

Abstract
Fixation of mutations in human immunodeficiency virus type 1 (HIV-1), such as those conferring drug resistance and immune escape, can result in a change in replication fitness. To assess these changes, a real-time TaqMan PCR detection assay and statistical methods for data analysis were developed to estimate sensitively relative viral fitness in competitive viral replication experiments in cell culture. Chimeric viruses with the gene of interest in an HIV-1NL4-3 backbone were constructed in two forms, vifA (native vif gene in NL4-3) and vifB (vif gene with six synonymous nucleotide differences from vifA). Subsequently, mutations of interest were introduced into the chimeric viruses in NL4-3VifA backbones, and the mutants were competed against the chimera with the isogenic viral sequence in the NL4-3VifB backbone in cell culture. In order to assess subtle fitness differences, culture supernatants were sampled longitudinally, and the viruses differentially quantified using vifA- and vifB-specific primers in real-time PCR assays. Based on an exponential net growth model, the growth rate of each virus was determined and the fitness cost of the mutation(s) distinguishing the two viruses represented as the net growth rate difference between the mutant and the native variants. Using this assay, the fitness impact of eight amino acid substitutions was quantitated at highly conserved sites in HIV-1 Gag and Env.
Keywords: HIV-1, replication fitness, competition, real-time PCR, cell culture
1. Introduction
In HIV-1 infection, emergence of some drug resistant (Martinez-Picado and Martinez, 2008; Nijhuis, van Maarseveen, and Boucher, 2007) and escape mutants from host humoral and cellular immune responses (Fernandez et al., 2005; Liu et al., 2007; Martinez-Picado et al., 2006; Troyer et al., 2009) have been associated with reduced replication fitness. Fitness can in some cases be restored by compensatory mutations (Brockman et al., 2007; Koval et al., 2006). Subtle changes in viral fitness can have substantial impacts on virus representation over time. To estimate the impact of viral mutations on replication capacity, it is therefore important to employ a sensitive method to determine replication fitness costs in surrogate assays of viral replication in cell culture.
Several methods have been developed to estimate the replication capacity of HIV-1 strains. Some studies measured viral replication using single cycle assays, by infecting indicator cell lines (Mammano et al., 2000) or by infection with pseudotyped virus particles that contain indicator genes (Petropoulos et al., 2000). These methods are simple and rapid but fail to interrogate the entire life cycle. Other studies estimated viral replication capacities by monitoring parallel mono-infections, with viral growth kinetics determined by measurements of p24 production (Doyon et al., 1996), RT activity (Resch et al., 2002) or the number of infected indicator cells (Brockman et al., 2006). Because of the difficulty in ensuring identical growth conditions, comparisons between parallel mono-infections may fail to detect more subtle differences in viral replication characteristics.
The gold standard for estimating the relative replication capacity of a viral strain is a dual infection competition assay, in which both experimental and reference strains are grown together to ensure equivalent growth conditions and during which they compete for resources, more akin to replication in vivo. In some studies, the production ratio of the two viruses in the competition was determined by bulk (Garcia-Lerma et al., 2004; Sharma and Crumpacker, 1997) or clonal (Martinez-Picado et al., 1999; Wang et al., 2006) sequencing; however, the breadth of range of detection was usually narrow for these methods. In other studies, a small reporter gene (Ali and Yang, 2006) or synonymous mutations (Anastassopoulou et al., 2009; Liu et al., 2007; Troyer et al., 2009; van Maarseveen et al., 2006) was introduced into the reference viral genome as a tag to facilitate the differential quantification of the two competing viruses. Synonymous mutations that avoid known sites of important RNA structures are less likely to disrupt viral genome function and thus are less likely to have an impact on viral replication capacity. The production of each virus can be determined by a heteroduplex tracking assay (HTA) involving radioactive probes (Liu et al., 2007; Quinones-Mateu et al., 2000; Troyer et al., 2009); oligonucleotide ligation assay (OLA) (Ellis et al., 2004; Lalonde et al., 2007; Troyer et al., 2008), or by real-time PCR (Ali and Yang, 2006; Anastassopoulou et al., 2009; van Maarseveen et al., 2006), reagents for the latter of which can be more expensive but the assay is more sensitive and involves fewer processing steps.
In one study (van Maarseveen et al., 2006), a tag composed of two synonymous mutations in the gag-p24 gene was used. Viruses with mutations in protease or RT were competed against the reference strain with the synonymously mutated gag-p24 tag in dual infections. Real-time PCR assays were used to monitor viral growth and detect replication fitness costs. In another study (Anastassopoulou et al., 2009), a tag composed of eleven synonymous mutations in the vif gene was used. Both studies used specific probes to quantify the mutant and reference viruses simultaneously in competitions. In the study using the gag-p24 tag (van Maarseveen et al., 2006), plasmids with wild type and mutant gag-p24 were mixed in different ratios in a total of 3×107 copies. The minor plasmid could be detected even when it was present in only 1% of the mixture. In the study using the vif tag (Anastassopoulou et al., 2009), no threshold for the detection of the minor strain was reported.
Previously, six synonymous mutations were introduced into the vif gene (Figure 1A) of the HIV-1 lab strain NL4-3 (Liu et al., 2007; Troyer et al., 2009). Using HTA to monitor viral growth in dual infections, no significant fitness impact was found associated with the introduction of these mutations. By competing chimeric mutant viruses (with the NL4-3 backbone containing the native vif) against chimeras with the isogenic viral sequences and the mutated vif tag, the replication fitness impact of CTL escape mutations in Gag and Env proteins was determined. Here, a real-time PCR based competition assay was developed that differentially recognized the vif tag with a detection threshold for the minor viral strain as low as 0.002%, and the replication fitness impact of eight Gag and Env mutations was determined.
Figure 1. Real-time PCR strategies.

A) Nucleotide sequences and the encoded amino acids of the vifA and vifB tags. B) Real-time PCR strategies that differentially quantify DNA containing vifA and vifB in mixtures. i) Use of a common primer set and vifA- and vifB-specific probes. Primers are located outside the vifA/vifB tag and the probes encompass the tags. ii) Use of a common sense primer and probe, and vifA and vifB specific antisense primers. The sense primer and probe are located upstream from the tag, while the vifA- and vifB-specific antisense primers encompass the tag.
2. Materials and methods
2.1 Wild type and mutant chimeric viruses
Recombinant plasmids containing the backbone of pNL4-3 and gag (nucleotides 1089–2022, HIV-1HXB2) or env (nucleotides 6347–7802, HIV-1HXB2) founder sequences derived from the PBMCs of a patient infected with HIV-1 subtype B were generated previously (Liu et al., 2007; Troyer et al., 2009, kindly provided by Dr. Eric J. Arts, Case Western Reserve University, Cleveland, Ohio). The founder sequence refers to the consensus sequence of viruses taken from the earliest time point sampled during infection, in this case, 8 days post onset of the acute symptoms of viral infection (Liu et al., 2006, ~3 weeks post exposure to the virus (Stekler and Collier, 2004)). For both gag and env founders, two forms of recombinant plasmids were generated, one in native pNL4-3 (pNL4-3VifA) and the other in pNL4-3 with six synonymous mutations introduced into the vif gene (pNL4-3VifB, Figure 1A). These six nucleotide differences were used to facilitate differential recognition in competition assays. In the current study, using recombinant vifA plasmids as templates, mutations at single sites in gag (n=2) and env (n=6) were introduced into the founder gag or env gene using the QuikChange II XL site-directed mutagenesis kit (Agilent, La Jolla, CA). Whole HIV-1 genome sequences in the resulting plasmids were determined to confirm the presence of the desired mutations and the absence of additional mutations. HEK293T cells (DuBridge et al., 1987) were then transfected with the founder or mutant recombinant plasmids using FuGENE®6 Transfection Reagent (Roche, San Francisco, CA) and cell-free supernatants were collected as viral stocks. The gag and env regions were then resequenced to confirm the presence of the desired mutations in the chimeric viruses following transfection.
2.2 p24 antigen capture assay and determination of TCID50
p24 production in transfection or culture supernatants was determined using an in-house double-antibody sandwich ELISA specific for the HIV-1 p24 antigen, as described previously (McClure et al., 2007). Viral infectivity was determined by 50% tissue culture infective dose (TCID50) in PBMCs (all from a single HIV-negative donor) using the p24 antigen capture assay, as calculated by the Reed-Muench method (Reed and Muench, 1938).
2.3 Viral competitions
Two chimeric viruses (NL4-3VifA or NL4-3VifB backbones) were mixed at a ratio of 1:1 based on TCID50, and 5×105 PBMCs that had been prestimulated for 2 days with PHA (1.5μg/ml) and then treated with IL-2 (20 U/ml) were infected at a multiplicity of infection (MOI) of 0.005 (lower MOIs resulted in more variable mutant frequencies in dual infections), in a total volume of 1 ml. After 24 h of infection (day 1), cells were washed with complete IMDM (Iscove’s Modified Dulbecco’s Media supplemented with 2mM L-glutamine, 100U/ml Penicillin, 100μg/ml streptomycin, and 10% FBS) and resuspended in fresh culture medium [complete IMDM supplemented with 20 U/ml human recombinant IL-2 (Roche, San Francisco, CA)] in a final volume of 2 ml. On days 2 through 9, 1 ml of culture supernatant was collected from each infection and 1 ml of fresh culture medium was added back. All dual infections were done in triplicate. As control, single replicate mono-infections were also performed in parallel for each competing virus at a MOI of 0.005.
2.4 Serial passage of chimeric viruses
Serial passages of chimeric viruses were set up as mono-infections in triplicate. At day 7 of each passage, 500 μl of culture supernatants were used to inoculate 5×105 PHA-IL-2 stimulated fresh PBMCs.
2.5 RT PCR, PCR and sequencing
Viral RNA was purified from transfection or culture supernatants using a QIAxtractor following the manufacturer’s recommendations (QIAGEN, Valencia, CA). For each extraction, 200 μl of viral supernatant was used and a final elution of ~ 70 μl RNA was obtained. The Turbo DNA free kit (ABI, Applied Biosystems, Foster City, CA) was used to remove DNA from a 44 μl aliquot of RNA, and 1μl of the resulting RNA was used for cDNA synthesis with SuperScript® III (Invitrogen, Carlsbad, CA). The primers used for cDNA synthesis were RT2 (HIV-1HXB2 nucleotides 3301–3321, Liu et al., 2006) for the gag fragments and gp120R2 (GTTGATCCTTTAGGTATCTTTCCACAGC, nucleotides 7968–7995) for the vif-env fragments. The gag fragment was PCR amplified using primer pair GAG-2 (nucleotides 794–817, Liu et al., 2006) and RSP15R (CAATTCCCCCTATCATTTTTGGTTTCC, nucleotides 2377–2403), and the vif-env fragment was amplified using primer pair envF (GCAGGTGATGATTGTGTGGCAAGT, nucleotides 5055-507) and envR (CCCTCAGCAAATTGTTCTGCTGCT, nucleotides 7873–7896), and the PCR products were sequenced directly.
2.6 Real-time TaqMan PCR
Viral variant representation was quantified using an ABI 7300 Real-Time PCR System (ABI). Single probe reactions contained 12.5 μl TaqMan® Gene Expression Master Mix (ABI), 5μM probe, 20μM each of the forward and reverse primers and cDNA template in a final volume of 25μl. Double probe reactions contained 5μM of each probe in a final volume of 50μl. Probes and primers are listed in Table 1. Probe and primers located in gag were used to quantify virus derived from transfection supernatants, while those located in vif were used to quantify virus variants in culture supernatants. PCR cycling parameters were 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. Standard curves for real-time PCR were generated using 30 to 3×106 copies of pNL4-3VifA or pNL4-3VifB, quantitated by spectrophotometry, as template in triplicate. The copy numbers of viral cDNA were determined in duplicate, using 1 μl of cDNA as starting material.
Table 1.
Primers and probes in real-time TaqMan PCR
| Name | Sequence | HXB2 location |
|---|---|---|
| To determine copy numbers of cDNA derived from transfection supernatants | ||
| gag B-S.1 (sense primer) | CAAGCAGCCATGCAAATGTT | 1372 to 1391 |
| gag B-A.1 (antisense primer) | CTAAAGGGTTCCTTTGGTCCTTGT | 1647 to 1670 |
| gag B.2 (antisense probe) | 6FAM-TAGTTCCTGCTATGTCACTT-MGBNFQ | 1488 to 1507 |
| Common primer set and specific probes to differentiate vifA and vifB | ||
| VifAB_sensePrimer2 | GGTCTGCATACAGGAGAAAGAGACT | 5251 to 5275 |
| VifAB_antisensePrimer | TGCAGATGAATTAGTTGGTCTGCTA | 5345 to 5369 |
| MGB_VifA_antisenseProbe | 6FAM-TACTTGTGTGCTATATCTCTTT-MGBNFQ | 5313 to 5334 |
| MGB_VifB_antisenseProbe | VIC-CACCTGCGTGCTATACC-MGBNFQ | 5318 to 5334 |
| Common sense primer and probe, and specific antisense primers to differentiate vifA and vifB | ||
| VifAB_sensePrimer2 | ||
| antisensePrimerVifA | AGGGTCTACTTGTGTGCTATATCTCTTTT | 5312 to 5340 |
| antisensePrimerVifB | CACCTGCGTGCTATACCTTTTCT | 5312 to 5334 |
| MGB_VifAB_antisenseProbe | 6FAM-CTCCATTCTATGGAGACTC-MGBNFQ | 5291 to 5309 |
2.7 Calculation of replication fitness costs
To determine the impact of an amino acid substitution on the replication fitness of the corresponding founder virus, the net growth rates between the mutant and the founder viruses were compared in competitions using a mathematical model adapted from Wu et al (Ma et al.; Wu et al., 2006). Because the turnover of free virus is typically fast in comparison with those of infected cells and a quasi steady state is established rapidly, this model assumes that the amount of free virus is proportional to the number of infected cells (Ma et al.; Wu et al., 2006). Findings of cell-to-cell transmission as the dominant mode of HIV-1 spread in T lymphocyte cultures also support this assumption (Dimitrov et al., 1993; Hubner et al., 2009). Therefore, serial sampling of supernatants (and consequent 2-fold dilution at each sampling) from our competition assays should have little effect on the rate of free virus production (and this was confirmed experimentally, c.f. Figure 6). In Wu’s model, viruses are assumed to reproduce continuously with an exponential net growth,
Figure 6. Lack of impact of daily sampling on viral growth.

The cDNA copy number of envVifA (A) and envVifB (B) derived from competition supernatants. envVifA was competed against envVifB and competition supernatants were sampled daily or only on days 4, 7 and 10 (no sampling or medium change on days 2, 3, 5, 6, 8, and 9). Reported are the means of triplicate competition experiments with standard deviations.
V(0) is the number of infectious particles in the inoculum at time zero, V(t) is the free viral production at time t after infection, and g is the net viral growth rate, a combined growth/decay parameter for viral production. Therefore, the net growth rate difference (d) between the mutant (m) and founder (f) viruses can be expressed as
and the ratio (r) of the net growth rates as
Here, copy numbers of cDNA derived from founder and mutant viruses in competition culture supernatants were used as the values of Vf (t) and Vm (t), respectively.
Our method to estimate growth rate parameters differed from Wu’s method as follows. To estimate d, the regression model was used:
| (1) |
where the outcome yi was ln[Vm (ti)/Vf (ti)], ti was the time at which the measurements were collected, and εi, the error term, was distributed normally. In this regression model, the estimated intercept, α̂, would provide an estimate of the log ratio of the number of infectious particles of the two competing viruses in the inoculum, ln[Vm(0)/Vf(0)], and the estimated coefficient of t, β̂, would provide an estimate of d. To estimate r, the growth rate ratio, the regression model with intercept as the only covariant was used:
| (2) |
where the outcome yi was
In this regression model, the intercept, α̂, provides an estimate of r. Similar to the estimation of d, gm and gf were obtained using a linear regression model with intercept, in which ln[Vm(t)] or ln[Vf(t)] was regressed on covariate t (time). The coefficient of t provides an estimate of net growth rate, gm or gf, and the intercept provides an estimate of the log number of infectious particles in the inoculum, ln[Vm(0)] or ln[Vf(0)]. All parameters were estimated using linear regression with generalized estimating equations (GEE) and exchangeable working correlation matrix to account for intra-replicate correlations. All confidence intervals (CI) were based on robust (empirical) standard errors.
Theoretically, a growth rate difference of d = 0 and a growth rate ratio of r = 1 indicates no replication fitness differences. A growth rate difference of d > 0 and a growth rate ratio of r > 1 indicates faster growth of the mutant virus, whereas a growth rate difference of d < 0 and a growth rate ratio of r < 1 indicates slower growth of the mutant virus. To account for any fitness differences between the vifA and vifB founders and assay variations between different experiments, estimates of d and r from competitions between a mutant and its vifB founder (experimental) were compared to those estimated from competitions between the corresponding vifA and vifB founders (control). To estimate the differences in d between the experimental and the control data, an extended regression model and the combined data from the control and experimental groups were used:
| (3) |
In this model two additional covariates were added to equation (1). The first new covariate, xi, was an indicator variable (xi = 1 for control and xi = 0 for experimental data) and the second new covariate, xi*ti, was an interaction between this indicator (xi) and the covariate t. The estimated coefficient of the interaction term, β̂, was used to determine whether the value of d estimated using the experimental data was significantly different from that estimated using the control data, i.e., when β̂ was significantly different than zero (p-value associated with the estimate β̂ < 0.05), the values of d in the experimental and control conditions were considered to be significantly different. Similarly, to estimate the differences in r between the experimental and the control data, regression equation (2) were modified:
| (4) |
The estimated coefficient, β̂, was used to determine whether the value of r estimated using the experimental data was significantly different from that estimated using the control data.
3. Results
3.1 Quantification of pNL4-3VifA and pNL4-3VifB in mixtures by real-time PCR
To establish a real-time TaqMan PCR assay that can quantify differentially DNA containing vifA or vifB in mixtures, our primers and probes were examined using plasmids pNL4-3VifA and pNL4-3VifB as templates. The strategy of a common primer set and vifA (FAM-labeled) and vifB (VIC-labeled) specific probes was tested first (Figure 1B.i, Table 1). The primers were located outside the vifA/vifB tag and the probes encompassed the tag sequences. In real-time PCR reactions, the vifA probe could detect 30 copies (the lower limit evaluated) of pNL4-3VifA in the absence of pNL4-3VifB, while binding to 3,000,000 copies of pNL4-3VifB in the absence of pNL4-3VifA was negative (Figure 2A). The same specificity was found for the vifB probe (Figure 2B). However, when both targets were present, such probes could not detect their specific targets when they were found at ≤ 1% of the total (data not shown).
Figure 2. Detection of pNL4-3VifA and pNL4-3VifB using a common primer set and vifA- and vifB-specific probes.

A) Detection of pNL4-3VifA (from 30 to 3×105 copies), or 3×106 copies of pNL4-3VifB, using the vifA-specific probe in the absence of the other plasmid. B) Detection of pNL4-3VifB (30 to 3×105 copies), or 3×106 copies pNL4-3VifA, using the vifB-specific probe in the absence of the other plasmid. The readings from 3×106 copies of pNL4-3VifB or pNL4-3VifA with the vifA- or vifB-specific probes and no template controls are all below the threshold. Results from triplicate reactions are shown.
Therefore, another real-time PCR strategy was tested using common sense primer and probe, and vifA- and vifB-specific antisense primers to quantify differentially DNA containing vifA or vifB in mixtures (Figure 1B.ii, Table 1). Both vifA and vifB antisense primers bound specifically to vifA and vifB DNA (data not shown). Although the amount of the different strains in mixtures had to be determined in separate reactions, this real-time PCR strategy was very sensitive. The presence of 30 copies of pNL4-3VifA or pNL4-3VifB could be detected in mixtures with 3×106 copies of the other plasmid (0.001% sensitivity) (Figure 3A and B). Furthermore, the presence of 3×106 copies of pNL4-3VifB did not interfere with the quantification of pNL4-3VifA (Figure 3C), and similar results were obtained for pNL4-3VifB quantification in the presence of 3×106 copies of pNL4-3VifA (data not shown). In the case of viral mixtures, 45 copies of gagVifA could be detected in the presence of 1.98×106 copies of gagVifB (0.002% sensitivity) (Figure 3D). Similar results were obtained when serially diluted gagVifB was mixed with 2.14×106 copies of gagVifA (data not shown). Therefore, for the rest of the study, this latter strategy was used to quantify viruses in competition assays.
Figure 3. Detection of pNL4-3VifA and pNL4-3VifB in mixtures using common sense primer and probe and vifA- and vifB-specific antisense primers.
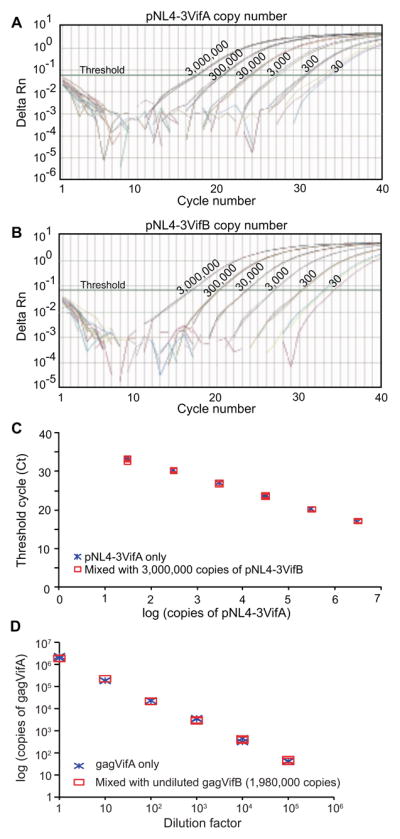
A) Detection of pNL4-3VifA (30 to 3×106 copies) using the vifA-specific primer in the presence of 3×106 copies of pNL4-3VifB. B) Detection of pNL4-3VifB (30 to 3×106 copies) using the vifB-specific primer in the presence of 3×106 copies pNL4-3VifA. C) Comparison of the threshold cycles in real-time PCR reactions containing differing amounts of pNL4-3VifA with or without 3×106 copies of pNL4-3VifB, using vifA-specific primer. For A) to C), results from triplicate reactions are shown. D) Quantification of serially diluted chimeric virus gagVifA (dilution factors ranged from 1 to 106) with or without undiluted gagVifB. Serially diluted transfection supernatants of gagVifA were mixed with undiluted transfection supernatants of gagVifB at a 1:1 ratio based on volume. RNA was purified from 50μl gagVifA only samples or gagVifA/gagVifB mixtures. After DNase treatment, cDNA was synthesized using 7μl DNA-free RNA and the PCR-amplifiable copy numbers of gagVifA in 1μ cDNA were determined in real-time PCR using the vifA-specific primer. In addition, the PCR-amplifiable copy number of gagVifB in the mixture was estimated from 1μ cDNA derived from 50μl of the gagVifB only sample using the vifB-specific primer. The copy numbers in the gagVifB and gagVifA only samples in the figure were adjusted (divided by 2) since there were only 25μl of gagVifA and gagVifB each in gagVifA/gagVifB mixtures. Results from duplicate reactions are shown.
3.2 Chimeric founder and mutant viruses
Chimeric viruses with the gag or env founder sequences derived from the PBMCs of a patient infected with HIV-1 subtype B (Liu et al., 2006) in both NL4-3VifA and NL4-3VifB backbones (gagVifA and gagVifB, envVifA and envVifB) were generated previously (Liu et al., 2007; Troyer et al., 2009). Two Gag and six Env mutations were introduced individually into the founder sequences in vifA backbones (Figure 4A) at amino acid sites that are over 98% conserved in HIV-1 group M sequences in the Los Alamos National Laboratory HIV Sequence Database (based on the alignments from (Rolland, Nickle, and Mullins, 2007)). The mutations introduced at these sites were observed at low frequency in PBMC-derived sequences from this patient (Troyer et al., 2009). cDNA copy number, p24 production and TCID50 derived from the transfection supernatants of the vifA founders (gagVifA and envVifA) and the env2, env3 and env6 mutants were similar to those of the corresponding vifB founders (gagVifB and envVifB). In contrast, the TCID50 of gag1, gag2, env1, env4 and env5 mutants were 1.5 to 2.5 log10 lower than their founders (Figure 4B&C).
Figure 4. Characterictics of gag and env chimeric founder and mutant viruses.

A) Mutants and location of mutations in proteins from HIV-1HXB2. B) gag mutants. C) env mutants. Viral RNA (~70μl) was purified from 200μl transfection supernatants. After DNase treatment, cDNA was synthesized using 1μl DNA-free RNA and the PCR-amplifiable copy numbers were determined in real-time PCR using specific gag primers and probe. p24 production was determined by ELISA (McClure et al., 2007). TCID50 were calculated using the Reed-Muench method (Reed and Muench, 1938) using PBMC and p24 antigen capture assays.
3.3 Viral growth in cell culture
vifA founders and mutant viruses were competed against the corresponding vifB founders in dual infections, and each competing virus also grown in mono-infections. All infections started with an MOI of 0.005 and all competitions were started with a virus ratio of 1:1 based on their TCID50. Because the TCID50 of many mutant viruses were substantially lower than their founder viruses, their cDNA copy numbers in the inocula were often higher (Figure 5, day 0). The copy numbers of day 2 viruses were <10% of those of the viral inocula, reflecting the eclipse phase of the initial round of infection. The founder-derived (gag or env) regions of the day 9 viruses were sequenced from all infections, and no additional mutations were found.
Figure 5. Growth kinetics of gag and env chimeric viruses in dual and mono infections.

Growth kinetics of gag chimeric viruses in dual (A–C) and mono-infections (D). Growth kinetics of env chimeric viruses in dual (E–K) and mono-infections (L). All dual infections were done in triplicate. In parallel, mono-infection of each competing virus was performed in a single replicate. Supernatants from infections were sampled daily, viral RNA were extracted and cDNA synthesized using 1μl DNase-treated RNA. cDNA copy numbers are reported using real time PCR in duplicate reactions along with standard deviations. The gray areas represent the data used for fitness calculations. Different symbols refer to different culture replicates. Different colored lines refer to different chimeric viruses.
In control dual infections (Figure 5A and E), the growth kinetics of gagVifA and gagVifB, as well as envVifA and envVifB, mirrored each other, indicating little fitness impact from the synonymous nucleotide differences between vifA and vifB. Virus particles (measured as viral cDNA copy number) derived from gagVifA and gagVifB increased rapidly during days 2 to 4 and then declined slowly after day 5 (Figure 5A). envVifA and envVifB, each of which derived their env from the patient’s founder sequence, grew slower, and the virion number increased steadily until day 7 or 8 (Figure 5E). Many mutant viruses showed slower growth kinetics compared to their founders (e.g., gag1 and env4, Figure 5B and I). Similar results were obtained with mono-infections (Figure 5D (gag) and L (env)).
3.4 Changes in replication fitness result from amino acid substitutions observed in vivo
In our competition assays, half the volume of supernatants were sampled and replaced with fresh media each day, which resulted in 2-fold dilution of the supernatants at each sampling. The model used here to determine viral growth assumes that the amount of free virus is proportional to the number of infected cells (see Methods); thus, daily sampling from supernatants should not reduce free viral production. To evaluate the impact of daily sampling on viral growth, envVifA was competed against envVifB and supernatants were sampled daily or only on days 4, 7 and 10 (no sampling or medium change on days 2, 3, 5, 6, 8, and 9). Indeed, daily sampling did not reduce viral production of envVifA or envVifB (Figure 6), thus validating the assumption above.
Data from the exponential growth period were used to calculate net growth rates, from days 2 to 5 for gag and days 3 to 7 for env chimeric viruses. Compared to individual net growth rates (g) in Figure 7A and B, the growth rate differences (d) estimated using the regression model (1) in Figure 7C and D, respectively, were consistent with individual g differences. However, in some cases, the growth rate ratios (r) estimated using the regression model (2) were quite different from those calculated based on the individual estimates of g and had wide 95% confidence intervals (env1, env4 and env5, Figure 7E and F).
Figure 7. Fitness impact of Gag and Env mutations in dual infections in cell culture.

Net growth rates (g) of individual chimeric gag (A) and env (B) viruses in competition assays. Net growth rate differences (d) between chimeric gag (C) and env (D) vifA viruses and the corresponding vifB founders in competition assays. Net growth rate ratios (r) between gag (E) and env (F) vifA viruses and the corresponding vifB founders in competition assays. See Methods for equations used to calculate each rate term. Reported are the means and 95% CI from triplicate competition experiments. * indicates a significant difference [p < 0.05, calculated using the extended regression model (3) for d and model (4) for r] relative to the corresponding vifA and vifB founders.
Consistent with previous findings (Liu et al., 2007; Troyer et al., 2009), the growth rate differences between vifA and vifB founders were small, especially the gag founders (ggagVifA − ggagVifB = −0.017 per day, <1% of the net growth rate of gagVifA; genvVifA − genvVifB = −0.091 per day, <10% of the net growth rate of envVifA, Figure 7A, B, C and D). The growth rate differences from different experiments were also small. For example, in repeated experiments, we obtained ggagVifA − ggagVifB = 0.019 (95% CI: −0.016 to 0.053) per day and genvVifA − genvVifB = 0.007 (95% CI: −0.004 to 0.018) per day. To further examine the impact of the six synonymous vif mutations, 4 passages of gagVifA and gagVifB were conducted, and sequences encompassing the vif tag were determined at day 7 of each passage. During this 28-day infection series (note that our competition assays were only 9 days long), no mutations or reversions at any position within the vif tags were observed, and no vifB or vifA viruses were detected in gagVifA or gagVifB cultures, respectively, by our real-time PCR assay.
The two gag mutations, gag2 and especially gag1, reduced viral replication fitness significantly (Figure 7C). For the env mutations, env4, env5 and env6 reduced viral fitness significantly, env1 had little fitness impact (p = 0.46), and env2 and env3 had increased viral fitness significantly (Figure 7D).
4. Discussion
A real-time TaqMan PCR based competition assay and statistical methods for data analysis were described to estimate the impact of amino acid substitutions on HIV-1 replication in cell culture. Native or mutant vif genes (vifA or vifB) in the NL4-3 strain backbone were used as tags for discriminating virus variants in competition assays. vifA and vifB had six synonymous nucleotide differences that had limited or no impact on viral replication. Chimeric viruses were generated with the mutants of interest in the NL4-3VifA backbone and competed against the chimera with the otherwise isogenic viral sequence in the NL4-3VifB backbone. Using primers binding specifically to vifA or vifB, the wild type and mutant viruses were quantified differentially, and the fitness costs of the mutations of interest were determined based on an exponential net growth model. Using this method, the fitness impact of eight mutations was determined at highly conserved amino acid sites, two in Gag and six in Env.
Real-time PCR strategies that use specific probes can quantify chimeric wild type and mutant viruses simultaneously in competitions (Anastassopoulou et al., 2009; van Maarseveen et al., 2006); however, this strategy did not work well in our study. Using vifA- and vifB-specific probes, neither pNL4-3VifA nor pNL4-3VifB could be detected sensitively in plasmid mixtures. In contrast, using vifA- or vifB-specific primers separately, the minor strain could be quantified even when it was present as only 0.001% of a plasmid mixture. In gagVifA and gagVifB virus mixtures, minor strains with a proportion of as low as 0.002% could also be detected.
One popular method to determine the replication fitness differences between two viruses in a competition is based on the relative fitness of the mutant, which is defined as the final proportion of the mutant in total viruses normalized by its initial proportion in the inocula, usually based on a prior TCID50 determination (Anastassopoulou et al., 2009; Liu et al., 2007; Quinones-Mateu et al., 2000; Troyer et al., 2009). A relative fitness of 1 indicates no fitness impact, < 1 indicates fitness cost and >1 indicates fitness gain from the mutation(s). This method is simple as it samples only once at the end of the competition. Because this method does not assume exponential growth, the restriction of sampling during exponential growth could be relaxed. However, one problem associated with this method is that the TCID50 values usually have large standard deviations, resulting a poor estimate of relative fitness. For example, in competitions of viral mutants env4 and env5 against their founder virus, the initial proportions of the mutants in the inocula based on TCID50 were 0.5 and the mean final proportions of the mutants on day 7 were 0.73 for env4 and 0.83 for env5 (Figure 5I and J). Using this method, the relative fitness would be 1.46 for env4 and 1.66 for env5, indicating a fitness gain, while our longitudinal data showed clearly fitness costs from the two mutations.
In the current study, the fitness impact of mutations was determined from longitudinal sampling and by an exponential growth model adapted from Wu et al (Ma et al., 2010; Wu et al., 2006). Their web-based tool vFitness (Ma et al., 2010) allows researchers to estimate growth rate differences (d) and ratios (r) between competing viruses. However, this tool is not designed for multiple experimental replicates and it is difficult to determine whether the estimated fitness change is significant. Based on the exponential growth model described in vFitness, our estimation procedure took measurements from all experimental replicates and accounted for intra-replicate correlations with generalized estimating equations (GEE). Using an interaction term in the linear regressions, whether the d and r estimated from the experimental group were significantly different from the control group could be determined. Although our data contained small variability between assays, our statistical comparisons of estimates of d and r from the experimental data to those estimates from the control data allowed us to account for this assay variability.
Because our model assumes exponential net growth of viruses, it is important to monitor viral growth in each dual infection and sample the supernatants within the exponential growth phase. Yet, different sets of mutants experienced exponential growth at different times following initiation of growth in culture. For example, chimeric gag founder viruses had high net growth rates and reached viral production peak in 5 days. Calculations based on samples after day 5 would violate the exponential growth assumption and underestimate the fitness impact of the examined mutations. In our earlier experiments, viral supernatants of days 4, 7 and 10 were sampled, and fitness costs associated with the gag1 and gag2 mutations were not detected (data not shown). In addition, the growth rate ratio r is a nonlinear combination of individual growth rates (r = gf/gm). The assumption of exponential growth is especially important for estimation of r from our regression model (2). Small departures from measurements of exponential growth can occur even in carefully monitored viral infections in cell culture conducted in the current study, which can have a major impact on point estimates of r (Figure 7F). Therefore, determination of fitness costs from mutations based on the estimation of d, which is less sensitive to small departures from exponential growth, is recommended.
The mean value of d can be used to determine how soon a mutant can be fixed or lost in the mixed population giving abundant target cells,
For example, in our competition assays, the d between gag1 and gagVifB (ggag1 − ggagVifB) was −0.85/day, and that between env5 and envVifB (genv6 − genvVifB) was −0.71/day. Thus, in the presence of abundant target cells, it would take 10.8 and 12.9 days, respectively, for gag1 or env5 virus to decline from 99% to 1% in a population mixed with their vifB founders.
In summary, our real-time PCR based competition assays and analysis methods can be used to estimate individual net growth rates of competing viruses and growth rate difference between the two, and determine the fitness impact of mutations in HIV with increased sensitivity and accuracy compared to previous methods. A web based tool that allows fitness to be calculated using our method is available at http://indra.mullins.microbiol.washington.edu/grc/.
Acknowledgments
We thank Brandon Maust for making the web based tool that allows fitness calculation using our model available. This work was supported by grants from the U.S. Public Health Service to YL (R21 AI081569), to JIM (R37 AI47734 and P01 AI57005), and the University of Washington Center for AIDS Research Computational Biology and Biostatistics Cores (P30 AI27757).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali A, Yang OO. A novel small reporter gene and HIV-1 fitness assay. J Virol Methods. 2006;133:41–7. doi: 10.1016/j.jviromet.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Anastassopoulou CG, Ketas TJ, Klasse PJ, Moore JP. Resistance to CCR5 inhibitors caused by sequence changes in the fusion peptide of HIV-1 gp41. Proc Natl Acad Sci U S A. 2009;106:5318–23. doi: 10.1073/pnas.0811713106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, Desouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E, Mulenga J, Goepfert PA, Walker BD, Allen TM. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol. 2007;81:12608–18. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman MA, Tanzi GO, Walker BD, Allen TM. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J Virol Methods. 2006;131:134–42. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA. Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol. 1993;67:2182–90. doi: 10.1128/jvi.67.4.2182-2190.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon L, Croteau G, Thibeault D, Poulin F, Pilote L, Lamarre D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J Virol. 1996;70:3763–9. doi: 10.1128/jvi.70.6.3763-3769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol. 1987;7:379–87. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis GM, Mahalanabis M, Beck IA, Pepper G, Wright A, Hamilton S, Holte S, Naugler WE, Pawluk DM, Li CC, Frenkel LM. Comparison of oligonucleotide ligation assay and consensus sequencing for detection of drug-resistant mutants of human immunodeficiency virus type 1 in peripheral blood mononuclear cells and plasma. J Clin Microbiol. 2004;42:3670–4. doi: 10.1128/JCM.42.8.3670-3674.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez CS, Stratov I, De Rose R, Walsh K, Dale CJ, Smith MZ, Agy MB, Hu SL, Krebs K, Watkins DI, O’Connor DH, Davenport MP, Kent SJ. Rapid viral escape at an immunodominant simian-human immunodeficiency virus cytotoxic T-lymphocyte epitope exacts a dramatic fitness cost. J Virol. 2005;79:5721–31. doi: 10.1128/JVI.79.9.5721-5731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Lerma JG, MacInnes H, Bennett D, Weinstock H, Heneine W. Transmitted human immunodeficiency virus type 1 carrying the D67N or K219Q/E mutation evolves rapidly to zidovudine resistance in vitro and shows a high replicative fitness in the presence of zidovudine. J Virol. 2004;78:7545–52. doi: 10.1128/JVI.78.14.7545-7552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner W, McNerney GP, Chen P, Dale BM, Gordon RE, Chuang FY, Li XD, Asmuth DM, Huser T, Chen BK. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science. 2009;323:1743–7. doi: 10.1126/science.1167525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval CE, Dykes C, Wang J, Demeter LM. Relative replication fitness of efavirenz-resistant mutants of HIV-1: correlation with frequency during clinical therapy and evidence of compensation for the reduced fitness of K103N + L100I by the nucleoside resistance mutation L74V. Virology. 2006;353:184–92. doi: 10.1016/j.virol.2006.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde MS, Troyer RM, Syed AR, Bulime S, Demers K, Bajunirwe F, Arts EJ. Sensitive oligonucleotide ligation assay for low-level detection of nevirapine resistance mutations in human immunodeficiency virus type 1 quasispecies. J Clin Microbiol. 2007;45:2604–15. doi: 10.1128/JCM.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, McNevin J, Cao J, Zhao H, Genowati I, Wong K, McLaughlin S, McSweyn M, Diem K, Stevens C, Maenza J, He H, Nickle DC, Shriner D, Collier AC, Corey L, McElrath MJ, Mullins JI. Selection on the human immunodeficiency virus type 1 proteome following primary infection. J Virol. 2006;80:9519–29. doi: 10.1128/JVI.00575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, McNevin J, Zhao H, Tebit DM, Troyer RM, McSweyn M, Ghosh AK, Shriner D, Arts EJ, McElrath MJ, Mullins JI. Evolution of human immunodeficiency virus type 1 cytotoxic T-lymphocyte epitopes: fitness-balanced escape. J Virol. 2007;81:12179–88. doi: 10.1128/JVI.01277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Dykes C, Wu T, Huang Y, Demeter L, Wu H. vFitness: a web-based computing tool for improving estimation of in vitro HIV-1 fitness experiments. BMC Bioinformatics. 2010;11:261. doi: 10.1186/1471-2105-11-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammano F, Trouplin V, Zennou V, Clavel F. Retracing the evolutionary pathways of human immunodeficiency virus type 1 resistance to protease inhibitors: virus fitness in the absence and in the presence of drug. J Virol. 2000;74:8524–31. doi: 10.1128/jvi.74.18.8524-8531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Picado J, Martinez MA. HIV-1 reverse transcriptase inhibitor resistance mutations and fitness: a view from the clinic and ex vivo. Virus Res. 2008;134:104–23. doi: 10.1016/j.virusres.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol. 2006;80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Picado J, Savara AV, Sutton L, D’Aquila RT. Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1. J Virol. 1999;73:3744–52. doi: 10.1128/jvi.73.5.3744-3752.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure J, van’t Wout AB, Tran T, Mittler JE. Granulocyte-monocyte colony-stimulating factor upregulates HIV-1 replication in monocyte-derived macrophages cultured at low density. J Acquir Immune Defic Syndr. 2007;44:254–61. doi: 10.1097/QAI.0b013e318030f5c5. [DOI] [PubMed] [Google Scholar]
- Nijhuis M, van Maarseveen NM, Boucher CA. HIV protease resistance and viral fitness. Curr Opin HIV AIDS. 2007;2:108–15. doi: 10.1097/COH.0b013e32801682f6. [DOI] [PubMed] [Google Scholar]
- Petropoulos CJ, Parkin NT, Limoli KL, Lie YS, Wrin T, Huang W, Tian H, Smith D, Winslow GA, Capon DJ, Whitcomb JM. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2000;44:920–8. doi: 10.1128/aac.44.4.920-928.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones-Mateu ME, Ball SC, Marozsan AJ, Torre VS, Albright JL, Vanham G, van Der Groen G, Colebunders RL, Arts EJ. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J Virol. 2000;74:9222–33. doi: 10.1128/jvi.74.19.9222-9233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Amer J Hyg. 1938;27:493–497. [Google Scholar]
- Resch W, Ziermann R, Parkin N, Gamarnik A, Swanstrom R. Nelfinavir-resistant, amprenavir-hypersusceptible strains of human immunodeficiency virus type 1 carrying an N88S mutation in protease have reduced infectivity, reduced replication capacity, and reduced fitness and process the Gag polyprotein precursor aberrantly. J Virol. 2002;76:8659–66. doi: 10.1128/JVI.76.17.8659-8666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland M, Nickle DC, Mullins JI. HIV-1 Group M Conserved Elements Vaccine. PLoS Pathog. 2007;3:e157. doi: 10.1371/journal.ppat.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma PL, Crumpacker CS. Attenuated replication of human immunodeficiency virus type 1 with a didanosine-selected reverse transcriptase mutation. J Virol. 1997;71:8846–51. doi: 10.1128/jvi.71.11.8846-8851.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stekler J, Collier AC. Primary HIV Infection. Curr HIV/AIDS Rep. 2004;1:68–73. doi: 10.1007/s11904-004-0010-2. [DOI] [PubMed] [Google Scholar]
- Troyer RM, Lalonde MS, Fraundorf E, Demers KR, Kyeyune F, Mugyenyi P, Syed A, Whalen CC, Bajunirwe F, Arts EJ. A radiolabeled oligonucleotide ligation assay demonstrates the high frequency of nevirapine resistance mutations in HIV type 1 quasispecies of NVP-treated and untreated mother-infant pairs from Uganda. AIDS Res Hum Retroviruses. 2008;24:235–50. doi: 10.1089/aid.2007.0138. [DOI] [PubMed] [Google Scholar]
- Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, Abraha A, Tebit DM, Zhao H, Avila S, Lobritz MA, McElrath MJ, Le Gall S, Mullins JI, Arts EJ. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 2009;5:e1000365. doi: 10.1371/journal.ppat.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Maarseveen NM, Huigen MC, de Jong D, Smits AM, Boucher CA, Nijhuis M. A novel real-time PCR assay to determine relative replication capacity for HIV-1 protease variants and/or reverse transcriptase variants. J Virol Methods. 2006;133:185–94. doi: 10.1016/j.jviromet.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Wang J, Dykes C, Domaoal RA, Koval CE, Bambara RA, Demeter LM. The HIV-1 reverse transcriptase mutants G190S and G190A, which confer resistance to non-nucleoside reverse transcriptase inhibitors, demonstrate reductions in RNase H activity and DNA synthesis from tRNA(Lys, 3) that correlate with reductions in replication efficiency. Virology. 2006;348:462–74. doi: 10.1016/j.virol.2006.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Huang Y, Dykes C, Liu D, Ma J, Perelson AS, Demeter LM. Modeling and estimation of replication fitness of human immunodeficiency virus type 1 in vitro experiments by using a growth competition assay. J Virol. 2006;80:2380–9. doi: 10.1128/JVI.80.5.2380-2389.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]