

Abstract
Ovarian cancer (OC) is the sixth most common cancer and the seventh cause of death from cancer in women. The etiology and the ovarian carcinogenesis still need clarification although ovulation may be determinant due to its carcinogenic role in ovarian surface epithelium. The link between ovarian carcinogenesis and DNA repair is well established and it became clear that alterations in DNA damage response may affect the risk to develop OC. Polymorphisms are variations in the DNA sequence that exist in normal individuals of a population and are capable to change, among other mechanisms, the balance between DNA damage and cellular response. Consequently, genetic variability of the host has a great role in the development, progression and consequent prognosis of the oncologic patient as well as in treatment response. Standard treatment for OC patients is based on cytoreductive surgery, followed by chemotherapy with a platinum agent and a taxane. Although 80% of the patients respond to the first-line therapy, the development of resistance is common although the mechanisms underlying therapy failure remain mostly unknown. Because of their role in oncology, enzymes involved in the DNA repair pathways, like DNA Ligase IV (LIG4), became attractive study targets. It has been reported that variations in LIG4 activity can lead to a hyper-sensitivity to DNA damage, deregulation of repair and apoptosis mechanisms, affecting the susceptibility to cancer development and therapy response. To overcome resistance mechanisms, several investigations have been made and the strategy to target crucial molecular pathways, such as DNA repair, became one of the important areas in clinical oncology. This review aims to elucidate the link between DNA repair and OC, namely which concerns the role of LIG4 enzyme, and how genetic polymorphisms in LIG4 gene can modulate the activity of the enzyme and affect the ovarian carcinogenesis and treatment response. Moreover, we try to understand how LIG4 inhibition can be a potential contributor for the development of new cancer treatment strategies.
Keywords: Ovarian Cancer, DNA Repair, DNA ligase IV, Polymorphisms, Susceptibility, Treatment response
INTRODUCTION
Ovarian cancer (OC) is the third most common gynecological cancer among women worldwide, with an estimate 225 000 new cases and 140 000 deaths due to this disease each year[1]. In Europe, OC is the fifth most incident cancer in women but the main cause of death among the gynecological tumors[2].
OC presents itself as a high heterogeneous disease, which may develop from three different cell types: epithelial cells, sex cord-stromal cells or germ cells. In spite of the fact that the ovarian epithelial layer only represents a small percentage of all ovarian cell types, epithelial ovarian carcinoma (EOC) comprises 85% to 90% of all malignant ovarian tumors in adult women[3-5].
One of the most interesting aspects about EOC is the fact that while the transformation processes occur, the ovarian epithelium becomes more differentiated with the capability to undergo metaplasia into Müllerian epithelium. This aberrant transformation occurs in the majority of EOC and allows its histological classification into four main sub-types: serous, mucinous, endometrioid and clear cell tumors according to their histological and secretory resemblance with fallopian tube, endometrium, endocervix or vagina, respectively[6-8].
EOC incidence is age related and is a characteristic of postmenopausal women. The majority of cases happen in women after age 40, with a median age of diagnosis of 63 years[4,9]. By the lack of adequate experimental models to the study of this neoplasm, the etiology and the ovarian carcinogenesis still need clarification although some reproductive and hormonal events can be determinant. In this way, there have been made some possible hypothesis, which based on epidemiological and biological observations, pretend to explain susceptibility to OC[8] (Table 1). However, one of the more important risk factors established to OC is family history. About 5% to 10% of all OC are attributed to inherited mutations in high penetrance genes associated with hereditary breast and OC, as in BRCA1 (3%-6%) and BRCA2 (1%-3%), and with Lynch Syndrome, generally attributed to MLH1 and MSH2 (1%-2%) gene mutations[10].
Table 1.
Hypothesis to epithelial ovarian cancer development
| Hypothesis | Biological mechanism proposed | Epidemiological evidence |
| Incessant ovulation[24] | Repetitive ovulation and quickly cellular proliferation in post-ovulation repair creates a propitious environment to carcinogenesis initiation by genetic alteration accumulation as well as inclusion cysts development | Events that suppress ovulation such as pregnancy, lactation and oral contraceptive use are protective factors |
| Ovulation inhibition results in gonadotropin and oxidative stress levels reduction, deceleration of ovarian follicle depletion and to a diminished inclusion cysts development in ovarian epithelium | ||
| Gonadotropins[91] | Excessive stimulation of ovarian epithelium by FSH and LH conducts to downstream genes activation as well as to stimulation of hormonal production by the ovary (as estrogen) in order to enhance cellular proliferation and consequently to malignant transformation and angiogenesis | Oral contraceptive use and pregnancy are protective. Hyper-gonadotropic conditions are common in infertile, in polycystic ovarian syndrome and in post-menopausal women |
| The formation of a protective progestagenic hormonal milieu can stimulate apoptosis in genetically damaged ovarian epithelial cells, preventing tumor development | ||
| Hormonal stimulation[92] | High androgen levels are harmful while an increase in progesterone levels is benefic | Protective effect due to multiparity and oral contraceptive use. Harmful effect is associated with higher androgen levels as in polycystic ovarian syndrome women |
| Inflammation[25] | Ovulation is accomplished by an inflammatory response: redox potential alteration, cellular infiltration, cytokine release that can introduce DNA damage in epithelial cells involved in ovary rupture/repair | Inflammatory gynecological diseases, as endometriosis, can enhance EOC risk. Non-steroid anti-inflammatory drugs can be a protective factor |
FSH: Follicle-stimulating hormone; LH: Luteinizing hormone; EOC: Epithelial ovarian cancer.
EOCs are themselves a heterogeneous group of tumors. Shih et al[11] and Kurman et al[12] proposed an EOC sub-classification according to its clinic behavior, tumor progression, morphologic and genetic features. In this way, EOCs are divided in two main groups named Type I (25% of all cases), considered as lower grade, and Type II (75% of all cases), considered as higher grade. Type I tumors include all major sub-types but exhibit low-grade nuclear and architectural features, slow growth and can be linked to well-defined benign ovarian precursor lesions. These tumors are characterized by a relative genetic stability, although mutations in KRAS, BRAF, PTEN and β-Catenin genes are common, and frequently associated with a worse therapy response. On the other hand, Type II ovarian tumors are infrequently associated with benign or borderline ovarian precursor lesions, arising in an aggressive and spontaneous manner, being usually sensitive to chemotherapy. They are comprised almost exclusively of high-grade serous carcinomas (90%) but also include two less common subtypes (mixed epithelial and undifferentiated carcinomas) and those associated with BRCA1 and BRCA2 hereditary tumors. Type II ovarian tumors are characterized by genetic instability being usual mutations in TP53 gene (50%-80%) and also amplification and overexpression of HER2/neu (10%-20%) and AKT2 (12%-18%) oncogenes[11,12]. This sub-classification of EOC can be the result of two divergent pathways in ovarian carcinogenesis although more studies need to be done to confirm this suggestion[13].
As mentioned above, OC is considered the most lethal gynecological cancer[2]. This high mortality is due, essentially, to late diagnosis since, in 75% of OC cases, it is only made in an advanced disease stage, when the tumor is no longer confined to ovary[14]. In spite of diagnosis and prevention strategies improvement, some barriers to early OC detection exist and are due to its low incidence, hidden location of ovaries, not existence of well defined pre-invasive lesions and for being usually asymptomatic[15].
Despite the high degree of phenotypic and genotypic variability between the sub-types of EOC, all patients are treated identically upon diagnosis[13]. Standard treatment for OC patients is based on cytoreductive surgery, followed by chemotherapy with a platinum agent (carboplatin or cisplatin) and a taxane (paclitaxel or docetaxel). Although 80% of the patients respond to the first-line therapy, the development of resistance is common and several patients eventually recur with a 5-year survival rate only around 45%[16-18].
Over the past several decades, great advances have been made in surgical techniques and chemotherapy regimens used to treat OC. However, despite the best achievements between clinic and research, these strategies have not yet been shown to have an impact on overall mortality from advanced-stage disease, which 5-year survival rate has improved only 8% in the last 30 years and remain mostly unknown the mechanisms underlying therapy failure[16,18].
Efforts to improve long-term results of first-line therapy through addition of a third cytotoxic agent have not been successful. An improvement in the understanding of OC biology has led to the identification of molecular targets and biological agents that interfere with DNA repair, growth factors, membrane-bound receptors and tumor-associated angiogenesis[18-20]. Emerging data regarding inhibition of vascular endothelial growth factor (VEGF)-mediated angiogenesis and inhibition of poly[adenosine diphosphate (ADP)-ribose] polymerase (PARP)-mediated DNA repair are promising[18,21,22].
OC AND DNA REPAIR
The traditional view of OC asserts that the majority of OC share a common origin within ovarian surface epithelium (OSE). OSE is a monolayer of uncommitted mesothelial cells that cover the exterior surface of the ovary[7]. During the monthly ovulation, the OSE is enzymatically degraded in order to allow the follicular rupture and oocyte release, creating a breach that must be posteriorly repaired (Figure 1)[7,23,24].
Figure 1.

Link between ovulation/ageing and ovarian cancer development. Adapted from Levanon et al[3]. ROS: Reactive oxygen species.
Over the course of a woman’s reproductive life, this process of damage and repair is repeated multiple times and may result in a stepwise accumulation of genomic alterations, as postulated by the incessant ovulation hypothesis (Table 1)[24]. In addition to physical trauma, OSE cells are subjected to ovulation-associated inflammatory cytokines, reactive oxygen species (ROS), and hormones (and its reactive metabolites) that are capable to damage DNA and conduct to a hormonal metabolism imbalance[3,19,23,25].
Ovarian epithelial inclusion cysts may develop as an ovulation result or due to ageing, becoming entrapped within the stroma (Figure 1). Once inside the ovary, epithelial cells lining the inclusion cysts are exposed to an environment of aberrant autocrine/paracrine stimulation by growth factors including hormones, phospholipids and VEGF[7,19,26]. If the epithelial cells harbor unrepaired DNA damage, they may be prime targets for neoplastic transformation[13].
As the link between DNA damage and ovarian carcinogenesis becomes stronger, it will become more important to completely understand the role of DNA damage response (DDR) proteins in OC prevention. Because defects in DNA repair genes involved in double-strand breaks (DSBs) repair, such as BRCA1 and BRCA2, are implicated in familiar OC, overall DNA repair capacity may have an effect on the risk of sporadic OC as well[27].
With the human genome sequencing, the identification of genetic variations and the understanding of how common variations can affect normal cellular processes has become possible[28]. Genetic polymorphisms are naturally occurring sequence variations, about 90% of which are single nucleotide polymorphism (SNP)[29]. SNPs are single base pair positions in genomic DNA at which different alleles exist in normal individuals in some population(s), wherein the least frequent allele has an abundance of 1% or greater[30]. SNPs occur every 100-300 bases along the human genome and several studies suggest that the risk to many complex diseases, like cancer, can be extensively affected by the individual’s SNP profile. Presumably, it will be the combination between the SNP profile and environmental factors that contribute to sporadic cancer development[30-32]. Genetic polymorphisms in DNA repair genes seem to determine the overall DNA repair capacity, which in turn may affect the risk of OC[15].
DDR pathways induce cell cycle arrest in response to DNA damage, in order to maintain genomic stability. In this way, these mechanisms are known to act as tumor suppressors and proteins involved in repair pathways are considered as genome caretakers. DDR pathways are controlled by specific sets of genes and although they are considered as good players in the cancer prevention, they can act as bad players in the treatment response[27,33]. The recognition of DNA damage and the consequent repair mechanism are crucial to the sensibility or resistance of cancer cells to treatment. This means that cells with proper DDR pathways are capable to efficiently repair the damage caused by chemo or radiotherapy, being responsible for the development of resistance in tumor cells[34].
Pharmacogenetics and pharmacogenomics are emerging areas that are essential to the development of personalized medicine, ultimately leading to drug prescription based on patient’s individual genetic and molecular profile[30-32,35]. The aim of these areas is to establish a relationship between the genotype (i.e., polymorphisms or mutations), gene expression profile and phenotype (both in drugs’ pharmacokinetic and pharmacodynamics), interpreted as the variability between individuals concerning the toxicity, effectiveness and therapy outcome[35-39]. Polymorphisms in genes involved in DNA repair could result in variations in efficacy and accuracy of DNA repair enzymes and consequently significantly affect the toxicity, effectiveness and therapy outcome. By their role in therapy response, genetic polymorphisms in these genes can influence the patient’s survival and be useful as prognostic and predictive markers in cancer[40-43].
Moreover, with the development of DDR protein inhibitors for cancer treatment, research on targeting molecular pathways, such as DNA repair, is becoming one of the most important areas in clinical oncology[43-48]. One of the enzymes involved in DDR is DNA Ligase IV (LIG4) enzyme, which is essential to catalyze the DNA phosphodiester bond formation, in the last step of one of the DNA repair mechanisms[49]. In this review, we explore the role of LIG4 in DDR, namely in OC carcinogenesis and treatment, as well as in the potential contribution to the development of new target therapies.
DDR
Along the cell cycle and during the lifetime of a cell, the genome is continuously exposed to a wide variety of agents and processes capable to damage the DNA[50]. Therefore, genetic stability is necessary and is maintained not only by precise replication mechanisms but also by accurate and redundant systems that detect and repair possible DNA lesions. Most of DNA injuries are transitory because after its recognition, a coordinated cellular response takes place in order to interrupt the cell cycle (allowing the repair) or to lead to cell death (if the damage is too serious), maintaining genomic stability[46,51].
There are several DNA repair mechanisms that use different enzymes to repair different kinds of damages[34]. One of the most deleterious DNA lesions is DNA DSBs. DSBs occur when the phosphodiester backbones of both strands are simultaneously broken and close enough to disrupt base pairing, whereas chromatin structure can not keep the ends juxtaposed[50,52]. The result is the release of two DNA ends that can get physically separated from each other, embarrassing the subsequent repair and providing an opportunity to inappropriate recombination[50]. These breaks can arise in all phases of the cell cycle from a wide range of agents and processes: as result of normal cellular metabolism, by the action of ROS, or as result of physiological processes like V(D)J recombination, DNA replication or meiosis. Exogenous factors can include ionizing radiation as well as chemotherapeutic agents[52-54].
In eukaryotic cells, DSBs can be repaired by two main pathways: Homologous Recombination (HR) and Non-Homologous End-Joining (NHEJ)[34,50,52] (Figure 2).
Figure 2.
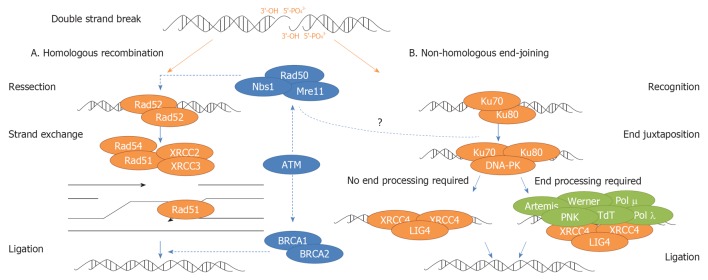
Simplified overview of homologous recombination and non-homologous end-joining. Homologous recombination (HR) pathway starts with break recognition and signaling by a complex containing NBS1, MRE11 and RAD50 (MRE11/RAD50/NBS1 - MRN complex). RAD51 and RAD52 catalyze and facilitate a strand exchange reaction. Assembly of RAD51 is facilitated by different RAD51 paralogs such XRCC2 and XRCC3. MRN complex also promotes activation of ATM, which in turn activates several DNA repair factors as BRCA1/2. HR finishes with DNA synthesis and final ligation. Non-homologous end-joining (NHEJ) pathway starts with the recruitment of Ku heterodimer (Ku70 and Ku80) to DNA ends. Once attached to double-strand breaks, Ku recruits and stimulates the DNA-PKcs, forming the DNA-PK holoenzyme. DNA-PK activates XRCC4-LIG4 complex, which links the broken complementary DNA ends together. If DNA ends are not ready to end joining, it is necessary a previous DNA end processing, which may involve numerous enzymes as Artemis, Werner, DNA Polimerases μ e λ, Polynucleotide kinase (PNK) and Terminal deoxynucleotidyl transferase (TdT), to conclude the NHEJ pathway. The role of MRN complex in NHEJ pathway it is still not clear.
In HR pathway, the break is repaired using the homologous chromosome or sister chromatid as template. This is considered an accurate repair pathway and it is thought that could be particularly important for DSB repair in S/G2 phases of the cell cycle, where replicated sequences are available to serve as repair templates[34,50,55]. In NHEJ pathway, the broken strands are crudely joined together at a site of micro-homology, frequently resulting in small alterations at the site of fusion, being often described as error-prone. Although been able to operate throughout cell cycle, NHEJ is the predominant pathway during G0, G1 and early S phases of the cell cycle[27,34,55-60].
NHEJ and HR proteins are highly conserved across all eukaryotes and ubiquitously expressed in multi-cellular organisms. HR appears to be the predominant DSB repair pathway in yeast although NHEJ is the main pathway in higher organisms such as mammals[50,52]. However, recent evidence suggests that these major repair pathways can cooperate and compete with each other at DSBs to promote efficient repair and genomic integrity[61].
LIGASE IV ENZYME AND ITS ROLE IN ONCOLOGY
DNA ligase enzymes are an evolutionary related protein family, involved in innumerous cellular processes such as DNA replication, genetic recombination and DNA repair. They are nucleotidyltransferase enzymes (NTases) that use an energetic source to catalyze phosphodiester bond formation in a three-step reaction mechanism[49,62,63] (Figure 3).
Figure 3.
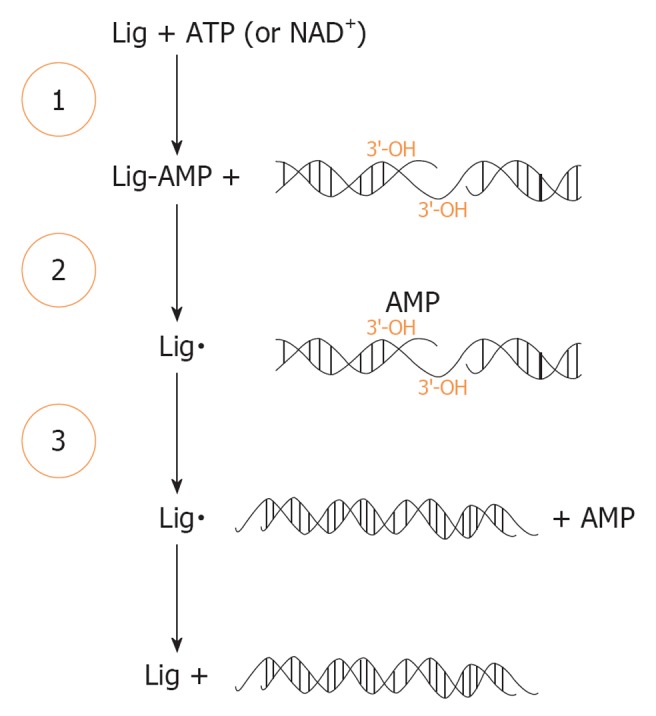
Enzymatic ligation of DNA by DNA ligase. The three-step reaction results in the sequential transfer of AMP (adenosine 5’-monophosphate) to an active-site lysine in Lig enzyme (step 1) then to DNA end (step 2), which results in the formation of a phosphodiester bond and consequently to a ligated DNA product (step 3). Lig: Ligase. Aapted from Ellenberger et al[49].
The ligation reaction has a high energetically yield, in which an adenylate group (adenosine 5’monophosphate - AMP) is sequentially transferred from ATP or NAD+ to a highly conserved lysine residue in the active site of the DNA ligase enzyme (Step 1), with the formation of a covalent enzyme-adenylate intermediate. This first step occurs independently of DNA whereas the subsequent steps involve interaction between the DNA ligase and its DNA substrat. Formerly, the AMP is transferred to the 5’-PO43- DNA end to generate a covalent DNA-adenylate intermediate (Step 2). In the final step, the non-adenylated DNA Ligase catalyzes the formation of a phosphodiester bond, in a reaction that involves a nucleophilic attack by a 3’-OH on the 5’ end of the DNA adenylate and the release of AMP (Step 3)[49,62]. By the high favorable reaction equilibrium, each chemical step makes this reaction sequence effectively irreversible, proving once again the importance of DNA repair[49].
DNA ligases have the capability to change their conformation during the DNA joining reaction in order to accommodate the multiple reactions that catalyze. These enzymes have multiple domains that provide the necessary flexibility to completely encircle their DNA substrates as well as the capacity to open and close around DNA[49].
In the human genome there are three genes that codify four DNA Ligases: LIG1, LIG3 and LIG4, with the DNA Ligases II and III being expressed by alternative splicing of mRNA from LIG3[49,64]. Consistent with the common evolution, all of the eukaryotic enzymes are ATP-dependent and are related in terms of sequence and structure[49,62,63].
LIG4 gene, located in 13q33 chromosome, codify an exclusively nuclear protein, with approximately 100 kDa, which shares homology with the other ligases in N-terminal region but not in the C-terminal region[62,65]. Its catalytic domain (CD) comprises six conserved sequence motifs (I, III, IIIα, IV, V, VI) that define the nucleotidyltransferase family. Motif I includes the lysine residue that is adenylated in the first step of the ligation reaction. The non-CD, which is poorly conserved between the different family members, does not have a known function yet[63,64].
LIG4 enzyme is characterized by a C-terminal extension that includes two tandem copies of the BRCT homology domain, which are found in other DNA repair and checkpoint-associated proteins[65-67]. These motifs are separated by a short linker sequence that contains a conserved binding site presumably necessary to the interaction with XRCC4 in NHEJ pathway[68-70]. XRCC4 is responsible for the stabilization and stimulation of the ligase activity by LIG4, such as adenylation, as well as to protect LIG4 from degradation[68,71]. Furthermore, the stability of LIG4 is also regulated by phosphorylation at a serine residue (Ser650)[49]. Structural studies suggest that in the XRCC4-LIG4 complex, the stoichiometric proportion is one molecule of LIG4 to two molecules of XRCC4[70,72].
Despite the fact of being essential in the DSB repair, the XRCC4-LIG4 complex is DNA-PK dependent and because of that is an exclusive complex of the NHEJ pathway. However, LIG4 appears to function in specialized cells of the immune system where it also completes V(D) J recombination[69].
Under normal conditions, the human genome is replicated and stabilized by highly accurate complex replication and repair machinery. The increased incidence of certain pathologies, like cancer, associated with DNA repair-deficient human syndromes, illustrates the crucial role of these pathways in protection against genomic instability[73]. The LIG4 importance in the maintenance of genomic stability appears to be associated with the fact that mutations in this gene are associated with a rare autosomal syndrome (OMIM 606593) characterized by microcephaly, several immunodeficiency, spontaneous genomic instability and a higher susceptibility to complex diseases such as cancer[50,60,74,75]. Cells from LIG4 patients display increased radiosensitivity and are defective in NHEJ DSB repair[60,75,76]. Knocking out DNA Ligase IV in mice results in late embryonic lethality with massive neuronal apoptosis and lymphocyte development arrest due to lack of V(D)J recombination[77,78].
To date, several polymorphisms have been identified in LIG4 gene, some of them potentially capable to modulate LIG4 activity. For example, LIG4 polymorphism rs1805388 C/T was found in N-terminal region and has been linked with a reduced adenylation and ligation activities of the enzyme[79]. Variations in enzymatic activity of LIG4 can conduct to a hyper-sensitivity to DNA damage, deregulation of repair and apoptosis mechanisms, affecting the susceptibility to cancer development as well as oncologic therapy response. The principal studies that have been developed to understand the LIG4 polymorphisms role in cancer are described in Table 2.
Table 2.
Some studies that evaluate LIG4 polymorphisms role in cancer
| Authors | LIG4 SNP identification | Tumor model | Ethinicity | Result |
| Jakubowska et al[84] | rs1805386 | Ovarian and Breast Cancer | Caucasian | The polymorphism was not associated with BRCA1-associated ovarian and breast cancer risk (P = 0.16 and P = 0.97, respectively) |
| Schildkraut et al[82] | rs10131 | Ovarian Cancer | Caucasian | The polymorphism was significantly associated with invasive serous ovarian cancer risk (P < 0.05) |
| Pearce et al[83] | rs1805386 | Ovarian Cancer | Mixed | The polymorphism was initially associated with ovarian cancer risk (P = 0.007) but replication results do not confirm this association |
| Yin et al[93] | rs1805388 | Non-small cell Lung Cancer | Mixed | The polymorphism was significantly associated with the risk of severe radiation pneumonitis in non-small cell lung cancer patients who received radio(chemo)therapy (P < 0.05) |
| Tseng et al[94] | rs1805388 | Non-small cell Lung Cancer | Asian | The polymorphism was significantly associated with lung cancer risk (P = 0.038) especially in smoking patients (P = 0.015), and with high fractional allelic loss (P = 0.016) |
| de las Peñas et al[89] | rs1805386 | Non-small cell Lung Cancer | Caucasian | The polymorphism was not associated with survival in cisplatin/gemcitabine-treated non-small cell lung cancer patients (P = 0.31) |
| Sakiyama et al[95] | rs2232641 | Lung Cancer | Japanese | The polymorphism was significantly associated with a diminish risk to develop lung cancer (P = 0.03) |
| Sobczuk et al[96] | rs2232641 | Breast Cancer | Caucasian | The polymorphism was not associated with breast cancer risk (P > 0.05) |
| Han et al[97] | rs1805386 | Breast Cancer | Mostly Caucasian | No statistically differences in breast cancer risk according LIG4 C299T or T1977C. The polymorphism T1977C was significantly associated with breast cancer risk if the patients had a first degree family history of breast cancer (P = 0.01) |
| rs4987182 | ||||
| Goode et al[98] | rs1805386 | Breast Cancer | Caucasian | The polymorphism was significantly associated with the breast cancer survival (P = 0.002) |
| Kuschel et al[99] | rs1805386 | Breast Cancer | Caucasian | The polymorphism was significantly associated with a decrease in breast cancer risk (P = 0.04) |
| Liu et al[100] | rs3093739 | Glioma | Asian | The polymorphism was significantly associated with glioma risk (P = 0.009) |
| Liu et al[101] | rs7325927 | Glioblastoma | Caucasian | The polymorphism was significantly associated with glioblastoma survival (P = 0.008) |
SNP: Single nucleotide polymorphism.
Radiotherapy and chemotherapy remain the core of conventional cancer treatment and it is necessary to understand how cells respond to DNA damage and determine whether DDR could be exploited or manipulated for therapeutic purposes. There is a growing interest in the identification of DNA repair inhibitors that will enhance the cytotoxicity of DNA damaging agents that, when used concomitantly, may have the capacity to increase the response to treatment[46,48].
Since DNA ligation is an ubiquitous stage in the majority of cellular processes and the last step of almost all DNA repair pathways, DNA ligases are attractive therapeutic targets since it is expected that cells defective in DSB repair will be more sensitive to chemotherapeutic agents[44,46,49].
Some studies suggest that LIG4 down-regulation could be a potential strategy to enhance the therapeutic effects of chemotherapy[44-46]. Kondo et al[45] designed a study to better understand the role of DSB repair pathways, including NHEJ, on cellular sensitivity to Temozolomide (TMZ) in glioblastoma. First, they evaluated the role of repair genes in the presence of TMZ-induced DNA damage. Within the cell lines evaluated, LIG4 -/- cells were the most sensitive to TMZ action. To test whether this result was pertinent to chemotherapy used against glioblastoma, LIG4 expression was silenced in A172 glioblastoma cells using siRNA. Results showed that LIG4 silencing increased cellular sensitivity to TMZ approximately three times. Therefore, the authors proposed that LIG4 down regulation can potentially be a useful strategy for enhance the therapeutic effects of TMZ, becoming LIG4 a new molecular target for chemotherapy[45]. In a study designed by Friesen et al[80], they investigated the role of LIG4 in deficient caspases activation by doxorubicin. The results showed that doxorubicin strongly induced apoptosis and caspases activation in LIG4 defective cells suggesting that LIG4, as a key enzyme for NHEJ repair, also plays an important role in deficient caspases activation in cancer cells[80].
In a last view, it may be useful the combination between LIG4 inhibitors with the individual’s LIG4 profile since observations suggest that in a partially defective genetic background, additional reduction in ligase levels additionally compromises the cellular ability to repair DSBs[81].
LIGASE IV IN OC
In which concerns to LIG4 polymorphisms and their role in OC risk just a few studies have been made[82-84] (Table 2). Due to contradictory results obtained to some polymorphisms and OC risk, Ovarian Cancer Association Consortium has been formed with the purpose to evaluate the evidence for association in SNPs, which had already been genotyped by multiple studies by combining the existing data. This collaboration has shown that 1977 T/C polymorphism in LIG4 gene (rs1805386) was not associated with OC risk, although the initial results proposed a significantly positive association[83].
OC remains a treatment challenge. Although the initial response of the OC patients to chemotherapy is good, many patients recur and develop, possibly, cell clones resistant to therapy. Platinum analogs, as cisplatin or carboplatin, are one of the most widely used anti-cancer drugs due to its broad-spectrum of activity against human tumors, namely OC. Platinum compounds react with DNA molecules, forming inter and intrastrand DNA crosslinks, and consequently blocking the movement of DNA replication and transcription machinery along DNA, which results in the arrest of the cell cycle and the activation of DNA repair pathways[85-88]. Nucleotide excision repair is the main mechanism responsible for platinum-DNA adducts removal although some studies proposed that these adducts can inhibit NHEJ repair pathway and consequently influence the patient’s overall survival since survival is longer in patients with higher levels of platinum-DNA adducts[18,52,89]. Clinical use of platinum compounds in OC treatment is conditioned by the development of resistance which can result from reduced intracellular accumulation, increased drug inactivation, increased repair of damaged DNA, increased activation of pro-survival pathways or inhibition of pathways that promote cell death[90]. Besides the importance of NHEJ, and specifically of LIG4, to platinum treatment response, to the best of our knowledge no study evaluated the association between LIG4 polymorphisms and the chemotherapy response of OC patients. It would be interesting to evaluate the role of LIG4 polymorphisms in platinum resistance and relate them to LIG4 mRNA expression in order to predict the clinical outcome of OC patients and possibly use this marker to guide chemotherapy selection in woman with OC.
Following the PARP inhibitors treatment applicability, LIG4 inhibitors may be concomitantly used with standard therapy for OC treatment in order to enhance its effect and to exploit intrinsic defects in specific DNA repair pathway. This approach might create a large therapeutic window and help to overcome chemotherapy failure in OC treatment. Potential strategies to inhibit the LIG4 action can be by the use of siRNA, as mentioned by Kondo et al[45], or by the use of small molecules in silico designed, as mentioned by Chen and collaborators[46].
CONCLUSION
Besides the strong link between DNA repair and OC, the knowledge about LIG4 role in ovarian carcinogenesis is still very limited and one of the aims of this review was to compile all the available information. In last years, translational research has reached an essential role in oncology and the identification of an individual SNP profile, which used in combination with risk factors, can lead to the establishment of potential susceptibility and prognosis factors. In this way, it became clear that the development of new studies are essential to better understand the functional role of polymorphisms in LIG4 gene and how they can be linked to OC development, namely which concerns with repair-associated ovulation. In other perspective, the definition of a SNP profile could be a useful manner to implement screening and prevention strategies and consequently decrease the OC mortality. To the best of our knowledge, no study has been done regarding LIG4 polymorphisms and their influence in OC treatment response. However, in this review, we described the dual role of LIG4 enzyme in cancer. Some studies have associated high levels of this enzyme with a good response to carcinogenic damage repair and the consequent genomic stability maintenance. However, in an opposite view, high levels of LIG4 enzyme can lead to worse treatment response due to the higher capability to repair the damage induced by chemo or radiotherapy. It is known that genetic polymorphisms are capable to affect the functional activity of DNA repair enzymes and affect significantly the effectiveness and therapy outcome. Besides this aspect, studies are made in order to discover and develop new treatment strategies to overcome therapy resistance and improve OC survival rates, namely using DNA repair as treatment target. The inhibition of LIG4 can possibly be an useful strategy to overcome the chemotherapy failure associated with OC standard treatment.
Footnotes
Supported by Research Department of Portuguese League against Cancer (NRNorte) and Minister of Health of Portugal (CFICS-45/2007)
P-Reviewers Altinisik J, Cheng WH S- Editor Wen LL L- Editor A E- Editor Zheng XM
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer. 2010;46:765–781. doi: 10.1016/j.ejca.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 3.Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol. 2008;26:5284–5293. doi: 10.1200/JCO.2008.18.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holschneider CH, Berek JS. Ovarian cancer: epidemiology, biology, and prognostic factors. Semin Surg Oncol. 2000;19:3–10. doi: 10.1002/1098-2388(200007/08)19:1<3::aid-ssu2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 5.Chen VW, Ruiz B, Killeen JL, Coté TR, Wu XC, Correa CN. Pathology and classification of ovarian tumors. Cancer. 2003;97:2631–2642. doi: 10.1002/cncr.11345. [DOI] [PubMed] [Google Scholar]
- 6.Bast RC, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001;22:255–288. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- 8.Fleming JS, Beaugié CR, Haviv I, Chenevix-Trench G, Tan OL. Incessant ovulation, inflammation and epithelial ovarian carcinogenesis: revisiting old hypotheses. Mol Cell Endocrinol. 2006;247:4–21. doi: 10.1016/j.mce.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 9.Leitao MM, Boyd J, Hummer A, Olvera N, Arroyo CD, Venkatraman E, Baergen RN, Dizon DS, Barakat RR, Soslow RA. Clinicopathologic analysis of early-stage sporadic ovarian carcinoma. Am J Surg Pathol. 2004;28:147–159. doi: 10.1097/00000478-200402000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Berchuck A, Schildkraut JM, Marks JR, Futreal PA. Managing hereditary ovarian cancer risk. Cancer. 1999;86:2517–2524. doi: 10.1002/(sici)1097-0142(19991201)86:11+<2517::aid-cncr8>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 11.Shih IeM, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511–1518. doi: 10.1016/s0002-9440(10)63708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurman RJ, Shih IeM. Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int J Gynecol Pathol. 2008;27:151–160. doi: 10.1097/PGP.0b013e318161e4f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karst AM, Drapkin R. Ovarian cancer pathogenesis: a model in evolution. J Oncol. 2010;2010:932371. doi: 10.1155/2010/932371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhoola S, Hoskins WJ. Diagnosis and management of epithelial ovarian cancer. Obstet Gynecol. 2006;107:1399–1410. doi: 10.1097/01.AOG.0000220516.34053.48. [DOI] [PubMed] [Google Scholar]
- 15.Fasching PA, Gayther S, Pearce L, Schildkraut JM, Goode E, Thiel F, Chenevix-Trench G, Chang-Claude J, Wang-Gohrke S, Ramus S, et al. Role of genetic polymorphisms and ovarian cancer susceptibility. Mol Oncol. 2009;3:171–181. doi: 10.1016/j.molonc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 17.Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM, Baergen R. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21:3194–3200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 18.Bookman MA. Developmental chemotherapy and management of recurrent ovarian cancer. J Clin Oncol. 2003;21:149s–167s. doi: 10.1200/jco.2003.02.553. [DOI] [PubMed] [Google Scholar]
- 19.Hennessy BT, Coleman RL, Markman M. Ovarian cancer. Lancet. 2009;374:1371–1382. doi: 10.1016/S0140-6736(09)61338-6. [DOI] [PubMed] [Google Scholar]
- 20.Banerjee S, Gore M. The future of targeted therapies in ovarian cancer. Oncologist. 2009;14:706–716. doi: 10.1634/theoncologist.2009-0013. [DOI] [PubMed] [Google Scholar]
- 21.Kling J. PARP inhibitors blaze a trail in difficult-to-treat cancers. Nat Biotechnol. 2009;27:784–786. doi: 10.1038/nbt0909-784. [DOI] [PubMed] [Google Scholar]
- 22.Barrena Medel NI, Wright JD, Herzog TJ. Targeted therapies in epithelial ovarian cancer. J Oncol. 2010;2010:314326. doi: 10.1155/2010/314326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murdoch WJ, McDonnel AC. Roles of the ovarian surface epithelium in ovulation and carcinogenesis. Reproduction. 2002;123:743–750. doi: 10.1530/rep.0.1230743. [DOI] [PubMed] [Google Scholar]
- 24.Fathalla MF. Incessant ovulation--a factor in ovarian neoplasia. Lancet. 1971;2:163. doi: 10.1016/s0140-6736(71)92335-x. [DOI] [PubMed] [Google Scholar]
- 25.Ness RB, Cottreau C. Possible role of ovarian epithelial inflammation in ovarian cancer. J Natl Cancer Inst. 1999;91:1459–1467. doi: 10.1093/jnci/91.17.1459. [DOI] [PubMed] [Google Scholar]
- 26.Smith ER, Xu XX. Ovarian ageing, follicle depletion, and cancer: a hypothesis for the aetiology of epithelial ovarian cancer involving follicle depletion. Lancet Oncol. 2008;9:1108–1111. doi: 10.1016/S1470-2045(08)70281-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006;5:1042–1048. doi: 10.1016/j.dnarep.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 28.Chakravarti A. To a future of genetic medicine. Nature. 2001;409:822–823. doi: 10.1038/35057281. [DOI] [PubMed] [Google Scholar]
- 29.Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8:1229–1231. doi: 10.1101/gr.8.12.1229. [DOI] [PubMed] [Google Scholar]
- 30.Brookes AJ. The essence of SNPs. Gene. 1999;234:177–186. doi: 10.1016/s0378-1119(99)00219-x. [DOI] [PubMed] [Google Scholar]
- 31.Baye TM, Wilke RA. Mapping genes that predict treatment outcome in admixed populations. Pharmacogenomics J. 2010;10:465–477. doi: 10.1038/tpj.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans WE, McLeod HL. Pharmacogenomics--drug disposition, drug targets, and side effects. N Engl J Med. 2003;348:538–549. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 33.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761, 763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 34.Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 35.Yan L, Beckman R. Pharmacogenetics and pharmacogenomics in oncology therapeutic antibody development. Biotechniques. 2005;39:S565–S568. doi: 10.2144/000112043. [DOI] [PubMed] [Google Scholar]
- 36.Santos AM, Sousa H, Portela C, Pereira D, Pinto D, Catarino R, Rodrigues C, Araújo AP, Lopes C, Medeiros R. TP53 and P21 polymorphisms: response to cisplatinum/paclitaxel-based chemotherapy in ovarian cancer. Biochem Biophys Res Commun. 2006;340:256–262. doi: 10.1016/j.bbrc.2005.11.176. [DOI] [PubMed] [Google Scholar]
- 37.Pinto D, Pereira D, Portela C, da Silva JL, Lopes C, Medeiros R. The influence of HER2 genotypes as molecular markers in ovarian cancer outcome. Biochem Biophys Res Commun. 2005;335:1173–1178. doi: 10.1016/j.bbrc.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Catarino R, Araújo A, Coelho A, Gomes M, Nogueira A, Lopes C, Medeiros RM. Prognostic significance of telomerase polymorphism in non-small cell lung cancer. Clin Cancer Res. 2010;16:3706–3712. doi: 10.1158/1078-0432.CCR-09-3030. [DOI] [PubMed] [Google Scholar]
- 39.Medeiros R, Pereira D, Afonso N, Palmeira C, Faleiro C, Afonso-Lopes C, Freitas-Silva M, Vasconcelos A, Costa S, Osório T, et al. Platinum/paclitaxel-based chemotherapy in advanced ovarian carcinoma: glutathione S-transferase genetic polymorphisms as predictive biomarkers of disease outcome. Int J Clin Oncol. 2003;8:156–161. doi: 10.1007/s10147-003-0318-8. [DOI] [PubMed] [Google Scholar]
- 40.Nogueira A, Catarino R, Coelho A, Araújo A, Gomes M, Medeiros R. Influence of DNA repair RAD51 gene variants in overall survival of non-small cell lung cancer patients treated with first line chemotherapy. Cancer Chemother Pharmacol. 2010;66:501–506. doi: 10.1007/s00280-009-1187-2. [DOI] [PubMed] [Google Scholar]
- 41.Nogueira A, Catarino R, Faustino I, Nogueira-Silva C, Figueiredo T, Lombo L, Hilário-Silva I, Pereira D, Medeiros R. Role of the RAD51 G172T polymorphism in the clinical outcome of cervical cancer patients under concomitant chemoradiotherapy. Gene. 2012;504:279–283. doi: 10.1016/j.gene.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 42.Silva J, Teixeira AL, Lobo F, Maurício J, Medeiros R. DNA repair system and prostate cancer progression: the role of NBS1 polymorphism (rs1805794) DNA Cell Biol. 2012;31:1182–1186. doi: 10.1089/dna.2011.1562. [DOI] [PubMed] [Google Scholar]
- 43.Moeller BJ, Pasqualini R, Arap W. Targeting cancer-specific synthetic lethality in double-strand DNA break repair. Cell Cycle. 2009;8:1872–1876. doi: 10.4161/cc.8.12.8743. [DOI] [PubMed] [Google Scholar]
- 44.Kondo N, Takahashi A, Mori E, Noda T, Su X, Ohnishi K, McKinnon PJ, Sakaki T, Nakase H, Ono K, et al. DNA ligase IV is a potential molecular target in ACNU sensitivity. Cancer Sci. 2010;101:1881–1885. doi: 10.1111/j.1349-7006.2010.01591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kondo N, Takahashi A, Mori E, Ohnishi K, McKinnon PJ, Sakaki T, Nakase H, Ohnishi T. DNA ligase IV as a new molecular target for temozolomide. Biochem Biophys Res Commun. 2009;387:656–660. doi: 10.1016/j.bbrc.2009.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM, MacKerell AD, Tomkinson AE. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68:3169–3177. doi: 10.1158/0008-5472.CAN-07-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinek I, Haldar K, Gaitskell K, Bryant A, Nicum S, Kehoe S, Morrison J. DNA-repair pathway inhibitors for the treatment of ovarian cancer. Cochrane Database Syst Rev. 2010;6:CD007929. doi: 10.1002/14651858.CD007929.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wicki A, Rochlitz C. Targeted therapies in breast cancer. Swiss Med Wkly. 2012;142:w13550. doi: 10.4414/smw.2012.13550. [DOI] [PubMed] [Google Scholar]
- 49.Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 2004;3:781–796. doi: 10.1016/j.dnarep.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Zhou J, Lim CU. The role of NBS1 in DNA double strand break repair, telomere stability, and cell cycle checkpoint control. Cell Res. 2006;16:45–54. doi: 10.1038/sj.cr.7310007. [DOI] [PubMed] [Google Scholar]
- 52.Pastwa E, Błasiak J. Non-homologous DNA end joining. Acta Biochim Pol. 2003;50:891–908. [PubMed] [Google Scholar]
- 53.Lee Y, McKinnon PJ. DNA ligase IV suppresses medulloblastoma formation. Cancer Res. 2002;62:6395–6399. [PubMed] [Google Scholar]
- 54.Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst) 2007;6:712–722. doi: 10.1016/j.dnarep.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 55.Adachi N, Ishino T, Ishii Y, Takeda S, Koyama H. DNA ligase IV-deficient cells are more resistant to ionizing radiation in the absence of Ku70: Implications for DNA double-strand break repair. Proc Natl Acad Sci USA. 2001;98:12109–12113. doi: 10.1073/pnas.201271098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bau DT, Fu YP, Chen ST, Cheng TC, Yu JC, Wu PE, Shen CY. Breast cancer risk and the DNA double-strand break end-joining capacity of nonhomologous end-joining genes are affected by BRCA1. Cancer Res. 2004;64:5013–5019. doi: 10.1158/0008-5472.CAN-04-0403. [DOI] [PubMed] [Google Scholar]
- 57.Biard DS. Untangling the relationships between DNA repair pathways by silencing more than 20 DNA repair genes in human stable clones. Nucleic Acids Res. 2007;35:3535–3550. doi: 10.1093/nar/gkm195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- 60.Pollard JM, Gatti RA. Clinical radiation sensitivity with DNA repair disorders: an overview. Int J Radiat Oncol Biol Phys. 2009;74:1323–1331. doi: 10.1016/j.ijrobp.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584:3703–3708. doi: 10.1016/j.febslet.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Timson DJ, Singleton MR, Wigley DB. DNA ligases in the repair and replication of DNA. Mutat Res. 2000;460:301–318. doi: 10.1016/s0921-8777(00)00033-1. [DOI] [PubMed] [Google Scholar]
- 63.Martin IV, MacNeill SA. ATP-dependent DNA ligases. Genome Biol. 2002;3:REVIEWS3005. doi: 10.1186/gb-2002-3-4-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shuman S, Schwer B. RNA capping enzyme and DNA ligase: a superfamily of covalent nucleotidyl transferases. Mol Microbiol. 1995;17:405–410. doi: 10.1111/j.1365-2958.1995.mmi_17030405.x. [DOI] [PubMed] [Google Scholar]
- 65.Tomkinson AE, Mackey ZB. Structure and function of mammalian DNA ligases. Mutat Res. 1998;407:1–9. doi: 10.1016/s0921-8777(97)00050-5. [DOI] [PubMed] [Google Scholar]
- 66.Wei YF, Robins P, Carter K, Caldecott K, Pappin DJ, Yu GL, Wang RP, Shell BK, Nash RA, Schär P. Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and recombination. Mol Cell Biol. 1995;15:3206–3216. doi: 10.1128/mcb.15.6.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilson TE, Grawunder U, Lieber MR. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- 68.Critchlow SE, Bowater RP, Jackson SP. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr Biol. 1997;7:588–598. doi: 10.1016/s0960-9822(06)00258-2. [DOI] [PubMed] [Google Scholar]
- 69.Grawunder U, Zimmer D, Leiber MR. DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Curr Biol. 1998;8:873–876. doi: 10.1016/s0960-9822(07)00349-1. [DOI] [PubMed] [Google Scholar]
- 70.Sibanda BL, Critchlow SE, Begun J, Pei XY, Jackson SP, Blundell TL, Pellegrini L. Crystal structure of an Xrcc4-DNA ligase IV complex. Nat Struct Biol. 2001;8:1015–1019. doi: 10.1038/nsb725. [DOI] [PubMed] [Google Scholar]
- 71.Calsou P, Delteil C, Frit P, Drouet J, Salles B. Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J Mol Biol. 2003;326:93–103. doi: 10.1016/s0022-2836(02)01328-1. [DOI] [PubMed] [Google Scholar]
- 72.Modesti M, Junop MS, Ghirlando R, van de Rakt M, Gellert M, Yang W, Kanaar R. Tetramerization and DNA ligase IV interaction of the DNA double-strand break repair protein XRCC4 are mutually exclusive. J Mol Biol. 2003;334:215–228. doi: 10.1016/j.jmb.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 73.Heinen CD, Schmutte C, Fishel R. DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol Ther. 2002;1:477–485. doi: 10.4161/cbt.1.5.160. [DOI] [PubMed] [Google Scholar]
- 74.Chistiakov DA, Voronova NV, Chistiakov PA. Genetic variations in DNA repair genes, radiosensitivity to cancer and susceptibility to acute tissue reactions in radiotherapy-treated cancer patients. Acta Oncol. 2008;47:809–824. doi: 10.1080/02841860801885969. [DOI] [PubMed] [Google Scholar]
- 75.O’Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 76.Schwarz K, Ma Y, Pannicke U, Lieber MR. Human severe combined immune deficiency and DNA repair. Bioessays. 2003;25:1061–1070. doi: 10.1002/bies.10344. [DOI] [PubMed] [Google Scholar]
- 77.Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. 1998;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- 78.Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, Kysela B, Beamish H, Plowman N, Arlett CF, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9:699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- 79.Girard PM, Kysela B, Härer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum Mol Genet. 2004;13:2369–2376. doi: 10.1093/hmg/ddh274. [DOI] [PubMed] [Google Scholar]
- 80.Friesen C, Uhl M, Pannicke U, Schwarz K, Miltner E, Debatin KM. DNA-ligase IV and DNA-protein kinase play a critical role in deficient caspases activation in apoptosis-resistant cancer cells by using doxorubicin. Mol Biol Cell. 2008;19:3283–3289. doi: 10.1091/mbc.E08-03-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Windhofer F, Wu W, Iliakis G. Low levels of DNA ligases III and IV sufficient for effective NHEJ. J Cell Physiol. 2007;213:475–483. doi: 10.1002/jcp.21120. [DOI] [PubMed] [Google Scholar]
- 82.Schildkraut JM, Iversen ES, Wilson MA, Clyde MA, Moorman PG, Palmieri RT, Whitaker R, Bentley RC, Marks JR, Berchuck A. Association between DNA damage response and repair genes and risk of invasive serous ovarian cancer. PLoS One. 2010;5:e10061. doi: 10.1371/journal.pone.0010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pearce CL, Near AM, Van Den Berg DJ, Ramus SJ, Gentry-Maharaj A, Menon U, Gayther SA, Anderson AR, Edlund CK, Wu AH, et al. Validating genetic risk associations for ovarian cancer through the international Ovarian Cancer Association Consortium. Br J Cancer. 2009;100:412–420. doi: 10.1038/sj.bjc.6604820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jakubowska A, Gronwald J, Menkiszak J, Górski B, Huzarski T, Byrski T, Tołoczko-Grabarek A, Gilbert M, Edler L, Zapatka M, et al. BRCA1-associated breast and ovarian cancer risks in Poland: no association with commonly studied polymorphisms. Breast Cancer Res Treat. 2010;119:201–211. doi: 10.1007/s10549-009-0390-5. [DOI] [PubMed] [Google Scholar]
- 85.Trimmer EE, Essigmann JM. Cisplatin. Essays Biochem. 1999;34:191–211. doi: 10.1042/bse0340191. [DOI] [PubMed] [Google Scholar]
- 86.Reed E. Cisplatin. Cancer Chemother Biol Response Modif. 1999;18:144–151. [PubMed] [Google Scholar]
- 87.Lawley PD, Phillips DH. DNA adducts from chemotherapeutic agents. Mutat Res. 1996;355:13–40. doi: 10.1016/0027-5107(96)00020-6. [DOI] [PubMed] [Google Scholar]
- 88.Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci. 2000;57:1229–1235. doi: 10.1007/PL00000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de las Peñas R, Sanchez-Ronco M, Alberola V, Taron M, Camps C, Garcia-Carbonero R, Massuti B, Queralt C, Botia M, Garcia-Gomez R, et al. Polymorphisms in DNA repair genes modulate survival in cisplatin/gemcitabine-treated non-small-cell lung cancer patients. Ann Oncol. 2006;17:668–675. doi: 10.1093/annonc/mdj135. [DOI] [PubMed] [Google Scholar]
- 90.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 91.Stadel BV. Letter: The etiology and prevention of ovarian cancer. Am J Obstet Gynecol. 1975;123:772–774. doi: 10.1016/0002-9378(75)90509-8. [DOI] [PubMed] [Google Scholar]
- 92.Risch HA. Hormonal etiology of epithelial ovarian cancer, with a hypothesis concerning the role of androgens and progesterone. J Natl Cancer Inst. 1998;90:1774–1786. doi: 10.1093/jnci/90.23.1774. [DOI] [PubMed] [Google Scholar]
- 93.Yin M, Liao Z, Liu Z, Wang LE, O’Reilly M, Gomez D, Li M, Komaki R, Wei Q. Genetic variants of the nonhomologous end joining gene LIG4 and severe radiation pneumonitis in nonsmall cell lung cancer patients treated with definitive radiotherapy. Cancer. 2012;118:528–535. doi: 10.1002/cncr.26214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tseng RC, Hsieh FJ, Shih CM, Hsu HS, Chen CY, Wang YC. Lung cancer susceptibility and prognosis associated with polymorphisms in the nonhomologous end-joining pathway genes: a multiple genotype-phenotype study. Cancer. 2009;115:2939–2948. doi: 10.1002/cncr.24327. [DOI] [PubMed] [Google Scholar]
- 95.Sakiyama T, Kohno T, Mimaki S, Ohta T, Yanagitani N, Sobue T, Kunitoh H, Saito R, Shimizu K, Hirama C, et al. Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int J Cancer. 2005;114:730–737. doi: 10.1002/ijc.20790. [DOI] [PubMed] [Google Scholar]
- 96.Sobczuk A, Smolarz B, Romanowicz H, Zadrozny M, Baszczyński J, Westfal B, Pertyński T. Analysis of the polymorphisms in non-homologous DNA end joining (NHEJ) gene Ku70 and Ligase IV in sporadic breast cancer in women. Pol J Pathol. 2010;61:27–31. [PubMed] [Google Scholar]
- 97.Han J, Hankinson SE, Ranu H, De Vivo I, Hunter DJ. Polymorphisms in DNA double-strand break repair genes and breast cancer risk in the Nurses’ Health Study. Carcinogenesis. 2004;25:189–195. doi: 10.1093/carcin/bgh002. [DOI] [PubMed] [Google Scholar]
- 98.Goode EL, Dunning AM, Kuschel B, Healey CS, Day NE, Ponder BA, Easton DF, Pharoah PP. Effect of germ-line genetic variation on breast cancer survival in a population-based study. Cancer Res. 2002;62:3052–3057. [PubMed] [Google Scholar]
- 99.Kuschel B, Auranen A, McBride S, Novik KL, Antoniou A, Lipscombe JM, Day NE, Easton DF, Ponder BA, Pharoah PD, et al. Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet. 2002;11:1399–1407. doi: 10.1093/hmg/11.12.1399. [DOI] [PubMed] [Google Scholar]
- 100.Liu Y, Zhou K, Zhang H, Shugart YY, Chen L, Xu Z, Zhong Y, Liu H, Jin L, Wei Q, et al. Polymorphisms of LIG4 and XRCC4 involved in the NHEJ pathway interact to modify risk of glioma. Hum Mutat. 2008;29:381–389. doi: 10.1002/humu.20645. [DOI] [PubMed] [Google Scholar]
- 101.Liu Y, Shete S, Etzel CJ, Scheurer M, Alexiou G, Armstrong G, Tsavachidis S, Liang FW, Gilbert M, Aldape K, et al. Polymorphisms of LIG4, BTBD2, HMGA2, and RTEL1 genes involved in the double-strand break repair pathway predict glioblastoma survival. J Clin Oncol. 2010;28:2467–2474. doi: 10.1200/JCO.2009.26.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]