

Abstract
Multi-molecular protein complexes are critical to many cellular functions, including signaling, DNA transcription and enzymatic reactions. In spite of their importance, current research techniques such as biochemistry and diffraction-limited microscopy cannot resolve the heterogeneity and nanoscale organization of protein complexes in intact cells. Here we describe a technique that enables the study of multi-molecular protein complexes at the single molecule level in intact cells. The technique uses photoactivated localization microscopy (PALM) to resolve individual proteins with a resolution down to 20nm in intact cells, and second-order statistics to study the spatial interactions of the proteins. We demonstrate the feasibility of this technique by studying signaling complexes that form in activated T cells. We first use single color PALM imaging and univariate second-order statistics to resolve the clustering of Linker for Activation of T cells (LAT) at the plasma membrane (PM) of the cells. We then use two color PALM and bivariate second-order statistics to resolve the interaction of LAT with key interacting proteins. We discuss potential caveats in studying molecular clustering and the robustness of the technique to study bimolecular interactions. Our proposed technique, combined with older techniques, could help shed new light on the nature of multimolecular protein complexes and their significance to cell function.
Keywords: Protein interactions, super resolution microscopy, photoactivated light microscopy, single molecule second-order statistics, T cell activation, microclusters
1. Introduction
Intracellular proteins frequently form multimolecular structures or complexes that convey signals or perform enzymatic reactions and other cellular functions [1–3]. Such protein complexes are generated through dynamic and stochastic processes that frequently give rise to heterogeneous structures of poorly defined content and structure. Unfortunately, current research techniques are ill-suited to study the heterogeneity of protein complexes, their fine structure and the process of their formation. Existing biochemical techniques and diffraction-limited light microscopy average important structural and functional properties of molecular ensembles, and as a result, the output of each technique suffers a critical loss of detail at the level of the individual complex. Likewise, electron microscopy and crystallography fail to resolve the heterogeneity and dynamics of these entities in situ.
Recently, super-resolution techniques that allow the visualization of details beyond the diffraction limit of visible light have revolutionized optical microscopy. In particular, techniques have been developed to locate single fluorescent molecules by activating a small number of molecules (less than one molecule per diffraction limited area), and then inactivating them or allowing them to photobleach. This cycle can be repeated until enough molecules have been imaged to produce a high resolution image or until all fluorescent molecules have been depleted. One of these methods, photoactivated localization microscopy (PALM) uses stochastic photoactivation or photoconversion of a few well-spaced molecules tagged with photoactivatable or photoswitchable fluorescent proteins (PAFPs or PSFPs, respectively), to generate an image with nanoscale resolution [4]. PALM has been used to study the fine details of several biological systems including mitochondria [4–5], dendritic spines [6–7] and focal adhesions [4, 8].
In this work, we describe an experimental approach and a statistical framework to study signaling complexes in intact cells at the single molecule level. We start by introducing a method to study protein clustering using PALM and second order statistics (i.e. statistics relating pairs of points). We demonstrate how this method can be used to study the clustering of the critical membrane molecule LAT at the PM of activated T cells. We then use two color PALM imaging and bivariate second order statistics to study the interactions of two molecular species in highly overlapping locations. Here, we demonstrate how this method enables the study of protein interactions within signaling complexes that are nucleated by LAT and that play a central role in T cell activation [2]. We further discuss various experimental factors and considerations related to the data analysis and their effects on the results. Finally, we discuss the pros and cons of our approach in comparison to existing methods for studying protein-protein interactions and the usefulness of the methods in the study of protein complexes of interest.
2. Materials and Methods
In 2006, it was shown that localization microscopy using photoactivatable fluorescent proteins was both possible and practical [4, 9]. Since then, many articles have been written detailing the procedures required for PALM imaging [10–15]. Our intent is not to review that information here but to indicate only briefly the requirements for PALM imaging in general. We then focus on methods needed for two color PALM imaging and on a statistical framework that allows the study of protein-protein interactions using PALM.
2.1 Instrumentation
PALM imaging relies on the ability to visualize the fluorescence produced by a single molecule, so sensitive and efficient detectors are needed. The most commonly used detector is an electron multiplying charge coupled device (EMCCD) camera capable of acquiring data at high speed. These cameras are now available from a variety of manufacturers with a range of sensitivity, speed and resolution. Moreover, the optical system should be optimized to capture as many photons as possible. Generally, a high numerical aperture (NA) objective is used along with high quality filters and dichroics to gather as much light as possible. High power lasers (10–100 mW) are needed to excite the activated molecules and to achieve fast frame rates. In addition, software that can synchronize lasers and camera is quite helpful. A computer capable of acquiring and storing a large number of frames is also needed.
Often, PALM imaging is performed using a total internal reflection system (TIRF) to increase z resolution and to reduce background fluorescence, however a widefield system could also be used. It is also possible to perform PALM using methods that deform the point spread function so that its shape depends on the z position. This allows increased z resolution to complement the increased lateral resolution of standard PALM imaging. A full discussion of three-dimensional PALM is beyond the scope of this article. While commercial systems are now available for PALM and other high-resolution methods, it is not difficult to modify a standard microscope to perform PALM imaging. Our system is based on a Nikon TIRF microscope equipped with a custom laser launch and an Andor iXon DU888 EM-CCD camera.
2.2 Experimental procedures
2.2.1 Labelling strategies
PALM depends upon the use of photoactivatable or photoswitchable fluorescent proteins (PAFPs and PSFPs respectively). In response to activating light, these proteins either change from a dark to fluorescent state or alter their emission spectra. Several recent publications contain lists of proteins that are suitable for PALM imaging [12–13, 16–17], although the number of usable probes seems to increase daily. Obviously, a pair of PAFPs with complementary properties is needed for two color work. One of the first studies of two color PALM used a combination of a photoactivatable protein, Dronpa, and a complementary photoswitchable protein, EosFP [10]. We choose to use the combination of Dronpa (MBL International Corporation) and PAmCherry [18] for two color PALM imaging as these proteins can be distinguished during imaging by both the intensity of the activating light and their spectral properties. Dronpa requires much less activation energy and has spectral properties similar to GFP (ex 488/em 505). PAmCherry requires higher intensity activating light and has spectral properties similar to mCherry (ex 561/em 565). Table 1 shows some of the combinations that have been used in published studies of two color PALM. Importantly, the study of protein-protein interactions requires low cross-talk between the two channels that serve to detect molecules of two types. This requirement is typically met by methods that rely on relatively large spectral separation between the emission of the imaged PAFPs (e.g. PAmCherry and PAGFP or Dronpa). To quantify the extent of cross-talk relevant to our studies, we imaged cells expressing either PAmCherryactin or Dropa-actin by two color PALM, imaging Dronpa first and then PAmCherry. We measured ~2% of Dronpa molecules, misidentified in the red channel and < 0.25% of misidentified molecules of PAmCherry in the green channel (Supp. Figure 1). PALM imaging with three colors has been demonstrated, but significant cross-talk between the spectrally overlapping channels in these studies seems problematic to the study of protein-protein interactions [19].
Table 1.
Examples of probes used for two color PALM
| Fluorescent Protein 1 | Activation | Fluorescent Protein 2 | Activation | Ref. |
|---|---|---|---|---|
| Dronpa | Dark to Ex503/Em 518 | EosFP | Ex506/Em 519 to Ex573/Em 584 | [5, 10, 32] |
| bs-Dronpa | Dark to Ex385+460/Em 505 | Dronpa | Dark to Ex503/Em 518 | [33] |
| PAGFP | Dark to Ex504/Em 517 | PAmCherry | Dark to Ex564/Em 595 | [34] |
| Dronpa | Dark to Ex503/Em 518 | PAmCherry | Dark to Ex564/Em 595 | [25] |
| PAGFP | Dark to Ex504/Em 517 | PATagRFP | Dark to Ex562/Em 595 | [35] |
When using genetically encoded probes, the details of the construct and expression system are important. In our work, we used the full length coding sequence of the protein of interest conjugated to a PAFP; however in some cases it might be preferable to use just the binding domains of particular proteins fused to the fluorescent probes to study protein-protein interactions. The conjugated proteins are usually cloned into plasmids that contain strong, constitutive promotors such as CMV. The level of overexpression generated by these promotors may be too high from some systems and it may be necessary to consider regulated gene expression to ensure appropriate protein levels. Our constructs were generated in EGFP-N1 or EGFP-C1 vectors (Clontech) that contain a CMV promotor. Different transfection approaches can be used depending on the cells being studied. For our studies, E6.1 Jurkat T cells or human lymphocytes were transfected with the DNA plasmids using a Nucleofector shuttle system, and the Amaxa T-kit (LONZA).
In two color PALM, it is necessary to insure that both proteins are being expressed. PALM imaging is a long process and the two colors are often collected sequentially, so the presence of singly transfected cells is a significant problem for data collection. One strategy for expressing two proteins is to produce a stable cell line expressing one PAFP and then supertransfect with the second construct. The extent of protein expression in the stable cells can be checked by flow cytometry to ensure stable and uniform expression. One disadvantage of this approach is that the levels of the two PAFPs tend to be inversely related. Alternatively, both constructs can be introduced at the same time, which generally gives proportional labeling of both proteins. We found that sorting was required to insure that all cells contain sufficient levels of both PAFPs no matter which transfection procedure was used. In addition, sorting the transfected cells allows control of the expression levels of the transfected proteins. We also used the fluorescence levels of the stable cell lines to set the gates that were used to sort our samples, thus controlling the expression level of the transiently expressed PAFPs. Transiently transfected cells were sorted 24 hours after transfection and imaged within 48–72 hours.
Conditions used for several PAFPs are as follows. Dronpa has enough basal fluorescence for detection using flow cytometry, so cells expressing Dronpa-tagged proteins were directly sorted for their GFP-like green emission (488 nm excitation, 500–520 nm emission). PAmCherry transfected cells were first activated using a light emitting diode (LED) source (a blue light source, CoolLED, PE-100, HITECH Instruments for 10 min). The emission of the LED light source was directed onto a single well of a 24-well plate that contained the cells, suspended in a buffer without phenol red. Cells were then sorted for red fluorescence (561 nm excitation, 560–600 nm emission). Sorting was performed by the NCI flow cytometry core facility using a Moflo Astrios high speed cell sorter (Beckman Coulter) and Summit software. The cells were then rested for at least 24 hours before imaging.
2.2.2 Fiduciary markers
In order to produce a super-resolution image, the capture frames must be aligned. In two color PALM, the images of the separate channels must also be superimposed. This requires the use of appropriate fiduciary markers that can be seen in both channels. The most commonly used markers are gold beads, between 50 and 100 nm in size, which reflect all wavelengths. Some studies have used fluorescent beads, particularly small (100 nm) multi-wavelength beads such as TetraSpeck beads from Invitrogen. In our studies, cleaned coverslips were coated with 100 nm gold beads (Microspheres-nanospheres) that had been sonicated and diluted in methanol. The coverslips with beads were dried and then coated with poly-L-lysine before a final coating with activating antibodies.
2.2.3 Data capture and localization of molecules
As with other forms of multi-color fluorescence microscopy, imaging of two color PALM samples can be performed simultaneously or sequentially. Simultaneous capture requires two separate detectors or the ability to image different wavelengths on different parts of the CCD chip. The use of a “chip splitter” can introduce non-linear distortions that are difficult to correct even with fiduciary markers. We used a sequential capture strategy. As a first step of the imaging sequence, Dronpa-tagged proteins were imaged using continuous and low intensity illumination of an arc lamp (DAPI cube, excitation 340–380 nm) and laser excitation at 488 nm in TIRF mode with optical elements suitable for visualizing GFP. After imaging Dronpa, the sample was illuminated with maximal intensity of the arc lamp illumination (CFP cube, excitation 426–446 nm) for 10–20 sec to activate a fraction of PAmCherry-tagged proteins. This light also bleached any remaining Dronpa molecules. Finally the PAmCherry images were collected in TIRF mode with optical elements suitable for visualizing red fluorescent proteins until the fluorescence of that set of molecules was depleted. The photoactivation and imaging of PAmCherry was then repeated multiple times.
The final step in PALM imaging is the identification and localization of individual molecules. A number of open source software packages are available for this task [15], including a dedicated ImageJ plug-in [20]. Proprietary software has also been developed by the microscope manufacturers who produce commercial localization microscope systems. We analysed our PALM images with the PeakSelector software that identifies individual molecules in the movie frames and renders the PALM images [4]. Using this software, individual molecules are presented in PALM images with intensity that corresponds to the probability density values of their fitted Gaussian distribution in respect to the maximal probability density values detected in the field. This is a common mode of presentation of PALM images.
2.2.3 Experimental system for activated T cells
To study protein interactions and clustering at the PM of cells by PALM, we activated T cells with immobilized stimulatory antibodies [21]. Briefly, we plated T cells expressing PAFPs on coverslips coated with a TCR-stimulating antibody (αCD3ε). Upon engagement with the coated coverslips, the cells become activated and spread on the coverslips, thus allowing the imaging of the PM using TIRF microscopy. Following three minutes of activation at 37°C, the cells were fixed with a low concentration (2.4%) of paraformaldehyde for 30 minutes. PALM imaging of the fixed cells was performed as soon as possible and the samples were protected from light to avoid both activation and photobleaching of the fluorescent proteins. We have found that photoactivation can be impaired by storage at 4°C, so samples were always kept at room temperature following fixation.
3. Data analysis
Once super resolution images of individual molecules are produced, they must be analyzed to understand the relationship between the molecules in the image. One important question is whether the molecules self-associate and thus show non-random distributions or clustering. We discuss this question in section 3.1. A more general question is the extent of interaction between two different types of molecules. This question is addressed in section 3.2.
3.1 Statistics for resolving molecular clustering
3.1.1 Univariate second order statistics and alternatives
Second order statistics relate the relative positions of pairs of points in a given point pattern. Thus, such statistics are useful to study protein-protein interactions in the molecular point patterns that are generated by PALM microscopy. Here, we consider two types of second order statistics - Ripley's statistics and the pair-correlation function (PCF). The two statistics are closely related as Ripley's statistics are the integral form of the PCF [22]. For that reason, Ripley's statistics report on significant interactions in point patterns up to a specific length-scale, whereas the PCF reports on the interactions at the specific length scale. To analyze our data using second order statistics, we used a published algorithm by Wiegand and Moloney [23]. This algorithm implements the statistical analyses by pixelizing the point patterns for practical reasons. This approach simplifies the analysis of patterns with irregular shapes and avoids problems in the analysis caused by study region boundaries. We used a pixel size of 20nm to match the resolution limit of our PALM imaging.
An alternative statistical approach to second order statistics is a clustering algorithm based on nearest neighbor proximity [24–25]. Briefly, in this method two proteins are considered to reside in the same cluster if they lie closer to each other than a specific distance threshold. The clustering algorithm then integrates proteins into existing clusters and coalesces clusters based on the distance criterion in an iterative process. Importantly, the clustering algorithm can provide information on the individual clusters, such as the number of molecules. This kind of information is lost in the second order statistics, and thus, these two approaches can be regarded as complementary. In spite of its usefulness, we will not discuss the clustering approach further and refer the interested reader to previous work for its implementation [24]. For brevity, we focus here on the use of second order statistics to analyze PALM images. Specifically, univariate PCF statistics are used to report on protein clustering for reasons that will be discussed in section 3.1.2. Additionally, we use bivariate PCF statistics to resolve the extent of protein-protein interactions, as detailed in section 3.2.
3.1.2 Resolving multiple levels of clustering
Molecular clustering, deduced by the statistics discussed above, is demonstrated by the significant deviation of local molecular densities from random distributions. The simplest random distribution that can be considered can be created by a homogeneous Poisson process. Through this modelling process, molecules are randomly distributed across the study region without preference for a particular sub-region. Thus, this process is useful in modelling complete spatial randomness (CSR). However, it is worth noting that multiple mechanisms can give rise to statistically significant molecular clustering, i.e. regions where the local concentration of molecules is higher than the concentration predicted by CSR. Specifically, in our example, the distribution of LAT in the PM of activated T cells is affected by the spreading process. As a cell approaches the stimulatory surface, lamellar protrusions make contact with the surface and LAT, along with other PM proteins, is confined within these lamellae. As the cell spreads, more of the PM makes contact with the stimulatory surface, but LAT clusters are retained within the irregular lamellar pattern at the sites of initial contact. This gives rise to an apparent clustering that is not related to protein-protein interactions. One way to overcome this problem is to analyze study regions without large holes where LAT molecules are absent (Figure 1D, yellow rectangle). Nevertheless, in most regions smaller holes are still visible and cannot be avoided entirely. As a better solution, we consider a heterogeneous Poisson process as our null hypothesis for molecular clustering. In this process, the local density of molecules is first detected across the study region (Figure 1F). Then, molecules are distributed across the study region through a random process in which the probability of localizing a molecule at any location is proportional to the detected local density. Clustering driven by molecular interactions can now be defined as molecular densities that exceed the densities generated by the heterogeneous Poisson process
Figure 1. Studying protein clustering by PALM imaging and univariate PCF.
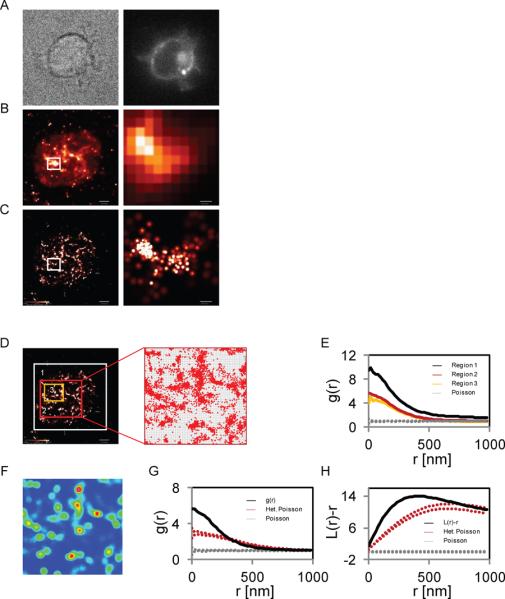
Images of a Jurkat E6.1 T cell expressing LAT-PAmCherry and spread on a TCR-stimulating coverlsip with (A) brightfield microscopy (left) or epiillumination (right), (B) TIRF microscopy (left) and zoomed region (right), and (C) PALM microscopy (left) and zoomed region (right). (D) Different study regions of the cell shown in panels A–C and an example of pixelization of one of the study regions. (E) Univariate pair-correlation function, g(r), (PCF) of the identified molecules within the study regions in panel D. (F) Heterogeneity in the density of molecules within one of the study regions in panel D. (G) PCF ,g(r), of the study region considering either a homogeneous Poisson process for calculating the 95% confidence interval (between the upper and lower gray dotted lines) or a heterogeneous Poisson process (between the upper and lower red dotted lines) corresponding to the density map in panel F. (H) Ripley's second order statistics, L(r)-r, with a similar study region and null models as in panel (G). Bars - 2 μm for whole cell images and 200nm for zoomed images.
3.1.3 Determination of significance using Monte-Carlo simulations
To quantify the significance of molecular clustering, we generated 19 random point patterns using Monte-Carlo simulations following the model that corresponds to a suitable null hypothesis, namely either a homogeneous or a heterogeneous Poisson process. Each random set had the same number of molecules as the point pattern under study. The highest and lowest values were taken at each length scale to determine the 95% confidence interval of the null model. If the PCF curve is located within this confidence interval, the null hypothesis is valid. If the PCF diverges from this interval, the null hypothesis can be rejected with a confidence level of >95%.
3.2 Statistics for resolving bimolecular interactions
3.2.1 Bivariate PCF and models for molecular interactions
Bivariate PCFs are a simple extension of univariate PCF discussed above. In this analysis, every molecule of type I is related to all of the molecules of type II. We can consider several models for molecular interactions that might serve as null hypotheses to interpret our data: (i) molecules of two species can efficiently interact, where all molecule of type I associate with molecules with type II. Here, the detected point patterns can be compared to random patterns where the molecular positions are kept but the labels of the molecules are randomly switched. We refer to this process as random labeling. If the interaction between the two molecules is efficient, the bivariate PCF will follow the 95% confidence interval produced by the random labeling model. (ii), the two types of molecules might not interact and show independence in their positioning within the study region. In this case, the bivariate PCF will be flat and equal to 1. (iii) the two types of molecules might demonstrate a partial interaction where a fraction of the molecules interact whereas the rest of the molecules do not. The bivariate PCF will then deviate significantly from either the random labeling or the model of independence. (iv) the molecules might demonstrate negative interactions or repulsion as the presence of one molecule decreases the probability of finding the second molecular species in its vicinity. Here, the bivariate PCF will show values smaller than 1. (v) the molecules might demonstrate attractive interactions beyond random mixing, hence giving rise to a bivariate PCF that exceeds the 95% confidence interval produced by the random labeling model. Finally, complex combinations of the interaction processes discussed above might exist. However, such processes lie outside the scope of this work.
3.2.2 The extent of molecular mixing
Individual cells give rise to different molecular patterns. Each of these patterns can be described with bivariate PCFs with unique 95% confidence intervals. Thus, the bivariate PCF curves and their confidence intervals cannot be readily averaged across multiple cells. To compare the efficiency of mixing of two types of molecules in multiple cells we defined a relative measure consisting of the ratio of the height (y-value) of the bivariate PCF curve to the y-value of the middle of the 95% confidence interval due to the random labeling model at the shortest length scales i.e., close to the intersection of the PCF curves with the y-axis(To reduce noise of the measurements, we actually considered averages of the curves of y-value ratios across 3 data points, between x=0nm and x=40nm) We refer to this measure as the extent of mixing between the two molecules of interest. This measure can be averaged for multiple cells and its values can be compared to the extent of mixing of other molecules or other cells under varying experimental conditions.
4. Results
4.1. Protein clustering
4.1.1 Univariate analysis and protein clustering
The self-association of proteins at the PM occurs at two levels, protein complexes at the nanoscale and larger clusters or microclusters visible at the diffraction-limited light microscopy level. Such clusters can be of great importance to cellular function as they increase the local concentration of molecules and enhance the effective activity of the clustered protein [3]. Here we focus on the example of microclusters that form at the PM of activated T cells and drive various cell functions by concentrating multiple effector proteins. Protein complexes are discussed below. LAT is a central adapter protein that forms microclusters, where it recruits proteins that are essential for multiple functions [2]. To study LAT clustering by PALM, we plated T cells expressing proteins conjugated to PAFPs on coverslips coated with a TCR-stimulating antibody. Upon engagement, the cells spread on the coverslips and microclusters containing the PAFP chimeric molecules formed (Figure 1A–C). We used PALM imaging to identify individual LAT molecules within the microclusters (Figure 1C) and employed PCF statistics to study the spatial organization of LAT molecules at the PM of the spread cells. The PCFs were calculated for study regions that covered most of the cell surface, but excluded edges (Figure 1D).
4.1.2 Considerations in data analysis
4.1.2.1 Selection of the study region
To calculate the PCF, the study region for the analysis must be defined. Including regions outside of the cell in the study region generates an a priori level of clustering since the molecules are confined only to a fraction of the considered area (the apparent surface of the cell; Figure 1D, white rectangle). This added clustering level is evident in the height of the PCF which is greater than the heights of PCF curves of study regions within the cell and further from the 95% confidence interval generated by a homogeneous Poisson process. In this analysis, either a large region within the cell surface or a smaller region (Figure 1D, red and yellow rectangles, respectively) gave rise to similar PCF curves (Figure 1E). A large study region better represents the surface of the cell as a whole, while a smaller region can capture or exclude local variations in the density of the molecules. Specifically, we can identify relatively large holes at the PM where molecules are absent and hypothesize that these regions represent membrane heterogeneities that developed during cell adhesion and spreading rather than features related to molecular interactions.
4.1.2.2 Considering multiple clustering levels with a heterogeneous Poisson process
Molecular confinement within distinct membrane parts and molecular binding are two distinct mechanisms that give rise to molecular clustering. These are difficult to separate by comparing PCFs to a homogeneous Poisson even when multiple small and homogeneous regions are used as study regions. Nevertheless, these two mechanisms might be distinguished by first capturing the heterogeneity of the membrane at a relatively large scale in comparison to molecular aggregates and then applying a heterogeneous Poisson distribution as a null hypothesis model, as detailed below. To capture the heterogeneity of the membrane (Figure 1F) we used a rolling window (Epanecnikov kernel [26–27]) that calculates the density of the molecules within 400nm around every point within the study region. Then, we modelled distributions of molecules according to the heterogeneous Poisson model by distributing the same number of molecules across the study region through a random process in which the probability of localizing a molecule at any location is proportional to the detected local density.
Figure 1G shows an example of a PCF [g(r)] plotted with a 95% confidence interval due a homogenous Poisson process and a 95% confidence interval due a heterogeneous Poisson process. The distance from the 95% confidence interval due to the heterogeneous Poisson process and the PCF of the study region that indicates the extent of significant clustering is smaller than the distance between the PCF and the 95% confidence interval due to the homogeneous Poisson process. This is because the first-order heterogeneity induced by cell spreading has been taken into account. The length scale of significant clustering also decreases from <1000nm or more to <320nm. In Figure 1H, we show that Ripley's second order statistics [L(r)-r] is highly sensitive to this first order clustering, as the difference between the two 95% confidence levels is even greater and the significance in reported clustering is greatly diminished when the sample PCF is compared to the 95% confidence interval due a heterogeneous Poisson process. The length scale of significant clustering is also greatly changed from clustering across all length-scales to only showing significance at length scales less than 760nm (Figure 1H). We further confirmed our results for additional rolling window sizes (Supp. Figure 2A,B) and found that Ripley's statistics overestimates clustering in comparison to the PCF statistics regardless of the choice of window size (Supp. Figure 2C). We conclude that the PCF is more robust in distinguishing multiple levels of molecular clustering.
4.2 Quantifying the extent of protein-protein interactions
We now turn to examples of the interactions of the critical adapter protein LAT with its activating protein, ZAP-70, one of its effectors, PLC-γ1 and an unrelated protein, TAC (the alpha chain of the IL-2 receptor). We chose these interactions to demonstrate how our method of two color PALM imaging combined with bivariate PCF can capture different strengths of interactions at the single molecule level.
4.2.1 Strongly interacting proteins
PLC-γ1 is a cytosolic kinase that binds LAT on one of its phosphotyrosines upon TCR stimulation and then triggers Ca++ influx. This protein has been shown to bind LAT by immunoprecipitation and Förster resonance energy transfer (FRET) studies [28]. The LAT-PLC-γ1 interaction is an example of a LAT-based protein complex. Using two color PALM imaging, we observed that PLC-γ1 and LAT efficiently mix in the same clusters (Figure 2A). The bivariate PCF closely follows the 95% confidence intervals of random mixing, with a slight deviation at the shortest length-scales. Similar analysis for multiple cells yields an extent of mixing of ~1, indicating that LAT and PLC-γ1 strongly associate at the PM of activated T cells. Notably, the extent of mixing between LAT and PLC-γ1 significantly exceeds the model of random labeling in multiple cases (Figure 2G). Such results mean that some PLC-γ1 molecules lie closer to LAT than the proximity expected by their random mixing within clusters. Such a result could indicate apparent structure within protein complexes, as further discussed in section 4.2.2.
Figure 2. Studying protein-protein interactions by two color PALM and bivariate PCF statistics.

(A) Two color-PALM image of a Jurkat E6.1 cell on a TCR stimulating coverslip. The cell expressed LAT-Dronpa and PLC-γ-PAmCherry. (B) Bivariate pair-correlation function (PCF) of the identified molecules within a study region of the cell. (C) Two color-PALM image of a Jurkat E6.1 cell on a TCR stimulating coverslip. The cell expressed LATDronpa and ZAP-70-PAmCherry. (D) Bivariate PCF of the identified molecules within a study region of the cell. (E) Two color-PALM image of a Jurkat E6.1 cell on a TCR stimulating coverslip. The cell expressed LAT-Dronpa and TAC-PAmCherry. (F) Bivariate PCF of the identified molecules within a study region of the cell. (G) The extent of mixing between LAT and TAC, ZAP-70 and PLC-γ1 in multiple cells (n=9). Bars - 2 μm for whole cell images and 200nm for zoomed images.
4.2.2 Partial mixing and structure
ZAP-70 is a kinase that is recruited to the phosphorylated intracellular chains of an activated TCR. Upon binding, ZAP-70 is activated and then serves to phosphorylate LAT on multiple tyrosines. Previously, we and others have found that microclusters containing LAT and ZAP-70 come close upon TCR activation, but do not completely overlap [25, 29]. Since ZAP-70 associates with both proteins, we expect only a part of its population to associate with LAT at any given time. Indeed, two color PALM imaging showed that these ZAP-70 and LAT reside in close proximity at the PM. Some molecules are visible within LAT clusters, yet others remain on the outside of these clusters (Figure 2C). The bivariate PCF due to the interaction of ZAP-70 and LAT lies below the 95% confidence intervals of random mixing, but it is not flat. Similar analysis for multiple cells yields an extent of mixing of ~0.4 in average. Taken together, these results show that LAT and ZAP-70 mix partially at the PM of activated T cells. Partial mixing of the molecules results here from a relatively complex process involving both attraction (or facilitation) of LAT and ZAP to proximal locations, partial repulsion in the smaller length scale within microclusters, and an overall non-trivial structural organization.
4.2.3 Independence
TAC is a membrane protein that is not involved in the TCR pathway. Thus, we use this protein to show that its location at the PM is independent of the position of LAT. Indeed, by two color PALM imaging the molecules do not seem to associate (Figure 2E). The bivariate PCF due to interactions between LAT and TAC seems flat and strongly depart from the null hypothesis of random mixing (Figure 2F). The extent of mixing in multiple cells scores a low value, close to 0. Taken together, these results show that the two molecules are independent of each other at the PM.
4.2.4 Robustness of bivariate statistics
4.2.4.1 Sensitivity to detection efficiency
PALM imaging typically cannot report on the exact number of molecules of interest for multiple reasons. For instance, endogenous untagged proteins cannot be detected, the detection efficiency of PA proteins is partial and remains unclear, and molecules can be disregarded during the analyses of PALM analyses. It is thus important to study the effect of the detection efficiency of PALM imaging on the results reported by our method. For that purpose we randomly selected subsets of molecules from a single cell with varying sizes. We then calculated the bivariate PCFs for each subset and compared these curves and their related values for the extent of mixing. We observe that the height of the bivariate PCFs was affected by incomplete detection of molecules (Figure 3A). However, the height of the 95% confidence intervals were affected in a similar fashion, resulting in values of the extent of mixing that had little sensitivity to the fraction of molecules considered in the analyses (Figure 3B).
Figure 3. Robustness of bivariate PCFs to experimental uncertainties and variables in the analysis.
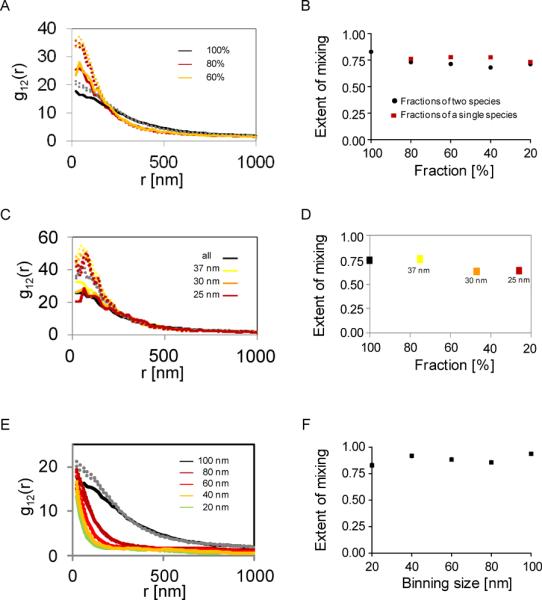
(A) Bivariate PCFs calculated for subsets of varying sizes from the detected pool of LATand PLC-γ1 molecules of the cell shown in Figure 2, panel A. (B). The extent of mixing of LAT and PLC-γ1 for the subsets of varying sizes. Subsets were considered for the two molecular species (black circles) or for a subset of only one species with the complete set of the other species (green squares). (C) Bivariate PCFs calculated for subsets of varying sizes from the detected pool of LAT and PLC-γ1 molecules of the cell shown in Figure 2, panel A. The molecules were chosen using various thresholds for their localization accuracy. (D) The extent of mixing of LAT and PLC-γ1 for the subsets of varying sizes due to the thresholds for localization accuracy used in panel A. (E) Bivariate PCFs calculated with varying pixel sizes for the cell shown in Figure 2, panel A. (F). The extent of mixing of LAT and PLC-γ1 as a function of the pixel size chosen in the analyses.
The presentation and analyses of PALM data often involves selection of molecules based on their localization accuracy. Thus, we tested the sensitivity of the PCF curves and the extent of mixing parameter to thresholds of localization accuracy for the selection of molecules (Figure 3C). We found a small effect of the threshold value on the sensitivity of the PCF curves and the extent of mixing (Figure 3D). This effect was comparable to the one due to random selection (Figure 3A,B). We conclude that our method for quantifying protein-protein interactions using two color PALM and bivariate PCFs is insensitive to factors that affect the number of molecules that are considered.
4.2.4.2 Sensitivity to distance thresholding (pixelization)
In these studies we used an algorithm that involves pixelization of PALM images to generate PCF statistics. Although image pixelization is unnecessary for that purpose, it is very convenient and avoids multiple pitfalls in the analysis [23]. In our analyses we used pixel sizes of 20nm that correspond to the typical error in molecular localization of our PALM imaging [25]. We examined the sensitivity of the bivariate PCF curves to the chosen pixel size. We observed again that the PCF curves decrease in height along with the 95% confidence intervals upon increasing the pixel size (Figure 3E). Thus, we also found little effect of the pixel size on the extent of mixing (Figure 3F). This allows us to conclude that our method is insensitive to the choice of the pixel size when conducting the bivariate PCF analyses.
5. Discussion
Protein-protein interactions in cells are commonly studied using methods such as immunoprecipitation, diffraction limited microscopy and spectroscopic methods involving resonance energy transfer, namely FRET and BRET. Unfortunately, these methods average many interactions of proteins across multiple signaling complexes, and across multiple cells. Thus, these methods lose information about potential heterogeneities in the interactions of proteins that could produce signaling complexes of varying structure and content. To overcome this issue, we employ the recently invented technique of PALM microscopy. This method allows the identification of single molecules in intact cells with resolution down to ~20nm. Using this method, the molecular organization of proteins can be studied in situ at their biologically relevant concentrations.
Multiple well-defined techniques exist for the quantification of colocalization in microscopy images. Pearson's or Mander's coefficients have been widely used for this purpose [30]. However, unlike conventional microscopy, PALM imaging generates maps of point patterns, where each point marks the putative location of an individual molecule. Two color patterns are generated by imaging two types of molecules. Here, in the absence of well defined pixels, standard techniques for quantification of protein colocalization cannot be readily used. One way to overcome this issue is to pixelize the PALM image first and then to apply the standard statistical tools (e.g. [31]). This method loses information about the precise locations of the molecules and the context and length scale of the interactions between the proteins. Second-order statistics are useful in overcoming these two problems [24]. First, they can be applied directly to point patterns, and second, they provide information at a range of length scales about the interactions between the proteins under study.
We have shown here how PALM imaging and second order statistics can be employed to study the clustering of the membrane protein LAT at the PM of activated T cells. Importantly, we provide practical considerations for using this technique and discuss its sensitivity to multiple experimental and analytical considerations. First, the study section should be defined within the apparent footprint of the cell to avoid erroneous levels of clustering. Second, we recommend the use of a heterogeneous Poisson process to account for multiple levels of clustering. Third, we recommend the use of PCF statistics rather than Ripley's statistics as the former seems more adequate to report on clustering due to molecular interactions in the presence of multiple clustering processes.
We further introduce a technique to study bimolecular interactions using two color PALM and bivariate second order statistics. Using this method, we were able to identify different levels of interactions of LAT with associated cytosolic proteins or an independent membrane protein. We further show that our approach is robust and insensitive to various experimental and analytical considerations. Nevertheless, our method of reporting on molecular interactions is statistical in nature and is limited to the resolution of the PALM data. In this sense, our method is complementary to methods that report on direct binding of molecules, such as immunoprecipitation and FRET.
6. Conclusions
In this work we introduced a method to study molecular clustering and protein-protein interactions in intact cells and at the single molecule level. We used PALM imaging in one and two colors to detect individual proteins at the PM of the cells and employed second-order statistics to study the resultant point patterns. We further demonstrated how this method can resolve molecular interactions in signaling complexes that form in the plasma membrane of activated cells. Importantly, we could describe interactions with a wide range of strength, from complete mixing to independence of interaction. We further discussed the sensitivity or the robustness of the method to various experimental and analytical factors. With the introduction of commercial microscopes for imaging single-molecules in cells (PALM, STORM, GDSIM and their variants), we expect that our methods will complement more traditional technique in the study of protein-protein interactions and multi-molecular protein complexes in cells.
Supplementary Material
Acknowledgements
The authors would like to thank Subhadra Banerjee and Barbara J Taylor at the FACS CORE Facility (NIH, NCI), Zeiss, Harald Hess (HHMI, Janelia Farm) for providing the PALM software, Wolfgang Losert (UMD) for multiple discussions on data analyses, Thorstan Wiegand (Helmholtz Centre for Environmental Research - UFZ) for providing us his point-pattern analyses software and Connie Sommers (NIH, NCI) for her comments on the manuscript. This research was supported by the Intramural Research Programs of the National Cancer Institute (The Center for Cancer Research).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. References
- 1.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 2.Samelson LE. Signal transduction mediated by the T cell antigen receptor: The role of adapter proteins. Annual Review of Immunology. 2002;20:371–394. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 3.Cebecauer M, et al. Signalling complexes and clusters: functional advantages and methodological hurdles. Journal of Cell Science. 2010;123(3):309–320. doi: 10.1242/jcs.061739. [DOI] [PubMed] [Google Scholar]
- 4.Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 5.Brown TA, et al. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol Cell Biol. 2011;31(24):4994–5010. doi: 10.1128/MCB.05694-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izeddin I, et al. Super-resolution dynamic imaging of dendritic spines using a low-affinity photoconvertible actin probe. PLoS One. 2011;6(1):e15611. doi: 10.1371/journal.pone.0015611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frost NA, et al. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron. 2010;67(1):86–99. doi: 10.1016/j.neuron.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shroff H, et al. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5(5):417–23. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91(11):4258–72. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shroff H, et al. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(51):20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shroff H, White H, Betzig E. Photoactivated localization microscopy (PALM) of adhesion complexes. Curr Protoc Cell Biol. 2008;Chapter 4:Unit 4 21. doi: 10.1002/0471143030.cb0421s41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gould TJ, Verkhusha VV, Hess ST. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat Protoc. 2009;4(3):291–308. doi: 10.1038/nprot.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown TA, et al. pproaches toward super-resolution fluorescence imaging of mitochondrial proteins using PALM. Methods. 2010;51(4):458–63. doi: 10.1016/j.ymeth.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galbraith CG, Galbraith JA. uper-resolution microscopy at a glance. J Cell Sci. 2011;124(Pt 10):1607–11. doi: 10.1242/jcs.080085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manley S, Gunzenhauser J, Olivier N. starter kit for point-localization super-resolution imaging. Curr Opin Chem Biol. 2011;15(6):813–21. doi: 10.1016/j.cbpa.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Suarez M, Ting AY. Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol. 2008;9(12):929–43. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 17.Patterson G, et al. Superresolution imaging using single-molecule localization. Annu Rev Phys Chem. 2010;61:345–67. doi: 10.1146/annurev.physchem.012809.103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subach FV, et al. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy (vol 6, pg 153, 2009) Nature Methods. 2009;6(4):311–311. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunewardene MS, et al. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys J. 2011;101(6):1522–8. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henriques R, et al. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods. 2010;7(5):339–40. doi: 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- 21.Bunnell SC, et al. High-Resolution Multicolor Imaging of Dynamic Signaling Complexes in T Cells Stimulated by Planar Substraes. Sci. STKE. 2003;2003(177):pl8. doi: 10.1126/stke.2003.177.pl8. [DOI] [PubMed] [Google Scholar]
- 22.Stoyan D, Stoyan H. Fractals, Random Shapes and Point Fields. Methods of geometrical statistics. John Wiley & Sons.; 1994. [Google Scholar]
- 23.Wiegand T, Moloney KA. Rings, circles, and null-models for point pattern analysis in ecology. Oikos. 2004;104(2):209–229. [Google Scholar]
- 24.Zhang J, et al. Characterizing the topography of membrane receptors and signaling molecules from spatial patterns obtained using nanometer-scale electron-dense probes and electron microscopy. Micron. 2006;37(1):14–34. doi: 10.1016/j.micron.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 25.Sherman E, et al. Functional nanoscale organization of signaling molecules downstream of the T cell antigen receptor. Immunity. 2011;35(5):705–20. doi: 10.1016/j.immuni.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Epanechnikov VA. Non-Parametric Estimation of a Multivariate Probability Density. Theory of Probility and Its Applications, Ussr. 1969;14(1):153–&. [Google Scholar]
- 27.Bowman AW, Azzalini A. Applied smoothing techniques for data analysis : the kernel approach with S-Plus illustrations. Oxford; 1997. (Oxford statistical science series 18). [Google Scholar]
- 28.Braiman A, et al. Recruitment and activation of PLC gamma 1 in T cells: a new insight into old domains. Embo Journal. 2006;25(4):774–784. doi: 10.1038/sj.emboj.7600978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lillemeier BF, et al. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat Immunol. 2010;11(1):90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manders EMM, Verbeek FJ, Aten JA. Measurement of Colocalization of Objects in Dual-Color Confocal Images. Journal of Microscopy-Oxford. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 31.Owen DM, et al. PALM imaging and cluster analysis of protein heterogeneity at the cell surface. J Biophotonics. 2010;3(7):446–54. doi: 10.1002/jbio.200900089. [DOI] [PubMed] [Google Scholar]
- 32.Hsu CJ, Baumgart T. Spatial association of signaling proteins and F-actin effects on cluster assembly analyzed via photoactivation localization microscopy in T cells. PLoS One. 2011;6(8):e23586. doi: 10.1371/journal.pone.0023586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andresen M, et al. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nature Biotechnology. 2008;26(9):1035–1040. doi: 10.1038/nbt.1493. [DOI] [PubMed] [Google Scholar]
- 34.Subach FV, et al. Photoactivation mechanism of PAmCherry based on crystal structures of the protein in the dark and fluorescent states. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(50):21097–21102. doi: 10.1073/pnas.0909204106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subach FV, et al. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptPALM of live cells. J Am Chem Soc. 2010;132(18):6481–91. doi: 10.1021/ja100906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.