anderson D, Schneewind O, Jabri B. Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell host & microbe. 2008;4:350–361. doi: 10.1016/j.chom.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 49.Lutz MB. Therapeutic potential of semi-mature dendritic cells for tolerance induction. Frontiers in immunology. 2012;3:123. doi: 10.3389/fimmu.2012.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathews WC, Caperna JC, Barber RE, Torriani FJ, Miller LG, May S, McCutchan JA. Incidence of and risk factors for clinically significant methicillin-resistant Staphylococcus aureus infection in a cohort of HIV-infected adults. J Acquir Immune Defic Syndr. 2005;40:155–160. doi: 10.1097/01.qai.0000179464.40948.b9. [DOI] [PubMed] [Google Scholar]
- 51.Zhao YX, Tarkowski A. Impact of interferon-gamma receptor deficiency on experimental Staphylococcus aureus septicemia and arthritis. J Immunol. 1995;155:5736–5742. [PubMed] [Google Scholar]
- 52.Tae YM, Park HT, Moon HG, Kim YS, Jeon SG, Roh TY, Bae YS, Gho YS, Ryu SH, Kwon HS, Kim YK. Airway activation of formyl peptide receptors inhibits Th1 and Th17 cell responses via inhibition of mediator release from immune and inflammatory cells and maturation of dendritic cells. J Immunol. 2012;188:1799–1808. doi: 10.4049/jimmunol.1102481. [DOI] [PubMed] [Google Scholar]
- 53.Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol. 2007;7:875–888. doi: 10.1038/nri2189. [DOI] [PubMed] [Google Scholar]
- 54.Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J Immunol. 2001;166:1141–1147. doi: 10.4049/jimmunol.166.2.1141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.