

Abstract
We carried out a genome-wide association study of serum aspartate aminotransferase (AST) activity in 866 Amish participants of the Heredity and Phenotype Intervention Heart Study and identified significant association of AST activity with a cluster of single nucleotide polymorphisms located on chromosome 10q24.1 (peak association was rs17109512; P=2.80E-14), in the vicinity of GOT1, the gene encoding cytosolic AST (cAST). Sequencing of GOT1 revealed an in-frame deletion of three nucleic acids encoding asparagine at position 389 c.1165_1167delAAC (p.Asn389del) in the gene. Deletion carriers had significantly lower AST activity levels compared with homozygotes for the common allele (mean±s.d.: 10.0±2.8 versus 18.8±5.2 U l−1; P=2.80E-14). Further genotyping of the deletion in other Amish samples (n=1932) identified an additional 20 carriers (minor allele frequency (MAF)=0.0052). The deletion was not detected in 647 outbred Caucasians. Asn at codon 389 is conserved among known mammalian cASTs. In vitro transient transfection of wild-type and mutant cAST indicated that mutant cAST protein was barely detectable in the cells. Furthermore, even after correction for cAST expression, mutant cAST had markedly diminished enzymatic activity. Remarkably, we did not find any association between the deletion and metabolic traits including serum fasting glucose or insulin, fasting and post-meal lipids, inflammatory markers, or sub-clinical markers of cardiovascular disease. In conclusion, we discovered a rare in-frame deletion in GOT1 gene, which inactivates cAST enzyme in the Old Order Amish. This finding will help us to understand structure and function of the enzyme and would be useful for predicting serum AST levels.
Keywords: Amish, AST, gene, GOT1, mutation
INTRODUCTION
Aspartate aminotransferase (AST; EC 2.6.1.1) catalyzes the reversible conversion between aspartate/α-ketoglutarate and oxaloacetate/glutamate, and has an important role in amino acid metabolism and in the urea and tricarboxylic acid cycles.1 AST is conserved from all prokaryotic to eukaryotic organisms and is expressed in many types of cells/tissues including liver, red blood cells and cardiac/skeletal muscles. Two highly similar isoenzymes are present in mammals, one located in cytoplasm (cAST), and the other in mitochondria (mAST). In serum, the enzyme activity is largely of cytosolic origin.2,3 Serum AST activity elevation is considered a surrogate marker of liver, heart and muscle injury.
The locus for soluble AST (GOT1) is on chromosome 10q24, whereas the mitochondrial form of AST (GOT2) is on 16q12. Twin studies4,5 suggest that serum AST activity is influenced by both genetic and environmental factors. However, to date, specific gene variants that influence serum AST activity and/or metabolic or other phenotypes that may be associated with AST activity have yet to be identified. We sought to determine the genetic determinants of serum AST levels by carrying out a genome-wide association study (GWAS) of serum AST in 866 Amish subjects from the Heredity and Phenotype Intervention (HAPI) Heart Study.
MATERIALS AND METHODS
Population
The HAPI Heart Study was initiated in 2003 to identify genes that interact with short-term environmental exposures to modify risk factors for cardiovascular disease. Details of recruitment and phenotyping have been reported previously.6 Briefly, subjects were members of the Old Order Amish (OOA) community aged 20 years or older and considered to be relatively healthy, based on exclusion criteria of severe hypertension (blood pressure greater than 180/105 mm Hg), malignancy, and kidney, liver or untreated thyroid disease.
HAPI Heart Study participants underwent measurement of cardiovascular disease risk factors and questioning about their prior history of cardiovascular disease. Physical examinations, including anthropometry were conducted at the Amish Research Clinic in Strasburg, PA, USA. Blood samples were obtained following an overnight fast. Those taking lipid-lowering medications at enrollment discontinued usage 7 days before examination. Serum lipid levels and high-density lipoprotein cholesterol were assayed by Quest Diagnostics (Horsham, PA, USA). All subjects had triglyceride levels less than 400 mg dl−1, and low-density lipoprotein cholesterol levels were calculated according to the Friedewald formula.7 Hypertension was defined as a systolic blood pressure of 140 mm Hg or greater, a diastolic blood pressure of 90 mm Hg or greater, or use of prescription blood pressure-lowering medications. Diabetes was defined as a fasting glucose of 126 mg dl−1 or greater, or current use of prescription diabetes medications. Smoking habits were recorded; current smoking status included use of cigarettes, pipes or cigars. Serum AST and alanine aminotransferase were assayed using spectrophotometry by Quest Diagnostics.
Additional Amish subjects who had DNA samples stored in our Biobank (n=1932) were screened for the mutation identified. Six hundred and forty seven non-Amish Caucasians were screened, who were participants in the type 2 diabetes case–control study conducted by Joslin Diabetes Center (Boston, MA, USA) and University of Maryland (Baltimore, MD, USA).
The study protocol was approved by the Institutional Review Board at the University of Maryland, and other participating institutions. Informed consent was obtained from each of the study participants.
Genotyping
Genotyping was performed in HAPI Heart Study participants with the Affymetrix GeneChip Human Mapping 500 K Array set (Affymetrix, Santa Clara, CA, USA), including a total of 500 568 single nucleotide polymorphisms (SNPs). The Affymetrix GeneChip Genotyping Analysis Software and BRLMM genotype-calling algorithm (Affymetrix) were used to generate SNP data files. Mean sample call rate in 866 samples was 98.3% after filtered by relationship check and call rate (>95%). A total of 373 825 SNPs that passed quality control (SNP call rate >95%, minor allele frequency (MAF) >0) and Hardy–Weinberg Equilibrium checks (at P>0.0001) were retained for analysis.
We confirmed the deletion c.1165_1167delAAC (p.Asn389del) by dideoxy sequence analysis (ABI 3730; Applied Biosystems, Foster City, CA, USA) in the exons of GOT1 gene, followed by genotyping using Custom TaqMan SNP Genotyping Assays (Applied Biosystems) in HAPI Heart Study participants, 1932 additional Amish DNA samples from our Biobank, and 647 non-Amish Caucasians.
Functional studies of recombinant cAST
Construction of expression vector of wild-type and mutant cAST
Full-length human cAST cDNA plasmid (IMAGE clone ID: 44420) was purchased from Open Biosystems (Huntsville, AL, USA). The plasmid served as template for PCR amplification of the full cAST protein-coding region with restriction enzyme HindIII-anchored upstream primer p2254 5′-cgatgacgacaagcttcatatgg-cacctccgtcagtctttgcc-3′ and downstream XbaI-anchored downstream primer p2255 5′-cccgggatcctctagtcactggattttggtgactgc-3′ with high-fidelity Phusion polymerase (New England Biolabs, Ipswich, MA, USA). The resultant PCR product was subcloned into pFlag-CMV2 precut with HindIII and XbaI by In-Fusion cloning (Clontech, Mountain View, CA, USA) to make wild-type expression vector pFlag-CMV2-cASTwt. For construction of cASTmut (c.1165_1167delAAC), three fragment In-Fusion cloning was carried out with mutant 5′-end fragment (PCR amplified with upstream primer p2254 and downstream primer p2323 5′-ccactcacgattcgaccacttggcagcag-3′ on wild-type cAST cDNA template), mutant 3′-end fragment (PCR amplified with upstream primer p2313 5′-ggtcgaatcgtgagtggcttaaccaccaaa-3′ and downstream primer p2255 on wtAST template), and fragment of HindIII and Xba-precut pFlag-CMV2, to make the mutant cAST expression vector pFlag-CMV2-cASTmut. Both cASTwt and cASTmut DNA inserts were verified by sequence analysis.
Expression and enzymatic assay of recombinant cASTwt and cASTmut proteins
HEK293 cells were grown in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and antibiotics ampicillin and streptomycin. The cells were plated into six-well plates. At ~95% confluency, the cells were transfected with empty vector pFlag-CMV2, pFlag-CMV2-cASTwt and/or pFlag-CMV2-cASTmut with LipoD293 (SignaGene, Gaithersburg, MD, USA) according to the manufacturer’s instruction. At 48 h post transfection, the cells were collected in 10 mM Tris-HCl buffer (pH 7.7) and lysed by sonication on ice. The lysates were centrifuged and supernatants were collected for protein quantification using the Bradford method, AST activity assay and western blot analysis.
AST activity was determined by using the AST kit (Catachem, Oxford, CT, USA) according to the manufacturer’s instruction. Briefly, 5 μl of cell lysate was incubated with a 0.2 ml mixture of reagent A and B containing L-aspartate, NADH, malate dehydrogenase and 2-oxoglutarate at 25 °C. Absorbance at 340 nm was recorded for 5 min at every 30 s interval after addition of the protein fraction. The slope of absorbance decrease is proportional to AST activity. Final AST activities were corrected by protein concentration of cell lysates. One unit of AST activity was defined as the amount of enzyme, which catalyzes the formation of 1 μmol l−1 of NAD per minute under conditions of the assay at 25 °C.
For western analysis, 50 μg of total proteins from reduced cell lysate were separated by electrophoresis on 7.5% polyacrylamide gels. Following electrophoresis, proteins were transferred onto polyvinylidene fluoride membranes and bound proteins were probed with primary anti-Flag antibody (Sigma, St Louis, MO, USA) or GAPDH (Millipore, Billerica, MA, USA), and developed with an alkaline phosphatase-conjugated second antibody.
Statistical analysis
Association analyses of quantitative traits were performed under a variance component model that assesses the effect of genotype, as an additive effect, on the quantitative trait, whereas simultaneously estimating the effects of age, age,2 sex, and a polygenic component to account for phenotypic correlation due to relatedness. The polygenic component was modeled using the relationship matrix derived from the complete pedigree structure as all the subjects are related. Association analysis using the complete pedigree structure was carried out using mixed model analysis for pedigree (MMAP) software developed by one of our colleague (JR O’Connell). AST, alanine aminotransferase, fasting glucose and fasting insulin were naturally log-transformed. Student’s t-test was used to examine differences between cASTwt and cASTmut activities.
RESULTS
We carried out a GWAS of serum AST activity levels in 866 Amish subjects from the HAPI Heart Study. Characteristics of study participants are summarized in Table 1. In the GWAS, the strongest SNP associations with serum AST levels were observed for a group of SNPs located on chromosome 10q24.1. The peak association was with rs17109512 (P-value=2.80E-14), in the vicinity of GOT1 (~1 Mb downstream), the gene encoding cAST (Figure 1). The frequency of the associated SNP is rare (MAF=0.006), with only 10 heterozygotes for rs17109512 found in the HAPI Heart Study. The 10 heterozygotes had significantly lower AST levels compared with the 856 homozygotes of the wide-type allele (mean±s.d. of AST: 10.0±2.8 versus 18.8±5.2 U l−1, P=2.80E-14).
Table 1.
Clinical characteristics of the HAPI Heart Study participants
| Men | Women | |
|---|---|---|
| n | 460 | 406 |
| Age, mean (s.d.), year | 42.2 (13.6) | 45.5 (14.2) |
| BMI, mean (s.d.), kg m−2 | 25.6 (5.2) | 27.7 (5.4) |
| Systolic blood pressure, mean (s.d.), mm Hg | 121.2 (12.5) | 121.0 (16.8) |
| Diastolic blood pressure, mean (s.d.), mm Hg | 77.6 (9.0) | 75.6 (8.4) |
| Total cholesterol, mean (s.d.), mmol l−1 | 5.25 (1.14) | 5.59 (1.27) |
| LDL-cholesterol, mean (s.d.), mmol l−1 | 3.54 (1.06) | 3.65 (1.19) |
| HDL-cholesterol, mean (s.d.), mmol l−1 | 1.37 (0.34) | 1.53 (0.39) |
| Triglyceride, median (s.d.), mmol l−1 | 0.72 (0.42) | 0.84 (0.51) |
| AST, mean (s.d.), U l−1 | 19.7 (5.1) | 17.6 (5.7) |
| ALT, mean (s.d.), U l−1 | 20.9 (7.5) | 16.9 (6.9) |
| Hypertension, n (%) | 52 (11.3) | 66 (16.2) |
| Diabetes, n (%) | 3 (0.7) | 2 (0.5) |
| Current smoker, n (%) | 91 (19.8) | 0 (0) |
| Lipid medication, n (%) | 5 (1.1) | 4 (1.0) |
Abbreviations: BMI, body mass index; HAPI, Heredity and Phenotype Intervention; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Figure 1.
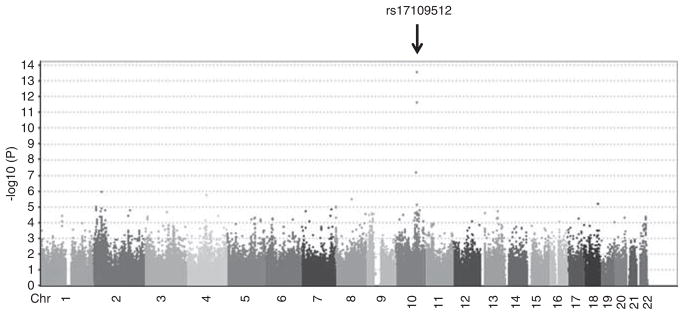
Genome-wide association analysis plot for serum AST levels in 866 Old Order Amish subjects. Arrow indicates that SNP rs17109512 on chromosome 10q24.1 is highly associated with serum AST activity (P=2.80E-14). A full color version of this figure is available at the Journal of Human Genetics journal online.
Given the low frequency of the associated SNP, its proximity to GOT1, and the very large effect size of the association, we hypothesized that rs17109512 might tag a functional SNP, most likely a mutation changing the protein sequence of cAST, in GOT1. We subsequently sequenced the gene and identified an in-frame deletion of three nucleic acids encoding asparagine at position 389 in GOT1 gene (Figure 2a). All of 10 heterozygotes for rs17109512 were heterozygotes for c.1165_1167delAAC. The deletion c.1165_1167delAAC (p.Asn389del) was in complete linkage with rs17109512. By constructing the pedigree, all the deletion carriers can be traced back to one most likely founder who was born in mid-18th century, suggesting a founder effect. Genotyping of 647 non-Amish Caucasians from Baltimore area did not identify carriers of this deletion.
Figure 2.

Detection of GOT1 (c.1165_1167delAAC) mutation. (a) Sequencing identified an in-frame deletion of three nucleic acids encoding asparagine at position 389 in GOT1 gene. (b) Asparagine (N, indicated by arrow) is conserved in cAST protein sequences among human, chimpanzee, mouse, xenopus, zebrafish and C. elegans. Peptide sequences are aligned using ClustalW212 with fully conserved amino acid marked with asterisk. Partial alignment of cAST is shown and amino acid positions are numbered to the left.
Glutamine at amino acid at 389 is conserved among known cAST of vertebrate species, including the human, chimpanzee, mouse and zebrafish (Figure 2b). Further genotyping of deletion c.1165_1167delAAC (p.Asn389del) in an additional 1,932 Amish samples identified 20 more carriers (MAF=0.0052), among which 14 had phenotype measurements. No homozygotes for the deletion were found. Phenotype comparison between the 24 (10 from the HAPI Heart Study plus 14 from the additional screening) c.1165_1167delAAC (p.Asn389del) carriers and 856 non-carriers (from the HAPI Heart Study) is shown in Table 2. The deletion carriers had serum AST activity levels that were approximately one-half that of normal homozygotes, suggesting that the deletion resulted in a complete loss of enzymatic function. Despite this large difference in AST activity, we did not find any association between the mutation and metabolic traits, including serum fasting glucose or insulin, fasting lipids (Table 2). Neither did we find any association with post-meal lipids, inflammatory markers or sub-clinical markers of cardiovascular disease (data not shown).
Table 2.
Clinical characteristics of the GOT1 c.1165_1167delAAC carriers and non-carriers
| Carrier | Non-carrier | P-value | |
|---|---|---|---|
| n | 24 | 856 | |
| Age, year | 50.6 (16.8) | 43.7 (14.0) | 0.03 |
| BMI, kg m−2 | 27.1 (4.8) | 26.6 (4.5) | 0.13 |
| Waist circumference, cm | 91.9 (11.2) | 87.3 (11.0) | 0.17 |
| AST, U l−1 | 10.0 (2.8) | 18.8 (5.2) | 2.80E-14 |
| ALT, U l−1 | 17.7 (3.9) | 19.1 (7.5) | 0.77 |
| Systolic blood pressure, mm Hg | 124.6 (17.6) | 121.0 (14.6) | 0.65 |
| Diastolic blood pressure, mm Hg | 78.2 (7.6) | 76.6 (8.8) | 0.25 |
| Total cholesterol, mmol l−1 | 5.51 (1.01) | 5.40 (1.22) | 0.87 |
| LDL-cholesterol, mmol l−1 | 3.75(0.88) | 3.59 (1.11) | 0.82 |
| HDL-cholesterol, mmol l−1 | 1.29 (0.34) | 1.45 (0.39) | 0.32 |
| Triglyceride, mmol l−1 | 1.05 (0.59) | 0.77 (0.47) | 0.19 |
| Fasting glucose, mmol l−1 | 5.43 (1.59) | 4.80 (0.72) | 0.15 |
| Fasting insulin, mmol l−1 | 0.67 (0.17) | 0.63 (0.31) | 0.22 |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; HAPI, Heredity and Phenotype Intervention; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Mean (s.d.), Adjusted for age, age-square, sex, and family structure for phenotypes other than age.
AST and ALT measurements were based on 10 GOT1 deletion carriers from HAPI Heart study. The rest of the phenotype measurements were based on 10 GOT1 deletion carriers from HAPI Heart Study and 14 additional identified GOT1 deletion carriers in Amish; 856 non-carriers were from HAPI Heart Study.
P-values for fasting glucose, fasting insulin and waist circumference were based on non-diabetics.
To further examine the functional consequences of p.Asn389del, we conducted in vitro functional studies by expressing the recombinant wild-type and mutant cAST (p.Asn389del) in the mammalian kidney cell line HEK293 and measuring AST activity. As shown in Figure 3a, the basal activity of the cells transfected with empty vector (lane 1) was barely detectable. AST activity levels were increased by >100 fold in a dose-dependent manner when the cells were expressing wild-type cAST (lanes 2 and 3). By contrast, lysates from cells transfected with mutant cAST (lanes 5 and 6) had almost no AST activity, suggesting that the mutant cAST is enzymatically inactive. Western analysis was conducted to determine the levels of recombinant protein expression in the cells. As shown in Figure 3b, mutant cAST only was barely detectable, compared with the wild type (lanes 5 and 6 versus 2 and 3). Interestingly, the activities from the cells transfected with wild-type and mutant cAST at a 1:1 ratio were less than half of that of wild-type AST (lane 4 versus 3; P<0.001), suggesting a dominant negative effect. Western analysis revealed that less mutant cAST protein was expressed after correction for GAPDH, suggesting a decreased expression, probably due to decreased RNA stability and/or increased protein degradation (Figure 3b). However, even after correcting for AST protein, mutant cAST had markedly decreased activity relative to wild-type cAST (lane 2 versus 5, P<0.001).
Figure 3.

Functional study of recombinant mutant and wild-type cAST proteins. (a) AST activities were measured in lysates of HEK293 cells transfected with empty vector (Vector), wild-type (cASTwt), or p.Asn389del mutant (cASTmut) at the indicated amount in a six-well plate dish. Data are expressed as mean±s.e. (n=4). (b) Representative western blot of flag-tagged wild-type and mutant cAST protein levels in HEK293 cells (same lane order as (a)) detected with anti-flag antibody. Anti-GAPDH was used as protein loading control.
DISCUSSION
Serum AST activity levels are determined by both genetic and environmental factors.4,5 Variation in serum AST activity has a moderate heritable basis (0.32–0.40) estimated from twin studies.4,5 A GWAS of AST protein levels, as one of the group of 42 proteins measured in 1200 in CHIANTI study participants, didn’t find any cis-effect associations with SNPs near or in GOT1 or trans-effect associations with SNPs elsewhere in the genome.8 In another GWAS, no SNP was associated with serum levels of AST at or exceeding genome-wide significance.9 Through a GWAS of serum AST activity in the OOA, we identified a rare in-frame 3 bp deletion in GOT1 that was strongly associated with low serum AST activity in the OOA. Subjects heterozygous for the GOT1 c.1165_1167delAAC had serum AST activity levels that were about one-half that of wild-type homozygotes. Consistent with this observation, functional studies found that AST activity of the mutant AST recombinant protein was totally abolished. This deletion was not present in 647 outbred Caucasians, consistent with negative GWAS findings of others. Also consistent with negative GWAS findings of others, our GWAS did not detect any other regions with genome-wide significant association to AST activity levels.
Elevated serum AST activity is a clinical marker for damage to hepatocytes, myocardial cells or skeletal muscle. By contrast, decreased serum AST and its clinical significance are less known. This loss-of-function mutation in AST provides a unique opportunity to determine if decreased AST is associated with a clinically significant phenotype. Although AST has a pivotal role in intermediary metabolism, deficiency might be expected to lead to abnormalities in glucose and/or lipid metabolism. Interestingly, we did not find any significant differences in a wide variety of metabolic traits in GOT1 c.1165_1167delAAC carriers compared with wild-type homozygotes. These findings suggest that in humans, AST is not a rate-limiting step in intermediary metabolism, and that a single functional allele is sufficient for normal metabolic function. Nevertheless, a total loss of this enzyme could be lethal, as we did not find any of the mutant homozygotes. Of possible relevance, fasting glucose levels tended to be higher in deletion carriers than that in non-carriers (P=0.15, after adjusting for age, sex and family structure). Recruitment and phenotyping of additional c.1165_1167delAAC carriers will improve the statistical power and help us understand this potential relationship more clearly.
Rare mutations or genetic diseases have been reported with higher frequency in founder populations, including the OOA. Through genetic drift, rare mutations may increase in frequency in founder populations. Advances in genetic technology have enabled the identification of many of these rare mutations and has provided the opportunity to glean insights into mechanisms underlying both rare and common phenotypes, which would otherwise be difficult in the general population.10,11 In conclusion, we discovered a rare inactivation mutation in GOT1 encoding cytosolic AST in the OOA. As AST is a widely used liver function marker, these mutant carriers will have disguisedly lower or near-normal AST levels in pathogenic liver conditions. The discovery of the GOT1 mutation will help to explain and alert physician about lower AST levels under actual liver injury in those carriers. It will also be interesting and clinically and diagnostically significant to examine if such or similar mutations exist in other populations.
Acknowledgments
The study was supported by grants of Maryland Clinical Nutrition Research Unit (P30DK072488), the Baltimore Diabetes Research and Training Center (P60DK079637) from the National Institutes of Health, and AHA Scientist Development Grant 0830146N. We gratefully acknowledge our Amish liaisons and field workers and the extraordinary cooperation and support of the Amish community without which these studies would not have been possible.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Rosenthal MD, Glew RH. Medical Biochemistry: Human Metabolism in Health and Disease. 1. Wiley; NJ, USA: 2009. [Google Scholar]
- 2.Rej R. A spartate aminotransferase activity and isoenzyme proportions in human liver tissues. Clin Chem. 1978;24:197–199. [PubMed] [Google Scholar]
- 3.Rej R. Measurement of aspartate aminotransferase activity: effects of oxamate. Clin Chem. 1979;25:555–559. [PubMed] [Google Scholar]
- 4.Rahmioglu N, Andrew T, Cherkas L, Surdulescu G, Swaminathan R, Spector T, et al. Epidemiology and genetic epidemiology of the liver function test proteins. PLoS One. 2009;4:e4435. doi: 10.1371/journal.pone.0004435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitfield JB, Zhu G, Nestler JE, Heath AC, Martin NG. Genetic covariation between serum gamma-glutamyltransferase activity and cardiovascular risk factors. Clin Chem. 2002;48:1426–1431. [PubMed] [Google Scholar]
- 6.Mitchell BD, McArdle PF, Shen H, Rampersaud E, Pollin TI, Bielak LF, et al. The genetic response to short-term interventions affecting cardiovascular function: Rationale and design of the heredity and phenotype intervention (HAPI) heart study. Am Heart J. 2008;155:823–828. doi: 10.1016/j.ahj.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 8.Melzer D, Perry JR, Hernandez D, Corsi AM, Stevens K, Rafferty I, et al. genome-wide association study identifies protein quantitative trait loci (pQTLs) PLoS Genet. 2008;4:e1000072. doi: 10.1371/journal.pgen.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet. 2008;83:520–528. doi: 10.1016/j.ajhg.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–1705. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen H, Damcott CM, Rampersaud E, Pollin TI, O’Connell JR, Horenstein RB, et al. Familial defective apolipoprotein B-100 and increased low-density lipoprotein cholesterol and coronary artery calcification in the old order amish. Arch Intern Med. 2010;170:1850–1855. doi: 10.1001/archinternmed.2010.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]