

Abstract
Inflammation is an obligatory attempt of the immune system to protect the host from infections. However, unregulated synthesis of pro-inflammatory products can have detrimental effects. Although mechanisms that lead to inflammation are well appreciated, those that restrain it are not adequately understood. Creating macrophage-specific L13a-knockout (KO) mice here we report that depletion of ribosomal protein L13a abrogates the endogenous translation control of several chemokines in macrophages. Upon LPS-induced endotoxemia these animals displayed symptoms of severe inflammation caused by widespread infiltration of macrophages in major organs causing tissue injury and reduced survival rates. Macrophages from these KO animals show unregulated expression of several chemokines e.g. CXCL13, CCL22, CCL8 and CCR3. These macrophages failed to show L13a-dependent RNA binding complex formation on target mRNAs. In addition, increased polyribosomal abundance of these mRNAs shows a defect in translation control in the macrophages. Thus, our studies provide the first evidence of an essential extra-ribosomal function of ribosomal protein L13a in resolving physiological inflammation in a mammalian host.
Introduction
Synthesis of pro-inflammatory products by animal hosts is an obligatory attempt of the immune system to protect the host from infections. Precise, rapid and temporal synthesis of inflammatory cytokines and chemokines by the monocytes and macrophages serves as an arsenal against the invading microorganisms (1, 2). However, unregulated synthesis of these pro-inflammatory products can have detrimental effects (3). Thus, the endogenous mechanisms that have evolved to restrict the cytokine storm and permit the resolution of inflammation are prime targets to search for novel anti-inflammatory molecules. In spite of its enormous importance, our understanding about inflammation is more orientated towards the mechanisms that accelerate the process, whereas those that restrain it remain limited (4). Here, we identify one such mechanism in a novel animal model that relies on the abrogation of ribosomal protein L13a-dependent translational silencing by creating viable macrophage-specific L13a-knockout (KO) mice (L13aflox/floxLysMCre+), in which termination of inflammation is severely compromised. Such unregulated inflammation is consistent with the series of our previous studies uncovering an L13a-dependent translational silencing mechanism in IFN-γ activated monocytic cells (5–10). This silencing is dependent upon the assembly of the Gamma-Activated Inhibitor of Translation (GAIT) complex on the GAIT element present in the 3′UTR of its target mRNAs upon the phosphorylation dependent release of L13a from the large ribosomal subunit (10). In addition, our studies have shown that P-L13a as a part of GAIT complex can bind the translation initiation factor eIF4G and prevent the formation of 48S preinitiation complex thereby blocking the translation initiation of GAIT element containing mRNAs (8). Our subsequent studies also showed that depletion of L13a by RNAi in monocytic cells abrogates GAIT element mediated translational silencing but interestingly overall protein synthesis was not inhibited (11). Apart from our studies, reports from other laboratories also provide compelling evidence that controlling the protein synthesis of many inflammatory molecules from the pre-existing mRNAs could be an effective cellular strategy to prevent their accumulation (12).
Monocytes and macrophages can perform pathogenic as well as protective functions during innate immune responses against infectious assaults and subsequent tissue homeostasis (13). Priming of these cells with IFN-γ plays a crucial role in the innate immune response relying in part on elevated synthesis of pro-inflammatory cytokines and mediators to kill the invading organisms (14). Using a genome-wide approach we showed that in IFN-γ induced monocytic cells protein synthesis from a novel post-transcriptional operon was severely inhibited; this operon was composed of mRNAs encoding several chemokines and their cognate receptors, viz. CCL22, CCL8, CCL21, CXCL13, CCR3, CCR4, CCR6 (9). These studies also identified functional GAIT elements in the 3′ untranslated regions (UTRs) of these mRNAs, which recruited the L13a containing GAIT complex(9). Emerging evidence suggests a critical role of chemokines and their receptors during the recruitment of mononuclear cells to the sites of inflammation (15). Based on the foregoing, we hypothesize that L13a-dependent translational silencing could be an endogenous defense mechanism against uncontrolled inflammation. This led us to predict that a deficiency of L13a in macrophages may promote runaway inflammation and associated pathology due to the abrogation of translational silencing of these inflammatory targets. To test this hypothesis, we generated macrophage-specific L13a KO mice using Cre-Lox system (16) and studied their response to the inflammatory assaults caused by LPS induced endotoxemia. The rationale for using the LPS-induced endotoxemia model is based on the fact that LPS stimulates macrophages to produce numerous pro-inflammatory cytokines and most importantly it is also a potent inducer of IFN-γ in the cells of myeloid lineage (17–19). Here, we show that macrophage-specific L13a KO mice are highly susceptible to endotoxemia demonstrating lower survival rate, invasion of myeloid cells in the peritoneal cavity and in the major organs with clear signs of organ damage in comparison to the control mice. Moreover, using macrophages harvested from these KO animals we also observed unregulated expression of several target proteins of GAIT complex probably by the abrogation of their translational silencing. To our knowledge these results provide the first in vivo evidence of an extra-ribosomal anti-inflammatory function of ribosomal protein L13a and its molecular basis.
Materials and Methods
Generation of Macrophage-specific KO mice (L13aflox/floxLysMCre+)
Embryonic stem (ES) cells from C57BL/6 mouse were transfected with the targeting construct and the recombinant ES cells harboring the lox P allele were screened by genotyping. Cre-dependent depletion was confirmed by infecting with Adenovirus expressing Cre (Ad-CMV-Cre). Two independent clones of the recombinant ES cells were injected into blastocytes. The chimeric mouse was generated in inGenious Targeting Laboratory Inc (Stony Brook, NY) by blastocyte injection of recombinant ES cells and the transfer of the blastocytes to the surrogate mother. The male chimera was further crossed with wild type C57BL/6 females for germline transmission. The neomycin gene from L13aflox-neo/+ was removed by crossing with ACTFLPe+ in C57BL/6 background. The F4 mice (L13aflox/flox) homozygous for the lox P and Neo deletion allele were confirmed by genotyping. The Cre-dependent depletion of L13a in the F4 mice was confirmed by infecting the isolated lung fibroblasts from these mice with Ad-CMV-Cre followed by immunoblot analysis with anti-L13a antibody. The macrophage specific KO mice (L13aflox/floxLysMCre+) were generated by crossing the L13aflox/flox mice with LysMCre+ mice (Jackson Laboratory, Bar Harbor) and identified by genotyping. The macrophage-specific depletion of L13a in L13aflox/floxLysMCre+ mice was confirmed by immunoblot analysis using anti-L13a antibody.
Antibodies for immunoblot analysis
Anti-L13a antibody was previously raised against a peptide NVEKKIDKYTEVLKTHG near the C-terminus of human L13a (10). This antibody recognizes a specific band both for human and mouse L13a between 28 kD and 21 kD. Anti-L28 and anti-actin antibodies were from Santacruz biotechnology Inc and Sigma-Aldrich Inc respectively. Anti-Cre recombinase antibody was from abcam.
Animal handling and isolation of leukocytes
All experiments involving mice were carried out in accordance with NIH guidelines and institutional IACUC. Age and sex-matched mice were challenged by intraperitoneal injection of thioglycollate (1.5 ml, 4% solution in distilled water) or LPS (15 mg/Kg of body weight). The peritoneal lavage was collected with ice-cold PBS after 72 hr for thiglycollate injected mice and 24 hr and 48 hr for LPS injected mice. The recovered cells were counted with a cell counter. Total leukocytes were isolated from spleen after isolation of cell pellet from single cell suspension and lysing the RBC from the cell pellet using RBC lysis buffer (eBioscience) following manufacturer’s suggested method. The mononuclear cells from blood were isolated by layering over Ficoll-Hypaque solution and centrifugation following established method (20).
FACS Analysis
Cells (106 per sample) were incubated with rat anti-mouse CD16/CD32 (BD Pharmingen) to block non-specific binding to Fcγ receptors. Cells were stained with the following antibodies: FITC conjugated rat anti-mouse CD11b IgG2b (BD Pharmingen), APC conjugated rat anti-mouse F4/80 IgG2b (Abd Serotec), PE conjugated rat anti-mouse Ly6G IgG2a (BD Pharmingen), FITC conjugated rat anti-mouse Mac2 IgG2a (Cedarlane Laboratories) and APC conjugated rat anti-mouse Ly6C (eBiosciences). Isotype-control antibodies consisted of PE-conjugated IgG2b (ebioscience) and FITC-conjugated IgG (ebioscience). For peritoneal cells and blood mononuclear cells, the cells were gated for leukocytes and for splenocytes the cells were gated for granulocytes. Isotype control antibodies were used in order to exclude background staining. Analysis of the stained cells was performed with a FACS Canto II flow cytometer (BD Biosciences) and data were analyzed by FACS Diva (BD BioSciences) and FloJo (Tree Star) softwares.
Quantification of cytokine and chemokine expression
Serum level of TNF-α and IFN-γ, IFN-γ secreted by total leukocytes and chemokines secreted by the primary macrophages were determined by ELISA using the commercially available detection kits (R&D Systems). Peritoneal macrophages derived from each mouse were seeded in three replicative wells (5 × 105 cells/well). Cells were incubated in RPMI at 37°C in 5% CO2 for overnight to allow the adherence. Non-adherent cells were removed by washing with PBS. The cells were incubated either alone or with murine IFN-γ (500 U/ml) (R&D systems) for different times. Conditioned medium was collected and subjected to ELISA analysis. Serum levels of IL-1β, IL-6, MCP-1, MIP-1α, RANTES and KC were determined using cytokine/chemokine ELISA array in a commercial facility (Quansys Biosciences, Utah, USA).
Histopathogical studies
Tissues were fixed in 10% neutral buffered formalin (Sigma-Aldrich Inc) for 16 hrs and stored in PBS overnight before paraffin embedding and sectioning. For Ym1 staining tissues were fixed with a solution of 60% acetic acid, 30% methanol and 10% H2O. 5 μ sections were stained with hematoxylin and eosin (H&E). Paraffin embedded tissue sections were deparaffinized with Trilogy (Cell Marque, Austin TX) in a steamer for 30 minutes. Tissues were stained for macrophages with purified anti-Mac2 antibody (rat anti-mouse) (Cedarland laboratories Ltd) and anti-Ym1 antibody (anti-rabbit) (Wako Chemicals USA Inc) followed by incubation with Biotin-SP conjugated appropriate secondary antibodies (Jackson Immunoresearch).
Determination of in vivo interaction of L13a with different chemokine mRNAs and 60S ribosomal subunit
The interaction of L13a with cellular mRNAs and 60S subunit was determined following our previously established methods (10). In short, macrophage lysate (500 μg of protein) were subjected to immunoprecipitation using 10 μl of affinity purified anti-L13a antibody. L13a-bound RNA was isolated by Trizol (Invitrogen). For mRNAs the reverse transcription was carried out using oligo-dT primer and for ribosomal subunit random primer was used. For PCR amplification specific primer pairs were used (Supplemental Table S1).
Determination of GAIT element mediated translational silencing activity of mouse macrophages using cell free in vitro translation system
cRNA of the reporter luciferase with 29-nt GAIT element was generated by in vitro transcription. The cRNA (100 ng) was subjected to in vitro translation using rabbit reticulocyte lysate (Promega), in the presence of [35S]methionine. Lysates were made from the peritoneal macrophages of control and KO mice and 4 μg of these lysates were used to test the translational silencing activity. A 10 μl aliquot of the translation reaction mixture was resolved by SDS-PAGE (7% polyacrylamide) followed by autoradiography.
Statistical analysis
To determine the significance of the differences in survival rates log-rank test (Mantel Cox) was used. Results are presented as mean ± s.d. The statistical significance of the differences between groups was determined by two-tailed Student’s t-test. All statistical analysis was performed using GraphPad Prism 5.0 software.
Results
L13aflox/floxLysMCre+ mice show the macrophage-specific depletion of L13a
Macrophages are the principal effectors of inflammatory and innate immune responses. To assess the role of L13a-dependent translational silencing in resolving physiological inflammation we have generated macrophage-specific L13a KO mice using Cre-Lox system (16). Lox P sites were introduced in the targeting construct after exon 1 and exon 8 of the mouse L13a genomic sequence. The targeting construct also harbors a Neomycin gene as a marker flanked by FRT sites to facilitate its removal (Fig. 1a left panel). Recombinant embryonic stem (ES) cells harboring the lox P allele were identified by PCR genotyping upon transfection of the ES cells from C57BL/6 mice (Fig. 1a right panel). Before injecting into the blastocytes the Cre-dependent depletion of L13a protein in the recombinant ES cells was confirmed by infecting with Adenovirus expressing Cre (Ad-CMV-Cre) (Fig. 1b). Germ line transmission from the male chimera was carried out by crossing with wild type C57BL/6 females. The neomycin gene from L13aflox-neo/+ was removed by crossing with ACTFLPe+ mice in C57BL/6 background (Jackson Laboratory, Bar Harbor). The F4 mice (L13aflox/flox) homozygous for the lox P and Neo deletion allele were confirmed by PCR genotyping (Fig. 1c). The Cre-dependent depletion of L13a in the F4 mice was confirmed by infecting the isolated lung fibroblasts from these mice with Ad-CMV-Cre followed by immunoblot analysis with anti-L13a (Fig. 1d). The macrophage-specific KO mice (L13aflox/floxLysMCre+) were generated by crossing the L13aflox/flox mice with LysMCre+ mice (Jackson Laboratory, Bar Harbor) and the mice homozygous for the flox allele and positive for the Cre transgene were identified by PCR genotyping (Fig. 1e). The immunoblot analysis using anti-L13a antibody shows the depletion of L13a in L13aflox/floxLysMCre+ mice only in macrophages but not in other organs such as liver and kidney (Fig. 1f). Consistent with this result, expression of Cre was observed only in the macrophages of these mice (Fig. 1f, left, middle panel). On the other hand, liver and kidney of the same animal showed no detectable expression of Cre (data not shown). This result confirmed the generation of macrophage-specific L13a KO mice. Throughout this manuscript we refer to these L13aflox/floxLysMCre+ mice as KO mice and L13aflox/flox mice as control mice.
Figure 1. Generation of mice homozygous for the lox P allele and the macrophage specific KO mice.

a, Design of the targeting construct and identification of the recombinant mouse embryonic stem (ES) cells harboring the conditional null allele. The positions of the lox P sites, Neo Cassette, long and short homology arms and the primers used in genotyping are shown (a, left panel). The recombinant ES cells were genotyped by PCR using Lox1/SDL2 (see above) primers and identified on the basis of the appearance of a doublet band of 325 bp/263 bp (a, right panel). b, Confirmation of the presence of conditional null allele in the recombinant ES cells. The recombinant ES cells were infected by Adenovirus expressing Cre recombinase (Ad-CMV-Cre, Vector Biolabs, USA). The Cre-dependent depletion of L13a was confirmed by immunoblot analysis of the infected cells using anti-L13a antibody. c, Identification of the F4 mice homozygous for the lox P allele (lox P+/+) and Neo deletion allele. Tail DNA samples of the pups were screened by PCR with the Lox1/SDL2 primer pair for the lox P allele (upper panel) and NDEL3/Anti AT2 primer pair for the Neo deletion allele (lower panel). The appearance of the 280 bp band shows the presence of Neo deletion allele. d, Identification of the F4 mice harboring the conditional null allele. Fibroblasts were isolated from the lung and infected with Adenovirus expressing Cre recombinase. The Cre dependent depletion of L13a was confirmed by immunoblot analysis with anti-L13a antibody. e, The macrophage-specific KO mice were generated by crossing the lox P+/+ with the lox P+/−LysM Cre+/− mouse and genotyping by PCR with the Lox1/SDL2 primer pair for the lox P allele and Cre specific primer pair (Jackson Laboratory) for Cre allele. f, Confirmation of macrophage specific depletion of L13a. Lysates were made from the peritoneal macrophages, liver and kidney harvested from L13aflox/floxLysMCre+, L13aflox/flox and wild type (WT) mice. Lysates were subjected to immunoblot analysis using anti-L13a antibody (10). The blot from the macrophage lysates was re-probed with anti-Cre recombinase antibody and anti-actin antibody.
The KO mice progeny from the mating of L13aflox/flox and L13aflox/+LysMCre+ obeyed Mendelian distribution with no detected embryonic lethality. The newborn KO animals were indistinguishable from the controls. Under unchallenged conditions, no visible sign of any pathology such as retardation of mobility, growth and fertility, decreased food intake, weight loss and any visible change in the major organs such as in liver, spleen, lung, kidney etc were observed over a 6 month observation period. Together these results suggest that macrophage-specific depletion of L13a does not cause any significant defect in animal development.
Macrophage-specific L13a KO mice show significantly enhanced susceptibility to endotoxin challenge
We determined the inflammatory responses of the KO and control group of mice using the LPS-induced endotoxemia model. The relevance of this model is based on the ability of LPS to rapidly induce IFN-γ and number of other pro-inflammatory cytokines in the cells of myeloid lineage (17–19). IFN-γ-dependent activation of myeloid cells is a critical component of inflammatory responses. We reasoned that if the translational silencing of the cohort of mRNAs encoding inflammatory proteins serves as a defense mechanism against uncontrolled inflammation then LPS-induced pathological outcomes would be significantly more severe for the KO mice compared to the control. To verify this, the KO and control mice were subjected to LPS injection with a dose of 20 mg/kg to induce systemic inflammation and their survival was monitored over 120 hrs post administration. The survival of the KO mice was significantly lowered compared to the control mice (45% vs. 78%) and Kaplan-Meier survival analysis showed the statistical significance of this result (p < 0.05) (Fig. 2a). Tachypnea or high breathing rate is also a visible symptom of sepsis (21). These KO animals showed significantly higher breathing rate even 24 hr after LPS treatment (Fig. 2b). Treatment with LPS is known to cause induction of lethargy and reduction of ambulatory activity in mice (22, 23). As a visible response to endotoxin treatment we quantified the ambulatory activity up to 24 hr post-LPS treatment using a device equipped to measure the number of sequential laser beam brakes in two dimensions. Rapid reduction of the ambulatory activity was observed in the KO group, in contrast to the control group upon LPS but not saline administration (Fig. 2c). To determine other markers of inflammation, we measured the serum levels of blood urea nitrogen (BUN), released liver enzyme aspartate amino transferase (AST) and TNF-α in the LPS-treated mice, and all three were markedly higher in KO mice (Fig. 2d). In addition to these markers of inflammation we also measured the serum level of a panel of other inflammatory cytokines e.g. IL-1β, IL-6, MCP-1, MIP-1α, RANTES and KC. Our result showed that serum levels of all of these cytokines were significantly higher in the LPS treated KO mice (Fig 3). Together, these results document that macrophage-specific deficiency of L13a leads to increased susceptibility to endotoxemia.
Figure 2. Macrophage-specific L13a KO mice show significantly enhanced susceptibility to endotoxin challenge.

a, Increased death rate of KO mice in response to endotoxin challenge. Age and sex-matched L13aflox/flox, LysMCre+ and L13aflox/floxLysMCre+ mice were challenged with sub-lethal dose of LPS (20 mg/kg) and observed for 120 hours for survival. The survival rate was plotted on Kaplan-Meier survival curves, n=19 in each group, P = 0.042, Log-rank test (Mantel Cox). b, Increase of breath rate of the KO mice upon endotoxin challenge. Control and KO mice were injected with LPS (15 mg/kg) or saline. After 3 and 24 hr post injection breath rate was measured. After 24 hr post saline injection the breath rates for control mice were considered as 1 and the breath rates of all other mice in both time points were plotted on an arbitrary scale. Results are mean ± s.d, n=5, *P = 0.018, two-tailed Student’s t-test. c, Endotoxin challenge causes significant reduction of the ambulatory activity in KO mice. After LPS challenge (15 mg/kg) the ambulatory activities of the KO and control mice were measured for a period of 25 hours using a device equipped to sense the number of sequential laser beam breaks in two dimensions. The ambulatory activities were plotted using arbitrary scale. Results are mean ± s.d, n=8, P = 0.049, unpaired two-tailed Student’s t-test. d, KO mice showed increase serum level of the markers of inflammation. Serum levels of BUN, AST and TNF-α in mice were measured after 8 hours of LPS challenge (15 mg/kg). Results are mean ± s.d, n=4, *P = 0.011, **P = 0.004, ***P = 0.032, two-tailed Student’s t-test.
Figure 3. Increased serum level of inflammatory cytokines in the KO mice upon endotoxin challenge.
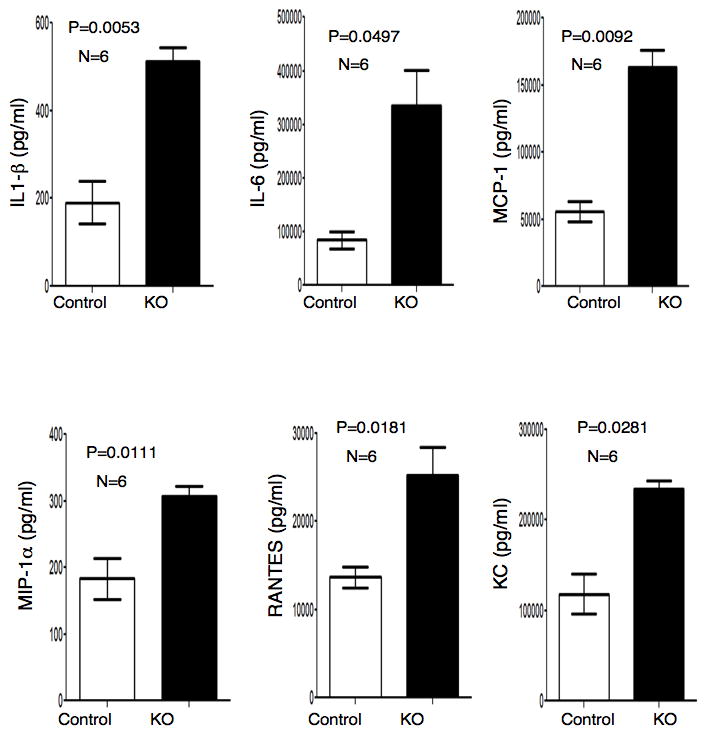
Control and KO mice were injected with LPS (15 mg/Kg). 12 hours after injection blood was collected through cardiac puncture. Serum was sent to a commercial facility (Quansys Biosciences, Utah, USA) and subjected to cytokine/chemokine ELISA array for IL-1β, IL-6, MCP-1, MIP-1α, RANTES and KC. Results are mean ± s.d, paired two-tailed Student’s t-test. n and P values for each are on the top of the figure.
Myeloid-specific depletion of L13a causes enhanced macrophage infiltration in the peritoneum and expansion of leukocyte populations in the spleen
Infiltration of leukocytes in the peritoneal tissue is a hallmark of inflammation and requires signal generated by chemokines and chemokine receptors (3, 24). We hypothesized that macrophage-specific deficiency of L13a would lead to greater macrophage influx by removing the endogenous translational silencing imposed on this cohort of chemokine and chemokine receptors (9). To measure mononuclear cell influx, we performed FACS analysis of the peritoneal cells isolated from KO and control mice using thioglycollate-induced peritonitis and LPS-induced endotoxemia models. We quantified Gr1-CD11b, F4/80-Gr1 and F4/80-CD11b double positive cells upon 48 and 24 hr of thioglycollate and LPS treatment to test the infiltration of macrophages and neutrophils. Thioglycollate treatment of KO mice for 48 hr showed significant enhancement of Gr1-CD11b and F4/80-Gr1 double-positive cell populations relative to control mice (Fig. 4a). On the other hand, LPS treatment for 24 hr showed significant enhancement of the double-positive cells of all three categories (Fig. 4b). Quantifications of these results from four independent experiments with statistical significance are shown in supplementary Fig. S1a. Mac2 is a carbohydrate binding protein and expressed on the surface of proinflammatory macrophages and monocytes (14, 25). Our studies show significant enhancement of the proinflammatory F4/80-Mac2 and Gr1-Mac2 double positive macrophages (Fig. 4c) in the KO mice upon LPS treatment for 24 hrs. These results have been quantitatively expressed in supplementary Fig S1b. Studies by others have found that the spleen can serve as a reservoir of leukocytes for rapid deployment of these cells to the sites of inflammation (26). Therefore, we measured populations of splenic leukocytes after RBC lysis of cell suspensions from spleens harvested from KO mice 48 hr post-LPS administration. Consistent with our expectation, we found increased abundance of F4/80-CD11b, Gr1-CD11b, F4/80-Gr1 (Fig. 4d and supplementary Fig. S1c) and Ly6Ghi/Ly6Chi (Fig. 5a and 5c) double-positive leukocytes in the LPS treated KO mice compared to the controls. Inflammation causes the release of Ly6Chi monocytes from bone marrow to the circulation and subsequent tissue recruitment and differentiation to macrophages (27). Therefore we also determined the abundance of Ly6Chi monocytes in circulation. Our results show these cells were significantly increased in the KO mice (Fig 5b and 5c). Together, these results show that the macrophage-specific deficiency of L13a leads to increased exudation of macrophages and neutrophils in the peritoneum, their increased abundance in the splenic reservoir and higher levels of inflammatory Ly6Chi mononuclear cells in the circulation in response to inflammatory stimuli.
Figure 4. Increased infiltration of immune cells in the peritoneum and higher abundance of splenic leukocytes in the KO mice upon endotoxin challenge.

a, b Enhanced infiltration of leukocytes in the peritoneum of the KO mice in response to thioglycollate and LPS treatment. Increased infiltration of Gr1-CD11b, F4/80-Gr1 and F4/80-CD11b double positive cells in the peritoneal cavity of the KO mice 48 hr after thioglycollate (a) and 24 hr after LPS treatment (15 mg/kg) (b). Results showing the quantitation of the infiltrates of the double positive cells 48 and 24 hr after thioglycollate and LPS treatment respectively from 4 independent experiments with statistical significance shown in supplementary Fig. S1a. c, Increased infiltration of F4/80-Mac2 and Gr1-Mac2 double positive cells in the peritoneum of the KO mice 24 hr after LPS treatment, for quantitation see supplementary Fig. S1b. d, Increased abundance of splenic leukocytes in the KO mice in response to LPS treatment. Control and KO mice were injected with LPS (15 mg/kg), 48 hr after LPS administration leukocytes were harvested from spleen following RBC lysis. F4/80-CD11b, Gr1-CD11b and F4/80-Gr1 double positive cells were quantified by FACS, for quantitation see supplementary Fig. S1c.
Figure 5. Increased abundance of Ly6Ghi-Ly6Chi leukocytes in spleen and Ly6Chi leukocytes in circulation of the KO mice upon endotoxin challenge.

a, Increased abundance of Ly6Ghi-Ly6Chi splenic leukocytes in the LPS treated KO mice. Control and KO mice were injected with LPS (15 mg/kg), 24 hr after LPS administration leukocytes were harvested from spleen following RBC lysis. Cells were stained and the Ly6Ghi-Ly6Chi and Ly6Chi leukocytes were quantified by FACS. b, Increased abundance of Ly6Chi leukocytes in circulation. Leukocytes from the serum of LPS treated (15 mg/kg) control and KO mice were harvested by Ficoll gradient centrifugation. Cells were stained and the Ly6Chi leukocytes were quantified by FACS. c, Quantifications of the results shown in a and b above. Results are mean ± s.d, n=4, two-tailed Student’s t-test. P values for each are on the top of the figure.
L13a deficiency in macrophages enhances endotoxemia induced tissue damage
Extensive tissue damage in the organs, widespread hemorrhage, intravascular congestion and tissue infiltration of immune cells are the essential features of systemic inflammation caused by LPS-induced endotoxemia (28, 29). Therefore we investigated if macrophage-specific deficiency of L13a could cause greater tissue damage and immune cell infiltrates following LPS administration. Histopathological analysis of the lung by H&E staining revealed extensive appearance of RBCs in bronchiolar and alveolar spaces of the KO mice whereas in the control animals the RBC are mostly intravascular (Fig. 6a). Immunohistological studies of the lungs showed significantly more Mac2 (Fig. 6b, upper panel) positive macrophages adherent to vascular endothelium. These results have been quantitatively expressed in Fig. 6b, lower panel. These data show enhanced adhesion of the macrophages to the vessel wall of KO mice in response to endotoxemia, a hallmark of inflammation. Similarly, analysis of the kidney sections showed conspicuous infiltration of Mac2 (Fig. 6c) and Ym1 (Fig. 6d) positive macrophages in the renal glomeruli of the LPS-treated KO animals. Quantification and statistical significance of these results have been presented in the right panel of these figures. No tissue injury and negligible macrophage infiltrates were found in these organs of the saline treated animals (data not shown). In summary, these results suggest that the loss of L13a expression in the macrophages leads to the enhanced infiltration of these cells and tissue injury in multiple major organs, consistent with the severe endotoxic shock observed in the KO animals.
Figure 6. Widespread infiltrates of immune cells in the major organs of the KO mice upon 24 hr of post-LPS challenge.

a, H&E staining of the bronchus and adjacent alveoli of lung. Slide prepared from formalin fixed lung tissue and methanol fixed tissue was used for Ym1 staining. Image taken at 40X magnification in 10 μ scale. In control mouse RBCs are largely intravascular with more open alveolar spaces. In the KO mouse the alveolar spaces are filled by RBCs, a sign of hemorrhage. b, Upper panel, Adhesion of macrophages in the blood vessel of lung. Formalin fixed lung sections were stained for macrophages using anti-Mac2 antibody. In the control mouse fewer Mac2 positive macrophages line the blood vessel whereas in the KO animal increased numbers of macrophages adhere to the blood vessel. Lower panel, Quantifications of the results shown above. Results are mean ± s.d, n=5, paired two-tailed Student’s t-test. P value is on the top of the figure. c and d, Infiltration of macrophages in the renal glomeruli. Kidney sections were stained with anti-Mac2 antibody (c) and anti-Ym1 antibody (d). Increased presence of Mac2 and Ym1 positive macrophages were observed in the glomeruli of KO mice. For Mac2 positive macrophages image taken at 20X (upper panel) and 40X (lower panel) magnification of two different fields of the same slides with 10 μ scale. For Ym1 positive macrophages image taken at 40X magnification with 10 μ scale. Quantification of the Mac2 and Ym1 positive macrophages per glomerulus is shown in the right panels. Results are mean ± s.d, n=5 per group, 70 glomeruli per animal were counted, paired two-tailed Student’s t-test. P value is on the top of the figure. The results shown in figures a to d are from one representative mouse in each group, similar results were found in all 5 mice per group.
Disruption of L13a in macrophages causes unregulated synthesis of GAIT target proteins in vivo
To understand the molecular underpinning of the inflammation in the L13a-KO animals we first tested whether the expression of the L13a target molecules indeed increased in the KO group upon endotoxin challenge. Ex vivo cultures of peritoneal as well as bone marrow derived macrophages harvested from the KO mice were subjected to 24 hr of IFN-γ treatment. The conditioned media of the cultures obtained from KO mice macrophages showed significantly increased levels of L13a-targets CCL22, CXCL13 and CCL8 (Fig. 7a and supplementary Fig. S2). In contrast, no significant difference between the control and L13a-KO macrophages was found following IFN-γ treatment for 8 hr. In the mouse model, administration of LPS can rapidly induce IFN-γ in the cells of myeloid lineage (17–19). Therefore, we investigated the ability of the macrophages harvested from LPS-treated KO mice to build up the steady state level of these target proteins of GAIT e.g. CCL22, CXCL13 and CCL8 in the conditioned medium of 24 hr ex vivo culture. Indeed, the levels of all three targets were significantly higher in the L13a-KO macrophages (Fig. 7b). We then investigated whether these high levels were possibly due to differences in the amounts of IFN-γ produced in response to LPS treatment. However, measurement of IFN-γ in the serum of LPS-treated control and KO mice and from the conditioned medium obtained from the ex vivo cultures of the total leukocytes harvested from these mice showed no significant difference (Fig. 7c). Therefore, these results demonstrate that in the mouse model, the deficiency of L13a in macrophages compromises their ability to control the synthesis of these GAIT target proteins and that the elevated levels of these proteins might contribute to the enhanced inflammatory response of the KO mice in response to endotoxin challenge.
Figure 7. Macrophage specific depletion of L13a causes over production of chemokine ligands CCL22, CXCL13 and CCL8 but not IFN-γ.

a, ex-vivo studies using the peritoneal macrophages of the KO mice showed higher accumulation (compared to control) of CCL22, CXCL13 and CCL8 in response to 24 hr of IFN-γ treatment (500 U/ml). Chemokine ligands were measured by ELISA from the conditioned medium after different times of IFN-γ treatment. Results are mean ± s.d, n = 5, *P = 2.5 × 10−5, **P = 5.2 × 10−6, ***P = 1.6 × 10−6, two-tailed Student’s t-test. b, Peritoneal macrophages harvested from LPS treated (15 mg/kg) KO mice showed higher accumulated level (compared to LPS treated control) of the same chemokine ligands in the conditioned medium upon 24 hr ex vivo culture. Results are mean ± s.d, n = 5, *P = 9.8 × 10−6, **P = 1.9 × 10−4, ***P = 2.7 × 10−6, two-tailed Student’s t-test c, Ability to induce IFN-γ in response to LPS treatment remains unchanged in KO mice. Serum level of IFN-γ (Left panel). Control and KO mice were injected with LPS (15 mg/kg) or saline. 16 hr post injection the serum levels of IFN-γ were measured using ELISA. n=9, difference between KO and controls are not significant. Level of IFN-γ secreted by leukocytes (Right panel). Leukocytes were isolated by RBC lysis of splenocytes from the control and KO mice after 3 hrs of LPS or saline administration. These leukocytes were cultured ex vivo for different times and IFN-γ was measured by ELISA from the culture supernatants. n=7, difference between KO and controls are not significant.
Macrophage-specific depletion of L13a abrogates translational silencing of the GAIT target proteins
Determination of polyribosomal abundance is a widely accepted method to determine the translational efficiencies of mRNAs (9, 30, 31). To directly test the deregulation of the translational silencing of these proteins in the macrophages of the KO mice we then investigated the polyribosomal abundance of the CCL22, CXCL13 and CCR3 mRNAs. Lysates from peritoneal macrophages from the LPS-treated KO and control mice were resolved by sucrose gradient centrifugation in order to separate the actively translated pool of polyribosome bound mRNAs from the pool of under-translated mRNAs. Translationally active polyribosomal pool and inactive pool of mRNAs were isolated from the fractions followed by determination of the GAIT target mRNAs by RT-PCR. Polyribosomal abundance of all three mRNAs was substantially increased in the peritoneal macrophages of KO mice, compared to control actin mRNA (Fig. 8a). The translational efficiencies of the target mRNAs have been quantitatively expressed by the ratio of the band intensities of the corresponding mRNAs and actin in polysomal and non-polysomal fractions (Fig. 8b). Essentially similar results were obtained from bone marrow derived macrophages of the KO mice upon treatment with IFN-γ for 24 hr in ex vivo cultures (supplementary Fig. S3). To directly test the abrogation of the GAIT element mediated translational silencing activity in the macrophages of the KO mice we have reconstituted the translational silencing ex vivo. In this experiment we have tested the translational efficiencies of a reporter RNA harboring an active GAIT element in the 3′UTR using cell free translation system of rabbit reticulocyte lysate. Result from this ex vivo study shows that treatment of LPS (but not saline) of the animal activates the translational silencing activity of the GAIT element containing mRNA in the macrophage. However in the macrophage-specific L13a KO animal LPS treatment failed to activate the translational silencing activity (Fig. 9a). Together, these results demonstrate that macrophage-specific deficiency of L13a may diminish the naturally imposed translational silencing on GAIT element containing mRNAs in vivo.
Figure 8. L13a controls translation of CCL22, CXCL13 and CCR3 mRNA in animal model.

a, Increased polyribosomal abundance of GAIT target mRNAs such as CCL22, CXCL13 and CCR3 in the macrophage of KO mice. Peritoneal macrophages harvested from 24 hr post LPS (15 mg/kg) injected control and KO mice were subjected to polyribosome fractionation by sucrose gradient. Fractions were subjected to RT-PCR analysis using specific primers (Supplemental Table S1). Increased association of the CCL22, CXCL13 and CCR3 but not actin mRNAs with the heavy polyribosome fractions were observed in KO mice compared to control mice. b, Quantifications of the results from a. Ratio of the abundance of the target mRNA and actin in polysomal and non-polysomal fractions were determined by measuring the intensities of the corresponding bands.
Figure 9. LPS treatment of the animal activates GAIT element mediated translational silencing activity, in vivo association of L13a with the target mRNAs and release of L13a from 60S ribosomal subunit.

a, The designs of the GAIT element containing reporter luciferase RNA and control T7 gene 10 RNA are at the bottom. These RNAs are in vitro translated in rabbit reticulocyte lysate in the presence of lysates made from the macrophage harvested from control or KO mouse after treatment with LPS or saline. Lysates made from U937 cells treated with IFN-γ for 8 or 24 hr were used as a positive control. An aliquot of the translation reaction mixture was subjected to SDS-PAGE followed by autoradiography to see the translated product of the reporter RNA. b, c, Macrophages from LPS treated wild type mice show in vivo association of CCL22 and CCR3 mRNA with L13a. RT-PCR of the total RNA isolated from the macrophages of LPS treated mice shows induction of CCL22 and CCR3 mRNA (b). Aliquots of these lysates were subjected to immunoprecipitation with anti-L13a or anti-L28 antibodies using Seize X immunoprecipitation kit (Pierce) to avoid contamination from light and heavy chain. RNA was extracted from the immunoprecipitates followed by RT-PCR with specific primers (c, upper panel). The efficiency of the immunoprecipitation was confirmed by immunoblot analysis (c, lower panel). d, Reduced association of L13a with 60S ribosomal subunit in the macrophages from LPS treated mice. Macrophages isolated from either LPS or saline injected mice were subjected to immunoprecipitation using anti-L13a antibody. An aliquot of the immunoprecipitate was subjected to RT-PCR with mouse 28S rRNA specific primer (upper panel). The other aliquot was subjected to immunoblot analysis using anti-L13a antibody (lower panel).
The ability of the L13a-containing GAIT complex to silence translation of its target mRNAs relies on it’s binding to functional GAIT elements in the 3′UTR (9, 10). Therefore, we investigated the in vivo interaction of these target mRNAs with L13a. Comparative RT-PCR analysis of the total RNA isolated from macrophages of the saline-and LPS-treated wild type (WT) mice showed the presence of CCL22 and CCR3 mRNA only upon LPS treatment (Fig. 9b). To test whether these mRNAs are bound to L13a, peritoneal macrophages from LPS-treated WT mice were subjected to immunoprecipitation using anti-L13a antibody followed by extraction of RNA. The presence of CCL22 and CCR3 mRNAs were detected by RT-PCR using specific primers; the control actin mRNA in spite of its cellular abundance, could not be detected in the immunoprecipitate. None of the GAIT target mRNAs were detected in the same extracts after immunoprecipitation using antibody against L28, another protein of the large subunit, which demonstrated specificity (Fig. 9c). Using U937 cells, a cellular model of human monocytes, our previous studies showed the regulated release of L13a upon treatment with IFN-γ (10). As LPS is a potent inducer of IFN-γ in vivo (17–19), we investigated the association of L13a with the 60S ribosomal subunit in the peritoneal macrophages harvested from LPS-treated mice. RT-PCR analysis of the anti-L13a immunoprecipitate using 28S rRNA-specific primers revealed significantly less association of L13a with 60S subunit in the macrophages isolated from LPS-treated animals when compared to those from saline-treated animals (Fig. 9d). This is consistent with the previous report demonstrating the release of L13a from the 60S ribosome in response to IFN-γ treatment (10). Taken together, our results demonstrate that macrophage-specific deficiency of L13a may diminish the naturally imposed translational silencing on GAIT target mRNAs and suggest the physiological significance of this mechanism as an endogenous defense against uncontrolled inflammation.
Discussion
The primary finding of this study is the identification of ribosomal protein L13a in macrophage as a physiological attenuator of endotoxin-induced inflammation. In this study using a new mouse model of macrophage-specific L13a depletion we identified a protective role of L13a-dependent translational silencing against endotoxic shock caused by uncontrolled inflammation. Targeted disruption of the mouse L13a gene in macrophages resulted in increased mortality during LPS-induced endotoxemia. The increased lethality was associated with widespread tissue damage and infiltration of macrophages in major organs such as liver, lung and kidneys. The clinical symptoms of increased inflammation in the KO mice included increased lethargy and high breath rate that were accompanied by significantly higher levels of serum markers of inflammation such as, BUN, AST, TNF-α and higher serum levels of a group of other inflammatory cytokines. The increased synthesis of several GAIT target proteins e.g. CCL22, CXCL13, CCL8, CCR3 is also consistent with our result that shows the abrogation of GAIT element mediated translational silencing in the macrophages harvested from LPS treated KO mice. Most interestingly, our studies also suggest that LPS treatment significantly compromises the association of L13a with the 60S ribosomal subunit in the macrophages of these KO animals. Together, these results demonstrate that macrophage-specific deficiency of L13a in animal model may diminish the naturally imposed translational silencing on GAIT element containing mRNAs. Thus we have identified a novel extra-ribosomal function of L13a as an endogenous defense mechanism against uncontrolled inflammation caused by endotoxin treatment.
Previous studies from our laboratory using in vitro models of monocytes showed the ability of the ribosomal protein L13a-dependent translational silencing to target a cluster of mRNAs encoding different chemokines and chemokine receptors. In addition, specific segments present in the 3′UTRs of these mRNAs contained significant folding homologies to the authentic GAIT element, and subsequent experiments showed their role in translation regulation driven by a L13a-containing RNA-binding complex (9). These results are consistent with the notion that folding homologies among the elements present in multiple transcripts could offer co-regulation of translation by a single RNA-binding complex demonstrating the existence of a post-transcriptional operon (32, 33). Our results presented here using macrophages harvested from the KO mice directly showed reduced polysomal abundance of a group of mRNAs encoding inflammatory proteins. To our knowledge this is the first report of the significance of a ribosomal protein-dependent translational silencing to control physiological inflammation at the level of a whole organism. In these studies we have tested a few representative targets e.g. CCL22, CXCL13, CCL8, CCR3 in our animal model although other targets of L13a identified from the polysome profile analysis of monocytes (9) are yet to be tested in this KO mouse model.
Post-transcriptional control of many mRNAs has been previously implicated in a variety of cellular events recognized as a signature of inflammation such as activation and chemoattraction of T lymphocytes, influx of neutrophils, NK cell-mediated cytotoxicity, monocyte adhesion and survival of macrophages in the injured tissue (12, 34). The patho-physiological significance of these control mechanisms has been tested using genetically engineered mouse models with deficiencies of the AU-rich element (ARE) sequence of TNF-α 3′UTR (35) and RNA-binding proteins such as TTP (36), TIA-1 (37) and AUF1 (38). All of these studies showed elevated inflammatory responses due to the overexpression of TNF-α caused by the absence of TNF-α mRNA destabilization with a constitutive signature of pathology even under unchallenged condition. However, macrophage specific L13a KO mice show no sign of any pathology under unchallenged conditions and no developmental defect in the newborn. On the other hand, significant enhancement of pathology compared to the control mice was observed upon endotoxin challenge. Recently several studies have appreciated the emerging role of miRNA in inflammation and LPS tolerance (39–42). However, in previous studies we have recapitulated the translational silencing of the target mRNA harboring the GAIT element in the 3′UTR using a cell free in vitro translation system of rabbit reticulocyte lysate by adding only purified recombinant L13a. In addition, using RNAhybrid tool we have not found any potential miRNA recognition sites in GAIT element (43). Therefore it would seem unlikely that a miRNA would have a direct role in this process. It is important to note that the L13a-dependent translational silencing mechanism is activated by LPS and also targets LPS-induced mRNAs encoding a cohort of inflammatory proteins. These findings are consistent with the emerging concept of resolution of inflammation by a self-limiting response (4) and clearly differentiate L13a-dependent translational silencing from previously identified post-transcriptional mechanisms to control inflammation (44).
Emerging evidence suggests that chemokines and their receptors play a cardinal role in directing the recruitment of mononuclear cells to the sites of inflammation. This process performs an essential step in innate immune responses and precise regulation of these molecules is required for efficient but not excessive immune system function (15). Our report shows that macrophages from LPS-challenged KO animals produced significantly higher levels of CCL22, CXCL13, CCL8 and CCR3 due to the abrogation of their translational silencing. All of these cytokines show specific roles in diverse aspects of inflammation. For example, controlling the trafficking of activated T lymphocytes (45) and regulatory T cells (Tregs) (46) to inflammatory sites by CCL22; the role of CXCL13 in B cell homing (47) and its correlation with the childhood-onset lupus (48); activation of a large cohort of immune cells by CCL8 (49) and its elevated level in intrinsic asthmatics (50) and finally promotion of LPS-induced lung inflammation (51) and ulcerative colitis (52) by CCR3. Our data from the KO mice showing the elevated levels of these cytokines by macrophage-specific L13a depletion and significant elevation of inflammation in response to endotoxin challenge are highly consistent with these previous reports. Another interesting aspect of our result is the presence of higher numbers of both the classically and alternatively activated macrophages that express Mac2- and Ym1 respectively in the kidney glomeruli of the LPS-challenged KO animals (Fig. 4e). Considering the role of Ym1-positive macrophages in wound healing activities (14) our observation could indicate that a fibrotic response is initiated at the onset of acute inflammation.
In humans, haploinsufficiency of many ribosomal proteins such as S19, S24, S17, S15, S7 L35A, L5, L11, L36 causes genetic disorders such as Diamond-Blackfan anemia (53) and in mice, deficiency of ribosomal protein L38 has been shown to cause a defect in tissue patterning (54). For many of these ribosomal proteins deficiencies lead to various abnormalities in ribosome biogenesis (53). In sharp contrast, our previous in vitro studies of human monocytes showed that the depletion of L13a caused no defect in ribosome biogenesis or overall protein synthesis (11). The role of other ribosomal proteins such as S6 (55) and L22 (56) has been implicated in innate immunity, whereby deficiencies of these proteins in T cells severely compromised T cell development by selectively upregulating p53 translation by the depleted ribosome. However, our results provide the first evidence for a novel extra-ribosomal function of ribosomal protein L13a in limiting inflammation. In macrophages, this mechanism is driven by the phosphorylation-dependent release of L13a from 60S ribosomal subunit (10, 57) to efficiently block the expression of a cohort of inflammatory proteins, e.g. a group of chemokine and chemokine receptors, directly at the level of translation (9). In addition, previous work from others identified the intermediate signaling molecules responsible for L13a phosphorylation and release from ribosome (58). Studies presented in this report show the ability of the L13a-dependent silencing mechanism to control physiological inflammation caused by endotoxemia. Together, our results suggest that genetic defect in this pathway may contribute to the progression of inflammatory diseases and that manipulations of this pathway by small molecules may offer novel therapeutic strategies.
Supplementary Material
Acknowledgments
These studies are supported by PHS grant NIH HL79164 and American Heart Association (AHA) G.I.A 0855555D to B.M., AHA Pre-doctoral Fellowship grant 11PRE7660008 to D.P., NIH P01AI087586 to W.B, NIH AG 033604 to R.K, NIH AI 059267 to S.B, and a Ohio Third Frontier grant to S.B.
We acknowledge Dr. Veronique Lefebvre for suggestions in the various steps of creating the conditional null allele, Dr. Lisa Aronov for technical assistance in the process of generating chimeric mouse and design of targeting construct, Dr. Tanmay Majumder for assistance in leukocytes isolation, Nina Dvorina for assistance in immunohistochemistry. We are thankful to Dr. Meredith Bond and Dr. Anton Komar for critical reading of this manuscript.
Abbreviations used in this article
- KO
Knockout
- GAIT
Gamma Activated Inhibitor of Translation
- LPS
Lipopolysaccharide
- BUN
Blood Urea Nitrogen
- AST
Aspartate amino transferase
- Ad-CMV-Cre
Adenovirus expressing Cre recombinase under the control of CMV promoter
- L13aflox/floxLysMCre+
Mice homozygous for loxP allele in L13a gene and heterozygous for the Cre recombinase transgene under the control of Lysozyme M-specific promoter.
- ACTFLPe+
Transgenic mice expressing Saccharomyces cerevisiae FLP1 recombinase under the control of human beta-actin promoter
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serbina NV, Pamer EG. Coordinating innate immune cells to optimize microbial killing. Immunity. 2008;29:672–674. doi: 10.1016/j.immuni.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–776. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 5.Mazumder B, Fox PL. Delayed translational silencing of ceruloplasmin transcript in gamma interferon-activated U937 monocytic cells: role of the 3′ untranslated region. Mol Cell Biol. 1999;19:6898–6905. doi: 10.1128/mcb.19.10.6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazumder B, Seshadri V, Fox PL. Translational control by the 3′-UTR: the ends specify the means. Trends Biochem Sci. 2003;28:91–98. doi: 10.1016/S0968-0004(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 7.Mazumder B, Seshadri V, Imataka H, Sonenberg N, Fox PL. Translational silencing of ceruloplasmin requires the essential elements of mRNA circularization: poly(A) tail, poly(A)-binding protein, and eukaryotic translation initiation factor 4G. Mol Cell Biol. 2001;21:6440–6449. doi: 10.1128/MCB.21.19.6440-6449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kapasi P, Chaudhuri S, Vyas K, Baus D, Komar AA, Fox PL, Merrick WC, Mazumder B. L13a blocks 48S assembly: role of a general initiation factor in mRNA-specific translational control. Mol Cell. 2007;25:113–126. doi: 10.1016/j.molcel.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vyas K, Chaudhuri S, Leaman DW, Komar AA, Musiyenko A, Barik S, Mazumder B. Genome-wide polysome profiling reveals an inflammation-responsive posttranscriptional operon in gamma interferon-activated monocytes. Mol Cell Biol. 2009;29:458–470. doi: 10.1128/MCB.00824-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazumder B, Sampath P, Seshadri V, Maitra RK, DiCorleto PE, Fox PL. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell. 2003;115:187–198. doi: 10.1016/s0092-8674(03)00773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaudhuri S, Vyas K, Kapasi P, Komar AA, Dinman JD, Barik S, Mazumder B. Human ribosomal protein L13a is dispensable for canonical ribosome function but indispensable for efficient rRNA methylation. Rna. 2007;13:2224–2237. doi: 10.1261/rna.694007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mazumder B, Li X, Barik S. Translation control: a multifaceted regulator of inflammatory response. J Immunol. 2010;184:3311–3319. doi: 10.4049/jimmunol.0903778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 16.Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- 17.Dai R, Phillips RA, Zhang Y, Khan D, Crasta O, Ahmed SA. Suppression of LPS-induced Interferon-gamma and nitric oxide in splenic lymphocytes by select estrogen-regulated microRNAs: a novel mechanism of immune modulation. Blood. 2008;112:4591–4597. doi: 10.1182/blood-2008-04-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le J, Lin JX, Henriksen-DeStefano D, Vilcek J. Bacterial lipopolysaccharide-induced interferon-gamma production: roles of interleukin 1 and interleukin 2. J Immunol. 1986;136:4525–4530. [PubMed] [Google Scholar]
- 19.Mattern T, Girroleit G, Flad HD, Rietschel ET, Ulmer AJ. CD34(+) hematopoietic stem cells exert accessory function in lipopolysaccharide-induced T cell stimulation and CD80 expression on monocytes. J Exp Med. 1999;189:693–700. doi: 10.1084/jem.189.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanof EM, Smith PD, Zola H. Isolation of whole mononuclear cells from peripheral blood and cord blood. In: Coligan AHKJE, Margulies DM, Shevach EM, Greene SW, editors. Current Protocol in Immunology. John Wiley & Sons Inc; 2001. pp. 7.1.1–7.1.3. [Google Scholar]
- 21.Balk RA. Severe sepsis and septic shock. Definitions, epidemiology, and clinical manifestations. Crit Care Clin. 2000;16:179–192. doi: 10.1016/s0749-0704(05)70106-8. [DOI] [PubMed] [Google Scholar]
- 22.Ogimoto K, Harris MK, Jr, Wisse BE. MyD88 is a key mediator of anorexia, but not weight loss, induced by lipopolysaccharide and interleukin-1 beta. Endocrinology. 2006;147:4445–4453. doi: 10.1210/en.2006-0465. [DOI] [PubMed] [Google Scholar]
- 23.Grossberg AJ, Zhu X, Leinninger GM, Levasseur PR, Braun TP, Myers MG, Jr, Marks DL. Inflammation-induced lethargy is mediated by suppression of orexin neuron activity. J Neurosci. 2011;31:11376–11386. doi: 10.1523/JNEUROSCI.2311-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011 doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong S, Hughes RC. Macrophage surface glycoproteins binding to galectin-3 (Mac-2-antigen) Glycoconj J. 1997;14:267–274. doi: 10.1023/a:1018554124545. [DOI] [PubMed] [Google Scholar]
- 26.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puneet P, Yap CT, Wong L, Lam Y, Koh DR, Moochhala S, Pfeilschifter J, Huwiler A, Melendez AJ. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science. 2010;328:1290–1294. doi: 10.1126/science.1188635. [DOI] [PubMed] [Google Scholar]
- 29.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 31.Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris AR, Mukherjee N, Keene JD. Systematic analysis of posttranscriptional gene expression. Wiley Interdiscip Rev Syst Biol Med. 2010;2:162–180. doi: 10.1002/wsbm.54. [DOI] [PubMed] [Google Scholar]
- 33.Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 34.Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol. 2010;10:24–35. doi: 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- 35.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 36.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 37.Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. Embo J. 2000;19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu JY, Sadri N, Schneider RJ. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev. 2006;20:3174–3184. doi: 10.1101/gad.1467606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El Gazzar M, McCall CE. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J Biol Chem. 2010;285:20940–20951. doi: 10.1074/jbc.M110.115063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El Gazzar M, Church A, Liu T, McCall CE. MicroRNA-146a regulates both transcription silencing and translation disruption of TNF-alpha during TLR4-induced gene reprogramming. J Leukoc Biol. 2011;90:509–519. doi: 10.1189/jlb.0211074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinn EM, Wang J, Redmond HP. The emerging role of microRNA in regulation of endotoxin tolerance. J Leukoc Biol. 2012;91:721–727. doi: 10.1189/jlb.1111571. [DOI] [PubMed] [Google Scholar]
- 43.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. Rna. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9:353–359. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 45.Chang M, McNinch J, Elias C, 3rd, Manthey CL, Grosshans D, Meng T, Boone T, Andrew DP. Molecular cloning and functional characterization of a novel CC chemokine, stimulated T cell chemotactic protein (STCP-1) that specifically acts on activated T lymphocytes. J Biol Chem. 1997;272:25229–25237. doi: 10.1074/jbc.272.40.25229. [DOI] [PubMed] [Google Scholar]
- 46.Riezu-Boj JI, Larrea E, Aldabe R, Guembe L, Casares N, Galeano E, Echeverria I, Sarobe P, Herrero I, Sangro B, Prieto J, Lasarte JJ. Hepatitis C virus induces the expression of CCL17 and CCL22 chemokines that attract regulatory T cells to the site of infection. J Hepatol. 2011;54:422–431. doi: 10.1016/j.jhep.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 47.Gunn MD, V, Ngo N, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- 48.Ezzat M, El-Gammasy T, Shaheen K, Shokr E. Elevated production of serum B-cell-attracting chemokine-1 (BCA-1/CXCL13) is correlated with childhood-onset lupus disease activity, severity, and renal involvement. Lupus. 2011;20:845–854. doi: 10.1177/0961203311398513. [DOI] [PubMed] [Google Scholar]
- 49.Proost P, Wuyts A, Van Damme J. Human monocyte chemotactic proteins-2 and -3: structural and functional comparison with MCP-1. J Leukoc Biol. 1996;59:67–74. doi: 10.1002/jlb.59.1.67. [DOI] [PubMed] [Google Scholar]
- 50.Dajani R, Al-Haj Ali E, Dajani B. Macrophage colony stimulating factor and monocyte chemoattractant protein 2 are elevated in intrinsic asthmatics. Cytokine. 2011;56:641–647. doi: 10.1016/j.cyto.2011.08.040. [DOI] [PubMed] [Google Scholar]
- 51.Li B, Dong C, Wang G, Zheng H, Wang X, Bai C. Pulmonary epithelial CCR3 promotes LPS-induced lung inflammation by mediating release of IL-8. J Cell Physiol. 2011;226:2398–2405. doi: 10.1002/jcp.22577. [DOI] [PubMed] [Google Scholar]
- 52.Manousou P, Kolios G, Valatas V, Drygiannakis I, Bourikas L, Pyrovolaki K, Koutroubakis I, Papadaki HA, Kouroumalis E. Increased expression of chemokine receptor CCR3 and its ligands in ulcerative colitis: the role of colonic epithelial cells in in vitro studies. Clin Exp Immunol. 2010;162:337–347. doi: 10.1111/j.1365-2249.2010.04248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, Ishijima J, Shiroishi T, Barna M. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145:383–397. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sulic S, Panic L, Barkic M, Mercep M, Uzelac M, Volarevic S. Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev. 2005;19:3070–3082. doi: 10.1101/gad.359305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson SJ, Lauritsen JP, Hartman MG, Foushee AM, Lefebvre JM, Shinton SA, Gerhardt B, Hardy RR, Oravecz T, Wiest DL. Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity. 2007;26:759–772. doi: 10.1016/j.immuni.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 57.Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, Ting SM, Dignam JD, Kim S, Driscoll DM, Fox PL. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell. 2004;119:195–208. doi: 10.1016/j.cell.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 58.Mukhopadhyay R, Ray PS, Arif A, Brady AK, Kinter M, Fox PL. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol Cell. 2008;32:371–382. doi: 10.1016/j.molcel.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.