

Abstract
The negative feedback mechanism is essential to maintain effective immunity and tissue homeostasis. 1,25-dihydroxyvitamin D (1,25(OH)2D3) modulates innate immune response, but the mechanism remains poorly understood. Here we report that vitamin D receptor (VDR) signaling attenuates Toll-like receptor-mediated inflammation by enhancing the negative feedback inhibition. VDR inactivation leads to hyper inflammatory response in mice and macrophage cultures when challenged with lipopolysaccharide (LPS), due to miR-155 overproduction that excessively suppresses SOCS1, a key regulator that enhances the negative feedback loop. Deletion of miR-155 attenuates vitamin D suppression of LPS-induced inflammation, confirming that 1,25(OH)2D3 stimulates SOCS1 by down-regulating miR-155. 1,25(OH)2D3 down-regulates bic transcription by inhibiting NF-κB activation, which is mediated by a κB cis-DNA element located within the first intron of the bic gene. Together these data identify a novel regulatory mechanism for vitamin D to control innate immunity.
Keywords: vitamin D, inflammation, macrophage, miR-155, SOCS1, negative feedback
Introduction
Macrophages play a key role in innate immune response. Upon stimulation activated macrophages release a panel of pro-inflammatory mediators including cytokines and chemokines to initiate inflammatory response. Inflammatory reaction protects the host from pathogenic microorganisms; however, over-sustained inflammation is detrimental and can cause tissue damage and even death of the host. Thus, negative feedback mechanisms are in place to control the duration and intensity of inflammatory reaction (1). The suppressor of cytokine signaling (SOCS) family of proteins are key components of the negative feedback loop that regulates the intensity, duration and quality of cytokine signaling (2). As feedback inhibitors of inflammation, SOCS proteins are up-regulated by inflammatory cytokines, and in turn block cytokine signaling by targeting the JAK/STAT pathway (3). SOCS1 is a well-established key negative regulator of lipopolysaccharide (LPS)-induced inflammation (4, 5) and inhibits the pro-inflammatory pathways of cytokines such as TNFα, IL-6 and INFγ (6). SOCS1 in T cells and macrophages has been shown to prevent lethal inflammation in mice (7, 8). SOCS1 can also inhibit LPS-induced inflammatory response by directly blocking Toll-like receptor (TLR)-4 signaling (4, 5) via targeting IRAK1 and IRAK4 (9).
MicroRNAs (miRNAs) are a class of naturally occurring small non-coding RNAs of ~22 nucleotides that control gene expression by translational repression or mRNA degradation (10). In recent years microRNA-155 (miR-155) has emerged as a critical regulator of innate immunity and TLR signaling (11). miRNA-155 is encoded by a non-coding gene known as bic (12) and highly inducible in macrophages in response to TLR ligands such as LPS and poly(I:C) (13, 14). As miR-155 targets SOCS1 in activated macrophages (15) leading to blockade of the negative feedback loop, the induction of miR-155 during macrophage activation serves to maximize and prolong inflammatory process. Suppression of miRNA-155, therefore, will de-repress the negative feedback mechanism resulting in attenuation of inflammatory reaction.
1,25-dihydroxyvitamin D [1,25(OH)2D3], the active metabolite of vitamin D, is a pleiotropic hormone. The activity of 1,25(OH)2D3 is mediated by the vitamin D receptor (VDR), a member of the nuclear receptor superfamily (16). 1,25(OH)2D3 has potent immunomodulatory activities in both innate and adaptive immunity (17, 18). The best-known activity of vitamin D in the regulation of innate immunity is to stimulate anti-microbial peptide production in macrophages (19). TLR4 activation by bacterial infection increases local production of 1,25(OH)2D3 and VDR in macrophages, and this local 1,25(OH)2D3-VDR signaling induces the expression of anti-microbial peptide cathelicidin to kill bacteria (19, 20). This mechanism, however, cannot explain the anti-inflammatory action of vitamin D, in which 1,25(OH)2D3 down-regulates pro-inflammatory cytokines and chemokines in macrophages and other cells (21, 22). Little is known about the molecular basis of this anti-inflammatory mechanism. In this study, we provide evidence that the VDR signaling in macrophages limits inflammatory response by targeting the miR-155-SOCS1 pathway, resulting in heightened negative feedback inhibition of TLR-4 signaling.
Experimental Procedures
Animals and treatment
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Chicago. VDR-/- mice were reported previously (23), and miR-155-/- mice (24) were purchased from Jackson Laboratory (Stock # 007745). All mice, including wild-type (WT) controls, were in C57BL/6 background. Mice were used experimentally at 2-4 months of age. To induce sepsis, mice were injected with one dose of lipopolysaccharide (LPS, O111:B4, Sigma L2630; 20 mg/kg, i.p.). To study the anti-inflammatory effect of vitamin D, mice were pre-treated daily with vehicle (propylene glycol:water:ethanol =60:30:10) or non-calcemic vitamin D analog paricalcitol (19-nor-1,25-dihydroxyvitamin D2, 200 ng/kg, provided by Abbott Laboratories) for one week (i.p injection) before LPS (20 mg/kg) challenge. Blood was collected from tail vein at indicated times after LPS treatment for serum cytokine measurement. Spleen and peritoneal macrophages were harvested at the end of the experiment for protein and RNA analyses.
Cell culture and treatment
HEK293T, L929 and RAW264.7 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Peritoneal macrophages were harvested and plated on 6-well plates in DMEM containing 10% FBS. Unattached cells were removed after overnight culture. Bone marrow derived macrophages (BMDMs) were cultured as described (25). Briefly, mouse bone marrow cells were plated in DMEM supplemented with 10% FBS after red blood cells were lysed with 10 mM NH4Cl (pH8.0). After overnight culture the unattached cells were replated and differentiated into BMDMs in 30% L929 condition media. Human peripheral blood mononuclear cells (PBMC) were isolated by Ficoll–Hypaque (Pharmacia) gradient centrifugation of buffy coats from healthy donors, and suspended in complete RPMI 1640 medium containing 10% FCS, 100 U/ml penicillin, 100 mg/ml streptomycin and 2.5 mg/ml amphotericin B. The cells were allowed to adhere in culture flasks for 1 h at 37°C in humidified 5% CO2. The nonadherent cells were removed and the remaining adherent cells were harvested and cultured at a density of 1× 106 cells/ml in complete RPMI 1640 medium. Macrophage cultures were usually treated with 100-200 ng/ml LPS for 0-72 hours with or without overnight pre-treatment with 20 nM 1,25(OH)2D3 as specified in each experiment, followed by isolation of total RNAs, lysates and/or media supernatants for various assays.
MicroRNA arrays
Total RNAs were extracted from RAW264.7 cells treated with LPS (100 ng/ml) in the presence or absence of 1,25(OH)2D3 (20 nM) overnight. MicroRNA profiling was performed using the miRCURY LNA microRNA Arrays (Exiqon, Vedvaek, Denmark) according to manufacturer’s standard protocols. The arrays were scanned with GenePix4000B scanner using the manufacturer’s recommended settings. The raw data was extracted using GenePix Pro and imported into GeneSpring GX10 for analyses. The raw and normalized microRNA array data were deposited in the GEO database (accession No: GSE43300. URL: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43300).
Cytokine quantitation
TNFα and IL-6 concentrations in the serum or culture media were determined by enzyme-linked immunoabsorbent assay (ELISA) using commercial ELISA kits obtained from BioLegend (San Diego, CA).
RT-PCR
Total cellular RNAs were extracted using TRIzol reagents (Invitrogen, Carlsbad, CA). First-strand cDNAs were synthesized using a ThermoscriptRT kit (Invitrogen) or a TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems) for miRNA analyses. Conventional PCRs were carried out in a BioRad DNA Engine (BioRad). Real time PCRs (qPCRs) were performed in a Roche LightCycler 480II Real-Time PCR System, using SYBR green PCR reagent kits (Clontech) for mRNA transcript quantitation or TaqMan OpenArray Real Time PCR Master Mix (Applied Biosystems) for miR-155 quantitation. Relative amount of transcripts was calculated using the 2-ΔΔCt formula as described previously (26). All PCR primers were reported in Table 1.
Table 1.
Primers and oligonucleotide probe sequences used in the study
| PCR primers | Sequences 5’>3’ |
|---|---|
| β2-microglobulin-1 | ACCGGCCTGTATGCTATCCAGAAA |
| β2-microglobulin-2 | ATTTCAATGTGAGGCGGGTGGAAC |
| Mouse TNFα-1 | TCAGCCTCTTCTCATTCCTG |
| Mouse TNFα-2 | CAGGCTTGTCACTCGAATTT |
| Mouse IL-6-1 | ATAGTCCTTCCTACCCCAATTTCC |
| Mouse IL-6-2 | CTGACCACAGTGAGGAATGTCCAC |
| Mouse IL1b-1 | CCAAAAGATGAAGGGGTGCTGCT |
| Mouse IL1b-2 | ACAGAGGATGGGCTCTTCTT |
| Mouse bic-1 | CTTGCTGAAG GCTGTATGCT |
| Mouse bic-2 | ATTCTCGACT GACATCTAGG |
| mmu-mir-155 | UUAAUGCUAAUUGUGAUAGGGGU |
| human TNF-α-F | CGAGTGACAAGCCTGTAGC |
| human TNF-α-R | GGTGTGGGTGAG-GAGCACAT |
| human IL-6-F | AAATGCCAGCCT-GCTGACGAAC |
| human IL-6-R | AACAACAATCTGAGGTGCCCATGCTAC |
| Human BIC forward | CTCTAATGGTGGCACAAA |
| Human BIC reverse | TGATAAAAACAAACATGGGCTTGAC |
| Human GAPDH1 | ACCACAGTCCATGCCATCAC |
| Human GAPDH2 | TCCACCACCCTGTTGCTGTA |
| Genotyping primers | |
| mi-155 KO typing-1 | GTG CTG CAA ACC AGG AAG G |
| mi-155 KO typing-2 | CTG GTT GAA TCA TTG AAG ATG G |
| mi-155 KO typing-3 | CGG CAA ACG ACT GTC CTG GCC G |
| Cloning primers | |
| bic promotor-5-Sal1 | CGGGTCGACTTGGGAATCAAACCTAGGC |
| bic promotor-3-Nco1 | CGGCCATGGCAGCATACAGCCTTCAGC |
| EMSA probes | |
| bic intronic κB-1 | TTCCTTGGTTTGGAAATTTCCAAGCCACGAT |
| bic intronic κB-2 | ATCGTGGCTTGGAAATTTCCAAACCAAGGAA |
| bic κB mutant-1 | TTCCTTGGTTTGGAGACTTCCAAGCCACGAT |
| bic κB mutant-2 | ATCGTGGCTTGGAAGTCTCCAAACCAAGGAA |
| ChIP primers | |
| bic-κB-1 | AGTCTTTCTTCATCCAATTAAA |
| bic-κB-2 | CTG CCC TTT CTT ATA GCT CCT |
Northern blot
Northern blot analysis of miR-155 was conducted according to previously published method (27). Briefly, total RNAs were separated on 6% polyacrylamide gels containing formaldehyde and transferred onto a nylon membrane. Hybridization was performed at 42°C using LNA mmu-mi-155 probe (Exiqon) labeled with 32P-dATP. The 32P-labelled U6 probe (Exiqon) was used as an internal control.
Western blot
Proteins were separated by SDS-PAGE and electroblotted onto Immobilon-P membranes. Western blotting analyses were carried out as previously described (28). The antibodies used in this study included: SOCS1 (Zymed laboratories, 38-5200, rabbit polyclonal), VDR (Santa Cruz, sc-1009, rabbit polyclonal), IκBα (Santa Cruz, sc-371, rabbit polyclonal), IKKα/β (Santa Cruz, sc-7607, rabbit polyclonal), p65 (Santa Cruz, sc-109, rabbit polyclonal), and β-actin (Sigma, A2066, rabbit polyclonal).
Cell transfection
Cell transfection was performed using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instruction. To overexpress miR-155 RAW264.7 cells were transfected with pCDH-miR-155 plasmid kindly provided by Dr. Mo (Southern Illinois University School of Medicine) or mmu-miR-155 oligo mimic (Thermo Scientific Dharmacon). RAW264.7 cells or peritoneal macrophages were transfected with 50 nM mmu-miR-155 hairpin inhibitor (Thermo Scientific Dharmacon) to knock down miR-155, or transfected with 50 nM p65-specific siRNA (Thermo Scientific Dharmacon) to silence p65.
Kinase assay
IKK kinase was performed as described (29). Cell lysates prepared from RAW264.7 cells treated with LPS in the presence or absence of 1,25(OH)2D3 were immunoprecipitated with anti-IKKγ antibodies (Santa Cruz). The precipitant was incubated with recombinant GST-IκBα (1-54) (Clontech) in the presence of γ-32P-ATP, and 32P-labelled GST-IκBα (1-54) was detected by autoradiography.
Luciferase reporter assays
The DNA fragment from -587 to +263 in mouse bic gene was PCR-amplified with specific primers (Table S1) and cloned into pGL3-Basic luciferase reporter plasmid (Promega), generating pGL-bic-Luc. Reporter plasmid carrying mutations at the +87 intronic κB site (pGL-bicmut-Luc, mutated from 5’-GGAAATTTCC-3’ to 5’-GGAgAcTTCC-3’) was generated by site-directed mutagenesis. RAW264.7 cells were transfected with these plasmids or pNF-κB-Luc (control) using Lipofectamine 2000 (Invitrogen). After overnight culture the transfected cells were treated with LPS in the presence or absence of 1,25(OH)2D3 (20 nM). Luciferase activity was measured after 24 hours using Promega luciferase assay system.
Electrophoretic mobility shift assays (EMSA)
To assess the activity of κB cis-element, p65 and p50 proteins were synthesized from pCI-HA-p65 and pCI-HA-p50 plasmids using in vitro Transcription and Translation System (Promega). These proteins were incubated with 32P-labeled bic intronic κB probe 5’-TTCCTTGGTTTGGAAATTTCCAAGCCACGAT-3’ (underlined is the κB site). Specificity of the protein-DNA interaction was confirmed by competition with an excess amount of the unlabeled probe, a mutant probe 5’-TTCCTTGGTTTGGAgAcTTCCAAGCCACGAT-3’ or a canonical κB probe 5’-AGTTGAGGGGCATTTCCCAGGC-3’ (Santa Cruz Biotechnology).
Chromatin immunoprecipitation (ChIP) assays
RAW246.7 pretreated with or without 20 nM 1,25(OH)2D3 for 24 hours were stimulated with LPS for 2 hours. ChIP assays were performed as described previously (30), using anti-p65 antibodies. PCR was carried out using primers flanking the intronic κB site in the bic gene as described in Table 1.
Statistical analysis
Data values were presented as means ± SD. Statistical analyses were performed using unpaired two-tailed Student’s t-test or one-way ANOVA as appropriate, with P< 0.05 being considered significant.
Results
VDR inactivation leads to hyperresponsiveness to LPS
To understand the role of VDR in innate immunity, we compared the response of wild-type (WT) and VDR-/- mice to LPS challenge. After one dose of LPS (20 mg/kg, i.p), >40% of VDR-/- mice died within 48 hours and all died by 96 hours; in contrast, 60% of WT mice still survived after 120 hours (Fig. 1A). Consistent with the hyperresponsiveness of VDR-/- mice, within 24 hours of LPS treatment the induction of serum TNFα and IL-6 was much more robust in VDR-/- mice compared to WT mice (Fig. 1B and 1C). Since macrophages play a critical role in innate immunity (31), we directly examined the impact of VDR inactivation on the response of macrophages to LPS. Time course studies showed that exposure of BMDMs to LPS led to much more robust and prolonged production of pro-inflammatory cytokines TNFα, IL-6 and IL-1β in VDR-/- BMDMs compared to the WT counterpart (Fig. 1D-F). These observations indicate that the innate immune response is dysregulated and over-sustained in macrophages when the VDR signaling is inactivated.
Figure 1.
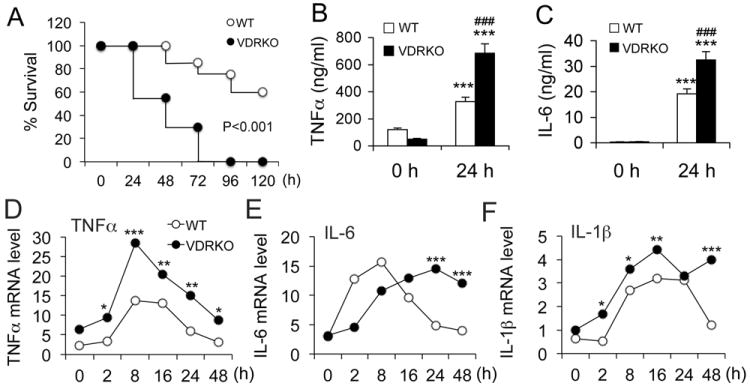
VDR inactivation leads to hyperresponsiveness to LPS. (A) Survival curves of wild-type (WT) and VDR-null (VDRKO) mice after intraperitoneal injection of LPS at 20 mg/kg; n=7-8. P<0.001 by logrank test. (B and C) Serum TNFα (B) and IL-6 (C) concentration in WT and VDRKO mice at 0 and 24 hours after LPS challenge. *** P<0.001 vs. 0 hour; ### P<0.001 vs. WT; n=7-8; (D-F) Time course of TNFα (D), IL-6 (E) or IL-1β (F) transcript induction following LPS (100 ng/ml) treatment in BMDMs derived from WT and VDRKO mice, n=3; * P<0.05, **P<0.01, *** P<0.001 vs. WT.
1,25(OH)2D3 and its analog suppress inflammatory response
To validate the importance of VDR in inflammatory regulation we examined the effects of VDR activation in mice and macrophages. WT mice were challenged with LPS following pretreatment with vehicle or a non-calcemic vitamin D analog paricalcitol for one week (1,25(OH)2D3 can induce hypercalcemia). Paricalcitol pretreatment substantially suppressed LPS-induced serum TNFα and IL-6 concentrations (Fig. 2A and 2B). Similarly, in LPS-treated WT BMDMs the time-dependent induction of TNFα and IL-6 release was markedly attenuated in the presence of 1,25(OH)2D3 (Fig. 2C and 2D). 1,25(OH)2D3 also inhibited LPS-induced TNFα, IL-1β and IL-6 expression in RAW264.7 cells, a murine macrophage cell line (Fig. S1A).
Figure 2.
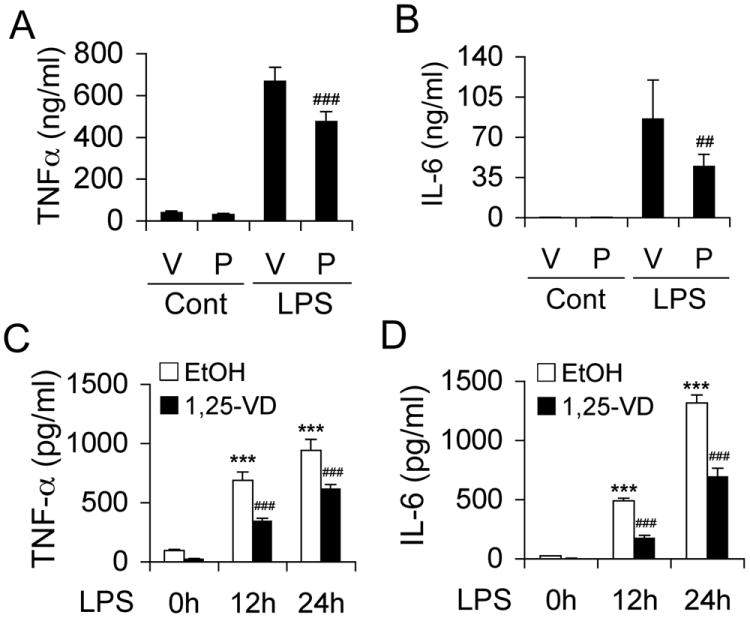
VDR activation inhibits LPS-induced inflammatory reaction. (A and B) WT mice were pretreated with vehicle (V) or paricalcitol (P, 200 ng/kg, daily i.p.) for one week before being challenged with saline control or LPS (20 mg/kg). Serum TNFα (A) or IL-6 (B) concentration was measured at 0 and 24 hours after LPS injection. ## P<0.01; ### P<0.001 vs. V; n=7. (C and D) WT BMDMs were pretreated with ethanol (EtOH) or 1,25(OH)2D3 (1,25-VD, 20 nM) for 24 hours before being exposed to LPS (100 ng/ml). The time course of TNFα (C) and IL-6 (D) release into the media was determined by ELISA. ***, P<0.001 vs. 0 hour; ### P<0.001 vs. corresponding EtOH; n=3.
Vitamin D-VDR signaling suppresses miR-155 in macrophages
As a growing number of miRNAs have emerged as regulators of immune response (32), we postulated that vitamin D down-regulates inflammation via targeting miRNAs. To test this hypothesis we analyzed miRNA profiling in RAW264.7 cells treated with LPS in the presence or absence of 1,25(OH)2D3. Among the miRNAs that were induced the most by LPS and suppressed the most by 1,25(OH)2D3, miR-155 was on the top of the list (Table 2), with 1,25(OH)2D3 suppressing about 50% of the LPS induction. Northern blot (Fig. 3A and 3B) and qPCR analyses (Fig. 3C) validated that 1,25(OH)2D3 substantially suppressed LPS-induced miR-155 in RAW264.7 cells. Similar regulations were seen for bic transcript (Fig. 3D). Time course studies confirmed that the time-dependent induction of bic transcript and miR-155 by LPS was suppressed by 1,25(OH)2D3 in RAW264.7 cells (Fig. 3E and 3F) as well as in BMDMs from WT mice (Fig. S1B and C).
Table 2.
Top 10 microRNAs that are induced by LPS and suppressed by 1,25-dihydroxyvitamin D identified by miRNA microarrays in macrophage cell line RAW264.7.
| Fold Induction over Control | Fold Suppression | ||
|---|---|---|---|
|
| |||
| Micro RNA ID | LPS | LPS+1,25VD | LPS+1,25VD/LPS |
|
| |||
| hsa-miR-155 | 18.5531352 | 9.261635464 | 0.499195169 |
| mmu-miR-155 | 17.9021754 | 9.179388553 | 0.512752688 |
| hsa-miR-146b-5p/mmu-miR-146b/rno-miR-146b | 4.988193783 | 3.615303291 | 0.724772021 |
| hsa-miR-146a/mmu-miR-146a/rno-miR-146a | 3.38135449 | 2.310883024 | 0.683419331 |
| hsa-miR-21/mmu-miR-21/rno-miR-21 | 3.313236092 | 1.661657289 | 0.501520943 |
| hsa-miR-494/mmu-miR-494/rno-miR-494 | 2.929190028 | 3.106773236 | 1.060625363 |
| hsa-miRPlus-F1024 | 2.850111992 | 1.186770578 | 0.416394367 |
| hsa-miR-1274a/mmu-miR-1274a | 2.666012187 | 3.03651172 | 1.138971433 |
| hsa-miRPlus-F1029 | 2.392788973 | 2.250856373 | 0.940683194 |
| mmu-miR-207/rno-miR-207 | 2.343471997 | 3.951941966 | 1.686361933 |
Figure 3.
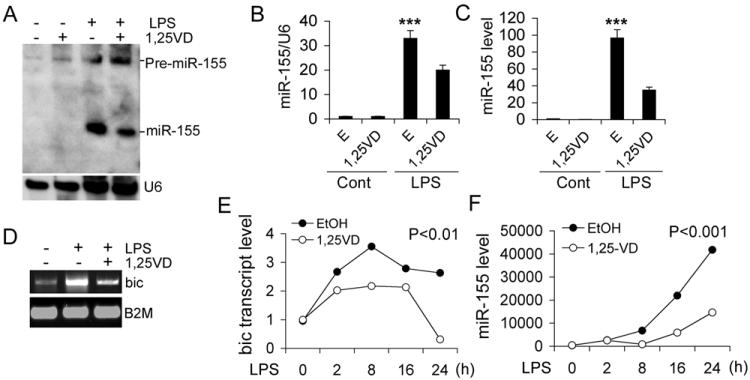
1,25-dihydroxyvitamin D down-regulates miR-155. (A and B) Northern blot (A) and PhosphoImaging quantitation (B) of pre-miR-155 and miR-155 in RAW264.7 cells stimulated with LPS for 24 hours in the presence of EtOH (E) or 1,25(OH)2D3 (1,25VD); (C) Real time PCR quantitation of miR-155 in RAW264.7 cells stimulated with LPS for 24 hours in the presence of EtOH or 1,25(OH)2D3; *** P<0.001 vs. the rest; n=3. (D) RT-PCR assessment of bic transcript in RAW264.7 cells stimulated by LPS in the presence EtOH vehicle or 1,25-VD; (E and F) Time course of bic transcript (E) and miR-155 (F) induction in RAW264.7 cells stimulated with LPS in the presence of EtOH or 1,25(OH)2D3. The bic transcript and miR-155 were quantified by qPCR.
We further examined the effect of VDR inactivation on miR-155. At baseline bic transcript and miR-155 levels were very low in WT peritoneal macrophages, but they were elevated in VDR-/- macrophages (Fig. 4A and 4B). After LPS stimulation the time-dependent induction of bic and miR-155 was much more robust and prolonged in VDR-/- BMDMs compared to WT BMDMs (Fig. 4C and 4D). Together these observations demonstrate that vitamin D-VDR signaling suppresses miR-155 in macrophages.
Figure 4.
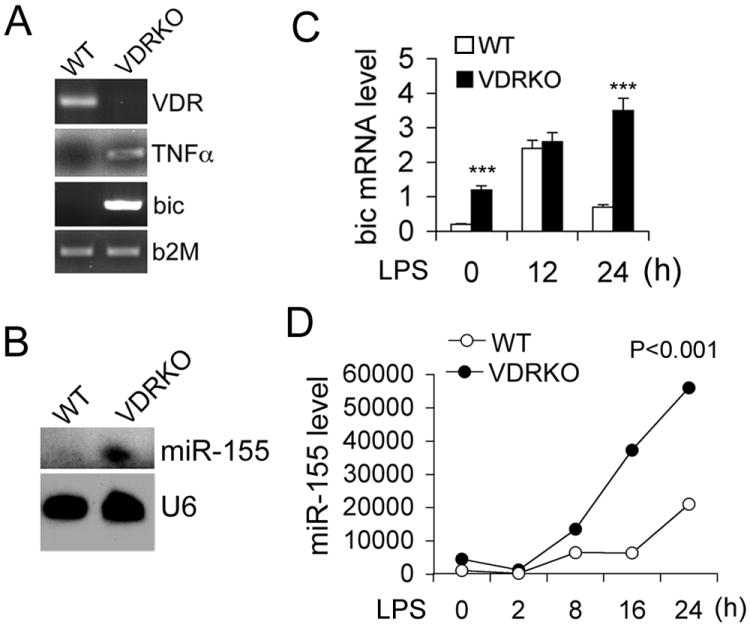
VDR inactivation leads to increased miR-155 expression. (A) RT-PCR analyses of baseline VDR, bic and TNFα transcripts in peritoneal macrophages harvested from WT and VDRKO mice; (B) Northern blot analysis of baseline miR-155 in peritoneal macrophages harvested from WT and VDRKO mice; (C and D) Time course of LPS-induction of bic transcript (C) and miR-155 (D) in WT and VDRKO BMDMs, quantitated by qPCR. *** P<0.001 vs. WT. n=3.
Vitamin D inhibits inflammatory cytokines and miR-155 in human PBMCs
As differences were reported in vitamin D’s immune-regulatory activities between mice and humans, we assessed the relevance of vitamin D regulation of miR-155 in human PBMCs. As shown in Figure 5, 1,25(OH)2D3 markedly blocked not only the induction of TNFα and IL-6 by LPS (Fig. 5A and B), but also that of bic transcript and miR-155 (Fig. 5C and D) in human PBMCs.
Figure 5.
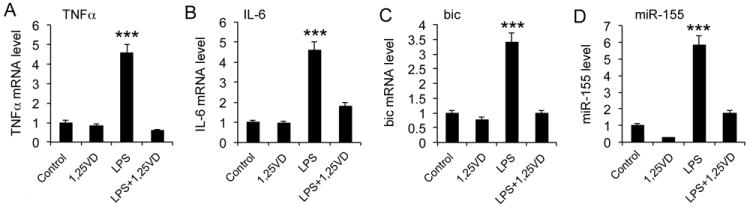
1,25-dihydroxyvitamin D down-regulates miR-155 and inflammatory cytokines in human peripheral blood mononuclear cells (PBMCs). Human PBMCs were pretreated with 1,25(OH)2D3 (20 nM) overnight followed by LPS stimulation of r4 hours. The transcript levels of TNFα (A), IL-6 (B), bic (C) and miR-155 (D) were then quantified by qPCR. *** P<0.001 vs. the rest.
Vitamin D-VDR signaling up-regulates SOCS1 via suppressing miR-155
Because SOCS1 is a proved target of miR-155, we expected that vitamin D inhibition of miR-155 would release the suppression on SOCS1, whereas VDR inactivation would depress SOCS1. Indeed we observed that the time-dependent induction of SOCS1 by LPS was further enhanced by 1,25(OH)2D3 treatment in RAW264.7 cells (Fig. 6A and B). In contrast, LPS was not able to induce SOCS1 protein in VDR-/- BMDMs (Fig. 6C and D), likely due to over-expression of miR-155. To assess the in vivo phenotype of macrophages after LPS challenge, we examined peritoneal macrophages immediately harvested from LPS-treated mice. SOCS1 was highly induced in the macrophages from WT mice as expected, but SOCS1 induction was hardly detectable in the macrophages obtained from VDR-/- mice (Fig. 6E and F). These results indicate that vitamin D-VDR signaling targets the miR-155-SOCS1 pathway in macrophages. Given the critical role of SOCS1 in the negative feedback regulation, these data suggest that vitamin D-VDR signaling limits inflammatory response by promoting the negative feedback action.
Figure 6.
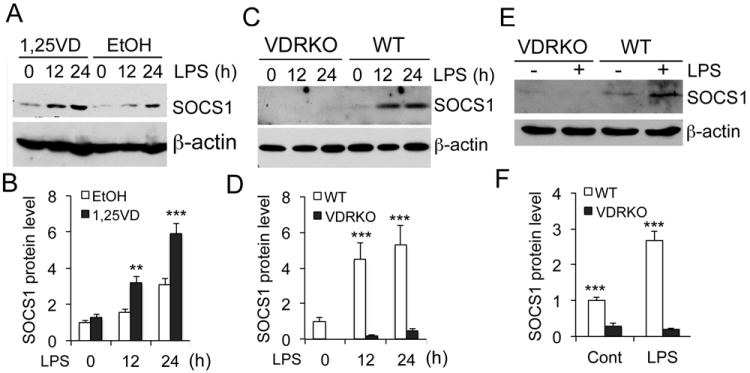
VDR signaling enhances SOCS1 induction. (A and B) Time course of SOCS1 protein induction (A) and its quantitation (B) in RAW264.7 cells stimulated with LPS in the presence of ethanol (EtOH) or 1,25(OH)2D3 (1,25VD). (C and D) Time course of SOCS1 protein induction (C) and quantitation (D) in WT and VDRKO BMDMs stimulated with LPS for 0-24 hours; (E and F) SOCS1 protein levels (E) and quantitation (F) in peritoneal macrophages immediately harvested from WT and VDRKO mice treated with saline (-) or LPS (+). ** P<0.01; *** P<0.001.
Up-regulation of miR-155 is required for hyper inflammatory reaction in VDR-null macrophages
To explore the role of miR-155 in vitamin D regulation of inflammation, we asked whether miR-155 up-regulation was responsible for the hyper-responsiveness of VDR-/- macrophages to LPS stimulation. As expected, in VDR-/- peritoneal macrophages LPS induced much more robust and more prolonged secretion (Fig. 7B and 7C) and mRNA expression (Fig. 7D and 7E) of TNFα and IL-6 compared to WT cells. When LPS-induced miR-155 up-regulation was attenuated using a miR-155-specific miRIDIAN hairpin inhibitor (Fig. 5A), the time dependent induction of TNFα and IL-6 was also markedly attenuated at the protein (Fig. 7B and 7C) and transcript (Fig. 7D and 7E) levels. The induction of bic, however, was not affected by miR-155 knockdown as expected (Fig. 7F). These results confirm that miR-155 overproduction is a major cause for the hyper inflammation seen in VDR-null macrophages.
Figure 7.
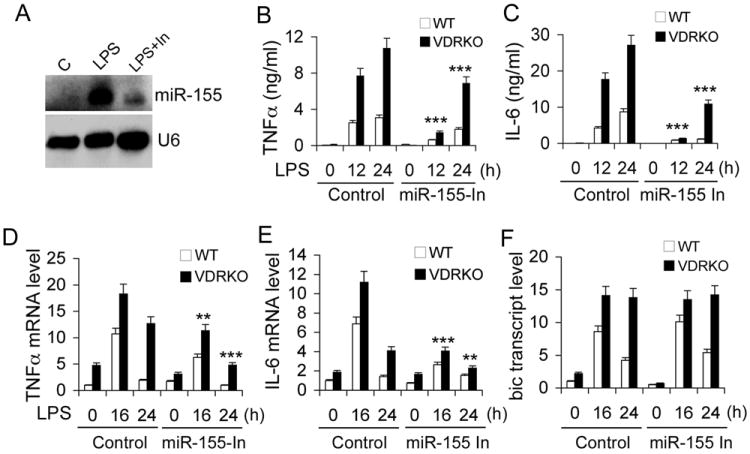
The up-regulation of miR-155 is required for hyper inflammatory response to LPS in VDR-null macrophages. (A) Knockdown of LPS-induced miR-155 in peritoneal macrophages using miR-155-specific hairpin inhibitor (In); (B and C) Time course of TNFα (B) and IL-6 (C) secretion from LPS-treated WT and VDRKO peritoneal macrophages transfected with control or miR-155-specific hairpin inhibitor. ** P<0.01; *** P<0.001 vs. corresponding control; n=3-5. (D-F) Time course of LPS-induced changes in TNFα (D), IL-6 (E) or bic (F) transcript in WT and VDRKO peritoneal macrophages transfected with control or miR-155-specific hairpin inhibitor.
We also asked whether ectopic overexpression of miR-155 could abrogate vitamin D inhibition of inflammation. Indeed, lentiviral overexpression of miR-155 in RAW264.7 cells led to up-regulation of TNFα, but this TNFα up-regulation could no longer be suppressed by 1,25(OH)2D3 (Fig. S2A-D). Together these data suggest a critical role of miR-155 in vitamin D regulation of macrophage inflammatory response.
miR-155 is required for vitamin D inhibition of inflammation
Next we used the miR-155-null mouse model (24) to further assess the role of miR-155 in vitamin D regulation of innate immunity. WT and miR-155-/- mice were pre-treated with vehicle or paricalcitol daily for one week before LPS challenge. As expected, paricalcitol substantially blocked the induction of serum TNFα and IL-6 in LPS-treated WT mice at 16 hours. In miR-155-/- mice, however, the induction of serum TNFα and IL-6 was significantly lower compared to WT mice; interestingly, paricalcitol failed to inhibit these pro-inflammatory cytokines (Fig. 8A and 8B). In vitro time course experiments showed that 1,25(OH)2D3 markedly suppressed LPS-induced TNFα and IL-6 transcripts in WT BMDMs. In miR-155-/- BMDMs the induction of these cytokines was less robust, but 1,25(OH)2D3 had little effects on the expression of these cytokines (Fig. 8C and 8D). As expected, LPS induction of SOCS1 was much more robust in miR-155-/- macrophages compared to WT macrophages (Fig. 8E). Whereas 1,25(OH)2D3 markedly elevated SOCS1 in WT BMDMs as expected, it failed to induce SOCS1 in miR-155-/- macrophages (Fig. 8F and 8G). Consistently, paricalcitol could induce splenic SOCS1 in WT mice but not in miR-155-/- mice (Fig. S3A and S3B). Together these data provide compelling evidence that vitamin D-VDR signaling maintains SOCS1 levels primarily via suppressing miR-155. As a result, the negative feedback inhibition loop is maintained. Therefore vitamin D limits inflammatory response by targeting the miR-155-SOCS1 pathway.
Figure 8.
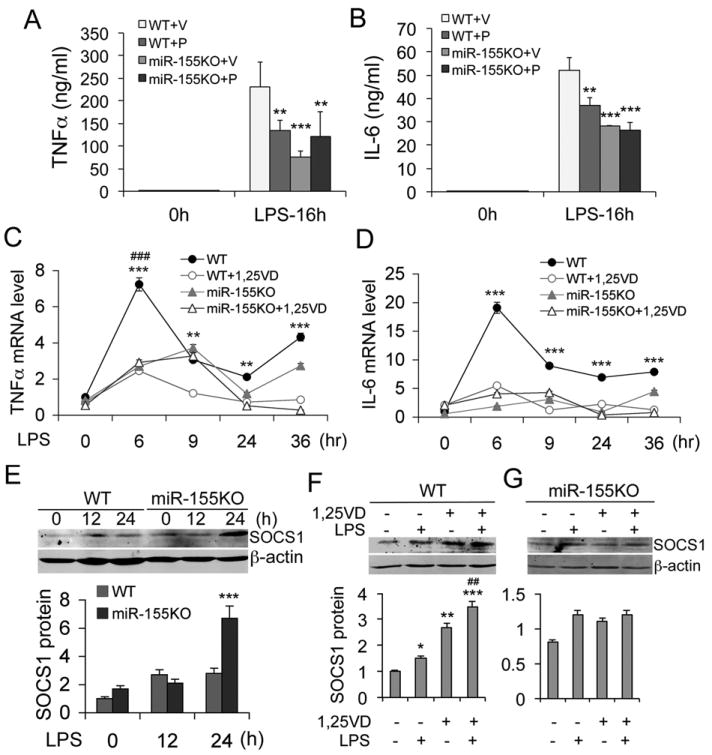
miR-155 is require for vitamin D suppression of inflammatory reaction. (A and B) WT and miR-155-null (KO) mice were pretreated with vehicle (V) or paricalcitol (P, 200 ng/kg, daily i.p.) for one week before an intraperitoneal LPS challenge. Serum TNFα (A) and IL-6 (B) concentration was determined at 0 and 16 hours after LPS treatment. ** P<0.01; *** P<0001 vs. WT+V; n=5-7; (C and D) Time course of TNFα (C) or IL-6 (D) transcript induction in WT and miR-155KO BMDMs treated with LPS (100 ng/ml) in the presence or absence of 1,25(OH)2D3; **P<0.01, *** P<0.001 vs. WT+1,25VD in (C) or vs. the rest in (D); ### P<0.001 vs. the rest; n=6. (E) Time course of SOCS1 induction (top panel) and quantitation (lower panel) in WT and miR-155KO peritoneal macrophages treated with LPS. *** P<0.001 vs. WT; (F and G) SOCS1 protein levels (top panel) and quantitation (lower panel) in WT (F) and miR-155KO (G) peritoneal macrophages stimulated with LPS in the presence or absence of 1,25(OH)2D3. * P<0.05; ** P<0.01; *** P<0.001 vs. untreated control; ## P<0.01 vs. LPS or 1,25(OH)2D3 alone.
1,25(OH)2D3 down-regulates bic transcription via blocking NF-κB activation
It has been speculated that NF-κB is involved in the up-regulation of miR-155 (33), but no functional κB cis-DNA elements have been identified in the mouse bic gene promoter. We found that LPS induction of bic transcript could be blocked by NF-κB inhibitor Bay 11-7082 (Fig. S4A), and that siRNA silencing of p65 abrogated LPS induced bic expression in RAW264.7 cells (Fig. S4B and S4C), confirming the importance of NF-κB activation in the induction of bic/miR-155. Through careful in silico analysis we identified a putative κB cis-element within the first intron of the bic gene (Fig. 9A) that shared >95% homology to the canonical κB sequence. ChIP assays showed that LPS induced p65 binding to this site, and the binding was attenuated by 1,25(OH)2D3 (Fig. 9B and 9C). EMSA showed that, like the canonical κB probe, this intronic κB probe interacted with p65/p50 (Fig. 9D, lanes 1 and 3), and the binding was competed off by an excess amount of unlabeled canonical κB probe (lanes 8 and 9) or WT bic intronic κB probe (lane 4 and 5), but not by a mutant intronic κB probe (lane 6 and 7, Fig. 9D).
Figure 9.
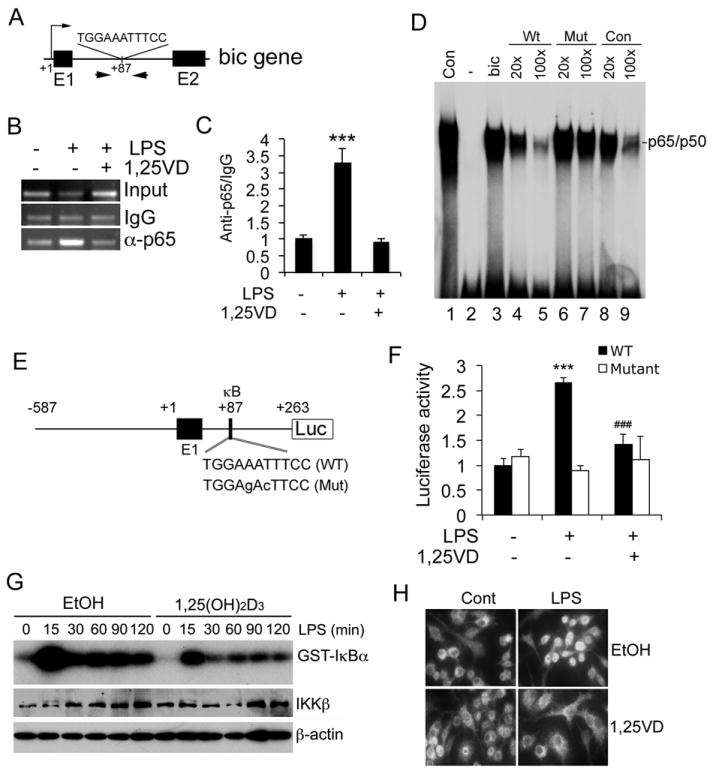
1,25(OH)2D3 down-regulates bic expression by blocking NF-κB activation. (A) Illustration of the putative κB cis-DNA element and ChIP primers within the first intron of the bic gene; (B and C) ChIP assays. LPS promotes p65 binding to the intronic κB site and 1,25(OH)2D3 blocks p65 binding in RAW264.7 cells. The DNA fragment precipitated by anti-p65 antibodies was assessed by regular PCR (B) and qPCR (C). *** P<0.001 vs. the rest. (D) EMSA. 32P-labelled canonical κB probe (Con, lane 1) or bic intronic κB probe (bic, lane 3) was incubated with in vitro translated p65 and p50. 32P-labelled bic intronic κB probe and NF-κB reaction was carried out in the presence of excess amount of unlabeled bic κB probe (lanes 4 and 5), mutant bic κB probe (lanes 6 and 7) or canonical bic κB probe (lanes 8 and 9). (E) Illustration of bic luciferase reporter constructs with WT and mutant intronic κB sequences. (F) Luciferase reporter assays. RAW264.7 cells were transfected with WT or mutant bic luciferase reporter plasmid followed by treatment with saline control (Cont) or LPS in the presence or absence of 1,25(OH)2D3. *** P<0.001 vs. Cont; ### P<0.001 vs. LPS; n=3. (G) IKK kinase assays and IKKβ protein levels in RAW264.7 cells treated with LPS in the presence of ethanol or 1,25(OH)2D3; (H) Immunostaining with anti-p65 antibodies of RAW264.7 cells treated with saline control (Cont) or LPS in the presence of ethanol (EtOH) or 1,25(OH)2D3.
We then cloned a DNA fragment covering -587 to +263 from the bic gene that contains the intronic κB site into luciferase reporter vector pGL3, and also generated a mutant construct that carried mutations at the intronic κB site (Fig. 9E). Luciferase reporter assays showed that LPS markedly stimulated luciferase activity in RAW264.7 cells transfected with the WT luciferase reporter, and the stimulation was attenuated by 1,25(OH)2D3; however, mutations at the κB site eliminated LPS induction as well as 1,25(OH)2D3 inhibition (Fig. 9F). These results confirmed the functionality of this intronic κB cis-element, and revealed that this κB element mediated the down-regulation of bic gene transcription by 1,25(OH)2D3. In fact, IκB kinase (IKK) assays showed that 1,25(OH)2D3 attenuated LPS-induced IκBα phosphorylation in macrophages (Fig. 9G), which explains why 1,25(OH)2D3 stabilized IκBα protein reported previously (34, 35), and immunostaining confirmed that 1,25(OH)2D3 blocked LPS-induced p65 nuclear translocation RAW264.7 cells (Fig. 9H). Therefore 1,25(OH)2D3 indeed is able to block NF-κB activation. Together these data demonstrate that 1,25(OH)2D3 down-regulates bic transcription by blocking NF-κB activation.
Discussion
Inflammatory response is regulated by negative feedback mechanisms to avoid excessive reaction. SOCS1 a critical negative regulator as it blocks the JAK/STAT signaling pathway of pro-inflammatory cytokines (2, 3, 6) as well as suppresses IRAK4 in the TLR4 signaling pathway (9, 36, 37). During endotoxin-induced inflammatory reaction the rapid increase in miR-155 suppresses SOCS1 translation, which allows inflammatory reaction to proceed and sustain without resistance (Fig. 10). In this study we demonstrated that vitamin D-VDR signaling down-regulates bic gene transcription by blocking NF-κB activation, leading to reduction in miR-155. As a result SOCS1 translation is increased, which enhances the negative feedback regulation of inflammatory response (Fig. 10). We conclude that this is the molecular basis of a novel anti-inflammatory mechanism whereby vitamin D-VDR signaling limits TLR4-mediated inflammation.
Figure 10.
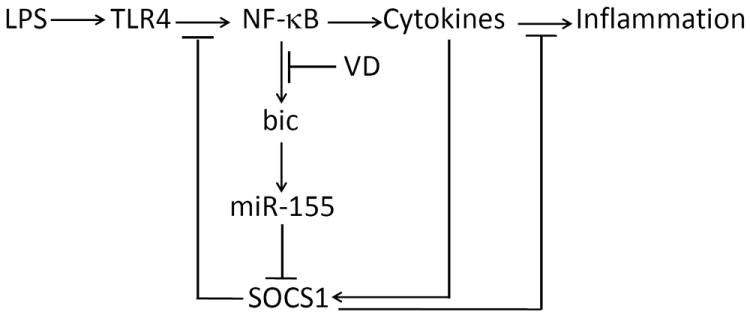
Proposed mechanism whereby VDR signaling down-regulates miR-155 and enhances the negative feedback inhibition of TLR4-mediated inflammatory response. VD, 1,25(OH)2D3 or its analogs.
Vitamin D is a well known immune modulator and affects both innate and adaptive immunities (18). 1,25(OH)2D3 inhibits T cell activation and proliferation (38) and suppresses IL-2 and IFN-γ production in TH1 cells (39, 40). On the other hand, 1,25(OH)2D3 promotes TH2 cell differentiation and increases IL-4 production (41, 42). 1,25(OH)2D3 inhibits antigen-presenting dendritic cell differentiation and suppresses the production of IL-12, a cytokine crucial for the development of TH1 cells (43). 1,25(OH)2D3 also inhibits TH17 cell response due to its capacity to inhibit IL-6 and IL-23 production (44, 45), and induces differentiation and expansion of FOXP3+ Treg cells (46). Recent studies reported that VDR is required for natural killer T cell development (47), TCR signaling and T cell activation (48). With regards to innate immunity, Liu et al have demonstrated an anti-microbial capability of the local macrophage vitamin D-VDR system. TLR4 activation triggered by bacteria induces VDR expression and local 1,25(OH)2D3 biosynthesis in macrophages, which in turn stimulate anti-microbial peptide cathelicidin expression to suppress bacterial growth (19). In this report we revealed a novel regulatory mechanism for the VDR signaling to control TLR4-mediated inflammation. This anti-inflammatory mechanism is mediated by miR-155 down-regulation, which de-represses SOCS1 leading to heightened negative feedback regulatory action. The importance of miR-155 in vitamin D regulation of innate immunity was confirmed by the experiments using the miR-155-null model and the miR-155 hairpin inhibitor in VDR-null macrophages. Consequently in the absence of VDR LPS triggers a hyper and sustained inflammatory response in macrophages, primarily due to de-regulation of the negative feedback loop. This is caused by miR-155 up-regulation leading to excessive suppression of SOCS1. Together these data demonstrate that the vitamin D-VDR signaling attenuates TLR4-mediated inflammatory reaction by upgrading the negative feedback regulation. This conclusion predicts potential detrimental effects of vitamin D-deficiency, under which condition inflammatory reaction may be unchecked and over-sustained. This may in part explain the association of vitamin D-deficiency and inflammatory disorders in humans (49-52).
Our data show that vitamin D-VDR signaling inhibits miR-155 by a transcriptional mechanism. We identified a functional κB cis-DNA element within the first intron of the mouse bic gene, and proved that this intronic κB site interacts with NF-κB and mediates the up-regulation of bic transcription upon TLR4 activation. 1,25(OH)2D3 suppresses LPS-induced bic transcription by blocking the activation of NF-κB, namely by inhibiting IKK activity leading to IκBα stabilization and arrest of p65/p50 nuclear translocation. However, exactly how vitamin D regulates NF-κB remains unclear, and the detailed molecular mechanism remains to be fully defined in future studies.
As an important regulator in the immune system, miR-155 has multiple targets involved in immune functions. In addition to SOCS1, miR-155 also represses SHIP-1 and C/EBPβ (53, 54), two key negative regulators of IL-6 signaling pathway. It has been reported that miR-155 can also attenuates TLR4 signaling by targeting IKKε (55). Thus it is postulated that vitamin D may modulate immune response by also regulating SHIP-1, C/EBPβ and/or IKKε via miR-155. This speculation warrants further investigations. Moreover, miR-155 has been detected in a variety of immune cells including B cells (24), T cells (56), macrophages (13), dendritic cells (57) and hematopoietic progenitor cells (58). Given the broad functionalities of miR-155, it is possible that vitamin D may regulate immune activities of other immune cells through miR-155. In this regard, this study has opened new avenues to explore various immunomodulatory mechanisms of vitamin D.
Supplementary Material
Acknowledgments
We thank Dr. Yinyuan Mo (Southern Illinois University School of Medicine) for providing pCDH-hmiR-155 plasmid.
Funding: This work was supported in part by NIH grant R01HL085793, R01DK092143 and CTSA grant UL1 RR024999 from National Center for Research Resources (NCRR).
Footnotes
Author contribution
Y.C., W.L., T.S. and D.Y. performed the research; Y.C. and Y.C.L. designed the research, analyzed data and wrote the paper; Y.W., D.K.D and J.K. provided research reagents and technical assistance; R.T. assisted in data analysis and manuscript preparation; and Y.C.L was responsible for the overall research design, data analysis and paper preparation.
Conflict of interest disclosure
The authors claim no competing interests in this work.
References
- 1.Ziegler-Heitbrock HW. Molecular mechanism in tolerance to lipopolysaccharide. Journal of inflammation. 1995;45:13–26. [PubMed] [Google Scholar]
- 2.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nature reviews. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 3.Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol. 2002;2:410–416. doi: 10.1038/nri818. [DOI] [PubMed] [Google Scholar]
- 4.Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 5.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, Seki E, Sato S, Takeuchi O, Takeda K, Akira S, Yamanishi K, Kawase I, Nakanishi K, Kishimoto T. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 6.Fujimoto M, Naka T. Regulation of cytokine signaling by SOCS family molecules. Trends in immunology. 2003;24:659–666. doi: 10.1016/j.it.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Chong MM, Metcalf D, Jamieson E, Alexander WS, Kay TW. Suppressor of cytokine signaling-1 in T cells and macrophages is critical for preventing lethal inflammation. Blood. 2005;106:1668–1675. doi: 10.1182/blood-2004-08-3049. [DOI] [PubMed] [Google Scholar]
- 8.Yang T, Stark P, Janik K, Wigzell H, Rottenberg ME. SOCS-1 protects against Chlamydia pneumoniae-induced lethal inflammation but hampers effective bacterial clearance. J Immunol. 2008;180:4040–4049. doi: 10.4049/jimmunol.180.6.4040. [DOI] [PubMed] [Google Scholar]
- 9.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 10.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 11.O’Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nature reviews. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 12.Tam W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene. 2001;274:157–167. doi: 10.1016/s0378-1119(01)00612-6. [DOI] [PubMed] [Google Scholar]
- 13.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruggiero T, Trabucchi M, De Santa F, Zupo S, Harfe BD, McManus MT, Rosenfeld MG, Briata P, Gherzi R. LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. Faseb J. 2009;23:2898–2908. doi: 10.1096/fj.09-131342. [DOI] [PubMed] [Google Scholar]
- 15.Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN, Tsatsanis C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31:220–231. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haussler MR, Whitfield GK, Haussler CA, Hsieh J-C, Thompson PD, Selznick SH, Dominguez CE, Jurutka PW. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone and Mineral Research. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 17.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart PH, Gorman S, Finlay-Jones JJ. Modulation of the immune system by UV radiation: more than just the effects of vitamin D? Nat Rev Immunol. 2011;11:584–596. doi: 10.1038/nri3045. [DOI] [PubMed] [Google Scholar]
- 19.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 20.Adams JS, Hewison M. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab. 2008;4:80–90. doi: 10.1038/ncpendmet0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Etten EV, Mathieu C. Immunoregulation by 1,25-dihydroxyvitamin D(3): Basic concepts. The Journal of steroid biochemistry and molecular biology. 2005;97:93–101. doi: 10.1016/j.jsbmb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Gurlek A, Pittelkow MR, Kumar R. Modulation of growth factor/cytokine synthesis and signaling by 1alpha,25-dihydroxyvitamin D(3): implications in cell growth and differentiation. Endocr Rev. 2002;23:763–786. doi: 10.1210/er.2001-0044. [DOI] [PubMed] [Google Scholar]
- 23.Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–9835. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 25.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Zhang Y, Ning G, Deb DK, Kong J, Li YC. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: blockade of compensatory renin increase. Proc Natl Acad Sci U S A. 2008;105:15896–15901. doi: 10.1073/pnas.0803751105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valoczi A, Hornyik C, Varga N, Burgyan J, Kauppinen S, Havelda Z. Sensitive and specific detection of microRNAs by northern blot analysis using LNA-modified oligonucleotide probes. Nucleic acids research. 2004;32:e175. doi: 10.1093/nar/gnh171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li YC, Bolt MJG, Cao L-P, Sitrin MD. Effects of vitamin D receptor inactivation on the expression of calbindins and calcium metabolism. Am J Physiol Endocrinol Metab. 2001;281:E558–E564. doi: 10.1152/ajpendo.2001.281.3.E558. [DOI] [PubMed] [Google Scholar]
- 29.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 30.Yuan W, Pan W, Kong J, Zheng W, Szeto FL, Wong KE, Cohen R, Klopot A, Zhang Z, Li YC. 1,25-Dihydroxyvitamin D3 Suppresses Renin Gene Transcription by Blocking the Activity of the Cyclic AMP Response Element in the Renin Gene Promoter. J Biol Chem. 2007;282:29821–29830. doi: 10.1074/jbc.M705495200. [DOI] [PubMed] [Google Scholar]
- 31.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 32.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nature reviews. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 33.Gatto G, Rossi A, Rossi D, Kroening S, Bonatti S, Mallardo M. Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-kappaB pathway. Nucleic acids research. 2008;36:6608–6619. doi: 10.1093/nar/gkn666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun J, Kong J, Duan Y, Szeto FL, Liao A, Madara JL, Li YC. Increased NF-{kappa}B activity in fibroblasts lacking the vitamin D receptor. Am J Physiol Endocrinol Metab. 2006;291:E315–322. doi: 10.1152/ajpendo.00590.2005. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z, Yuan W, Sun L, Szeto FL, Wong KE, Li X, Kong J, Li YC. 1,25-Dihydroxyvitamin D(3) targeting of NF-kappaB suppresses high glucose-induced MCP-1 expression in mesangial cells. Kidney Int. 2007;72:193–201. doi: 10.1038/sj.ki.5002296. [DOI] [PubMed] [Google Scholar]
- 36.Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–268. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Janssens S, Burns K, Tschopp J, Beyaert R. Regulation of interleukin-1- and lipopolysaccharide-induced NF-kappaB activation by alternative splicing of MyD88. Curr Biol. 2002;12:467–471. doi: 10.1016/s0960-9822(02)00712-1. [DOI] [PubMed] [Google Scholar]
- 38.Rigby WF, Stacy T, Fanger MW. Inhibition of T lymphocyte mitogenesis by 1,25-dihydroxyvitamin D3 (calcitriol) J Clin Invest. 1984;74:1451–1455. doi: 10.1172/JCI111557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rigby WF, Denome S, Fanger MW. Regulation of lymphokine production and human T lymphocyte activation by 1,25-dihydroxyvitamin D3. Specific inhibition at the level of messenger RNA. J Clin Invest. 1987;79:1659–1664. doi: 10.1172/JCI113004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemire JM. Immunomodulatory role of 1,25-dihydroxyvitamin D3. J Cell Biochem. 1992;49:26–31. doi: 10.1002/jcb.240490106. [DOI] [PubMed] [Google Scholar]
- 41.Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O’Garra A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol. 2001;167:4974–4980. doi: 10.4049/jimmunol.167.9.4974. [DOI] [PubMed] [Google Scholar]
- 42.Mahon BD, Wittke A, Weaver V, Cantorna MT. The targets of vitamin D depend on the differentiation and activation status of CD4 positive T cells. J Cell Biochem. 2003;89:922–932. doi: 10.1002/jcb.10580. [DOI] [PubMed] [Google Scholar]
- 43.D’Ambrosio D, Cippitelli M, Cocciolo MG, Mazzeo D, Di Lucia P, Lang R, Sinigaglia F, Panina-Bordignon P. Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J Clin Invest. 1998;101:252–262. doi: 10.1172/JCI1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorman S, Kuritzky LA, Judge MA, Dixon KM, McGlade JP, Mason RS, Finlay-Jones JJ, Hart PH. Topically applied 1,25-dihydroxyvitamin D3 enhances the suppressive activity of CD4+CD25+ cells in the draining lymph nodes. J Immunol. 2007;179:6273–6283. doi: 10.4049/jimmunol.179.9.6273. [DOI] [PubMed] [Google Scholar]
- 45.Penna G, Amuchastegui S, Cossetti C, Aquilano F, Mariani R, Sanvito F, Doglioni C, Adorini L. Treatment of experimental autoimmune prostatitis in nonobese diabetic mice by the vitamin D receptor agonist elocalcitol. J Immunol. 2006;177:8504–8511. doi: 10.4049/jimmunol.177.12.8504. [DOI] [PubMed] [Google Scholar]
- 46.Penna G, Roncari A, Amuchastegui S, Daniel KC, Berti E, Colonna M, Adorini L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood. 2005;106:3490–3497. doi: 10.1182/blood-2005-05-2044. [DOI] [PubMed] [Google Scholar]
- 47.Yu S, Cantorna MT. The vitamin D receptor is required for iNKT cell development. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5207–5212. doi: 10.1073/pnas.0711558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Essen MR, Kongsbak M, Schjerling P, Olgaard K, Odum N, Geisler C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol. 2010;11:344–349. doi: 10.1038/ni.1851. [DOI] [PubMed] [Google Scholar]
- 49.Gombart AF, Bhan I, Borregaard N, Tamez H, Camargo CA, Jr, Koeffler HP, Thadhani R. Low plasma level of cathelicidin antimicrobial peptide (hCAP18) predicts increased infectious disease mortality in patients undergoing hemodialysis. Clin Infect Dis. 2009;48:418–424. doi: 10.1086/596314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loftus EV, Jr, Sandborn WJ. Epidemiology of inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31:1–20. doi: 10.1016/s0889-8553(01)00002-4. [DOI] [PubMed] [Google Scholar]
- 51.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 52.Lim WC, Hanauer SB, Li YC. Mechanisms of Disease: vitamin D and inflammatory bowel disease. Nature Clinical Practice Gastroenterology & Hepatology. 2005;2:308–315. doi: 10.1038/ncpgasthep0215. [DOI] [PubMed] [Google Scholar]
- 53.Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, Ciarlariello D, Neviani P, Harb J, Kauffman LR, Shidham A, Croce CM. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009;114:1374–1382. doi: 10.1182/blood-2009-05-220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 56.Haasch D, Chen YW, Reilly RM, Chiou XG, Koterski S, Smith ML, Kroeger P, McWeeny K, Halbert DN, Mollison KW, Djuric SW, Trevillyan JM. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cellular immunology. 2002;217:78–86. doi: 10.1016/s0008-8749(02)00506-3. [DOI] [PubMed] [Google Scholar]
- 57.Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, Pierre P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2735–2740. doi: 10.1073/pnas.0811073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, Paquette RL, Baltimore D. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. The Journal of experimental medicine. 2008;205:585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.