

Abstract
A photocatalytic system involving [Ru(bpyrz)3](PF6)2·2H2O, visible light, and air has been developed for cleavage of the N–N bonds of hydrazines and hydrazides. This catalytic system is generally effective for N,N-disubstituted hydrazine and hydrazide derivatives, including arylhydrazides, N-alkyl-N-arylhydrazines, and N,N-diarylhydrazines. The utility of this cleavage reaction has been demonstrated by synthesizing a variety of secondary aromatic amines.
Keywords: nitrogen–nitrogen bonds, visible light, cleavage, ruthenium, amines
As our society becomes increasingly aware of the issue of environmental sustainability, green chemistry is gaining acceptance in industry and academia. The use of renewable and abundant sources such as visible light is becoming one of the key criteria for developing future ‘green’ methods. As such, the field of visible light photocatalysis has started to attract growing attention from the synthetic organic community.1 Seminal works from MacMillan, Stephenson, Yoon, and others have shown the versatility of visible light induced reactions in organic synthesis by using ruthenium(II) or iridium(III) complexes or dyes as photocatalysts.2–6
Amines are routinely used as a sacrificial oxidant in these processes to reduce photoexcited organometallic complexes to ruthenium(I) or iridium(II) complexes while being oxidized to nitrogen radical cations (Scheme 1). Then carbon radicals generated through reduction by the ruthenium(I) or iridium(II) complexes undergo C–C bond formation in most of the reported reactions. On the other hand, the fate of the nitrogen radical cations is relatively unknown. We recently became interested in harnessing the synthetic potential of these nitrogen radical cations generated by visible light photocatalysis. Hydrazines and hydrazides were chosen initially in our studies because they are more easily oxidized than amines. Furthermore, they are useful precursors to generate amines or amides after cleavage of the N–N bonds.7 This cleavage is often accomplished under strong reductive,8 oxidative,9 or other10 relatively harsh conditions. Herein, we report cleavage of the N–N bonds of hydrazines and hydrazides using visible light photocatalysis under extremely mild conditions.11
Scheme 1.

General catalytic cycle for visible light photocatalysis
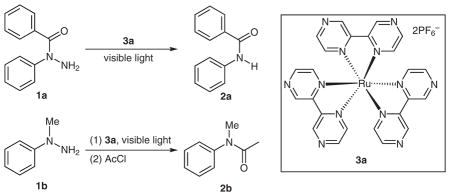
Our preliminary studies focused on the cleavage of the N–N bond of N-phenyl-N-benzoyl hydrazine (1a; Table 1). [Ru(bpyrz)3](PF6)2·2H2O (3a)12,13 was first examined as the photocatalyst using a 13 watt GE compact fluorescent light bulb as the light source. The reactions were carried out under an air atmosphere. Because the photocatalyst 3a is insoluble or sparingly soluble in most solvents except acetonitrile, the reaction was initially carried out in acetonitrile with 2 mol% of 3a. It was incomplete after 24 hours, providing 42% of the desired cleavage product, N-phenylbenzamide (2a) along with 44% of recovered 1a (Table 1, entry 1). Addition of methanol to the reaction mixture (MeCN–MeOH, 1:1 v/v) greatly facilitated the cleavage and provided 2a in 71% yield (100% conversion) after 24 hours (entry 2). The ratio of MeCN versus MeOH had little effect on the rate of cleavage, and 2a was generated in similar yields (entry 3). Reducing the catalyst loading (1 mol% 3a) reduced the yield of 2a to 59%, with 6% of recovered 1a (entry 4). Little or no conversion was observed when the reaction was conducted either in the absence of 3a or in the dark (entries 5 and 6). Use of degassed MeCN and MeOH dramatically slowed down the reaction, and 2a was isolated in only 13% yield after 40 hours (entry 7). This result hinted that oxygen might play a significant role in the cleavage of the N–N bond. Use of O2 in the place of air did not improve the reaction and a lower yield (54% yield, 100% conversion) of 2a was obtained after 24 hours (entry 8). Because the N–N bond cleavage of some electron-rich hydrazines is known to be realized by simple exposure to light, we subjected N-methyl-N-phenyl hydrazine (1b) to similar studies to those of 1a. As expected, the cleavage was faster. Using only MeCN and 2 mol% 3a, the desired cleavage product, N-methylaniline was obtained as its N-acetyl derivative 2b in 82% yield after 24 hours (entry 9).14 The use of MeOH as a co-solvent did not provide any benefit to either the reaction rate or the yield of 2b (entry 10). The background reaction in the absence of the catalyst 3a was faster than that of 1a, yet it was still much slower than the catalyzed reaction (12% conversion after 48 h, entry 11).
Table 1.
Optimization of the Catalyst Systems based on [Ru(bpyrz)3](PF6)2·2H2O (3a)
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Catalyst (mol%) | Conditions | Time (h) | Yield (%) |
| 1 | 1a | 3a (2) | air, MeCN | 24 | 42 (44)a |
| 2 | 1a | 3a (2) | air, MeCN–MeOH (1:1) | 24 | 71 |
| 3 | 1a | 3a (2) | air, MeCN–MeOH (2:1) | 24 | 74 |
| 4 | 1a | 3a (1) | air, MeCN–MeOH (1:1) | 24 | 59 (6)a |
| 5 | 1a | 3a (0) | air, MeCN–MeOH (1:1) | 120 | trace |
| 6b | 1a | 3a (2) | air, MeCN–MeOH (1:1) | 48 | 0 |
| 7 | 1a | 3a (2) | N2, degassed MeCN–MeOH (1:1) | 40 | 13 |
| 8 | 1a | 3a (2) | O2, MeCN–MeOH (1:1) | 24 | 54 |
| 9 | 1b | 3a (2) | air, MeCN | 24 | 82 |
| 10 | 1b | 3a (2) | air, MeCN–MeOH (2:1) | 24 | 83 |
| 11 | 1b | 3a (0) | air, MeCN–MeOH (2:1) | 48 | 12 (88)a |
Yield of recovered starting material.
No light.
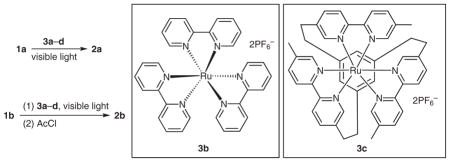
We then compared [Ru(bpyrz)3](PF6)2·2H2O (3a) against two other known ruthenium complexes (3b4a and 3c15) and Rose Bengal (3d)16 in the cleavage of the N–N bonds of 1a and 1b (Table 2). For hydrazides such as 1a, among the three ruthenium complexes 3a–c (entries 1–3), only 3a showed sufficient efficiency to catalyze the cleavage reaction. Rose Bengal showed similar activity to 3a but gave more side products (entry 4). For hydrazines such as 1b, the three ruthenium complexes (3a–c) and Rose Bengal (3d) all led to completion of the cleavage of the N–N bond. Surprisingly, 3a was the least active catalyst among the three ruthenium complexes (entry 5). Catalyst 3b was more active than 3a and 3c but also gave more side products than the latter (entries 5–7). Rose Bengal (3d) was also more active than 3a (entry 8). Similar to 1a, the reaction catalyzed by Rose Bengal (3d) gave more side products than 3a. Overall, 3a was the optimal catalyst for both the hydrazines and hydrazides.
Table 2.
Comparison of Ruthenium Catalysts 3a–c and Rose Bengal (3d)
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Catalyst (mol%) | Conditions | Time (h) | Yield (%) |
| 1 | 1a | 3a (2) | air, MeCN–MeOH (1:1) | 24 | 71 |
| 2 | 1a | 3b (2) | air, MeCN–MeOH (1:1) | 110 | 10 (80)a |
| 3 | 1a | 3c (2) | air, MeCN–MeOH (1:1) | 110 | 8 (73)a |
| 4 | 1a | 3d (2) | air, MeCN–MeOH (1:1) | 36 | 49 |
| 5 | 1b | 3a (2) | air, MeCN–MeOH (2:1) | 24 | 83 |
| 6 | 1b | 3b (2) | air, MeCN–MeOH (2:1) | 4 | 65 |
| 7 | 1b | 3c (2) | air, MeCN–MeOH (2:1) | 10 | 85 |
| 8 | 1b | 3d (2) | air, MeCN–MeOH (2:1) | 10 | 56 |
Yield of recovered 1a.
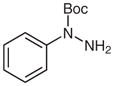
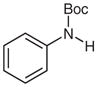
With the catalyst system optimized, we next applied it to a variety of hydrazine and hydrazide derivatives (Table 3). This catalytic system was found to be generally effective for N,N-disubstituted hydrazine and hydrazide derivatives including arylhydrazides, N-alkyl-N-arylhydrazines, and N,N-diarylhydrazines. Between the two substituents on the nitrogen atom, one needed to be an aryl group while the second could be an aryl, alkyl, or acyl group. Most of the substrates that meet this criterion gave the desired products in satisfactory yields. Electron-withdrawing substituents on the aromatic rings, such as a trifluoromethyl group, had little effect on the cleavage because 1c behaved very similarly to 1d (entries 1 and 2). Functional groups such as olefins and Cl were compatible with the reaction conditions (entries 3 and 6). Hydrazide derivatives containing heterocycles worked under the optimized conditions although they were not as effective as their counterparts with only aromatic rings (entry 4). The cleavage of a hydrazine substituted with a biaryl group and an alkyl group also afforded the desired secondary amine in modest yield (entry 7). In this case, it appeared that the cleavage product decomposed under the cleavage reactions. To our surprise, tetra-substituted hydrazine derivatives were found to be inert under the optimized conditions (entry 8). Trisubstituted hydrazine derivatives, depending on the pattern of substituents, underwent one of the two types of oxidations to give either hydrazones or azo compounds. One was oxidation of N(H)R to form hydrazones (entries 9 and 10). The other was oxidative cleavage of the N-Me group followed by oxidation of the forming N–N bonds to provide azo compounds (entry 11).
Table 3.
Ruthenium(II)-Catalyzed Photoinduced Cleavage of N–N Bondsa

| |||
|---|---|---|---|
| Entry | Substrate | Product | Yield (%) |
| 1 |
 1c |
 2c |
82 |
| 2 |
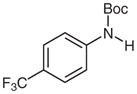
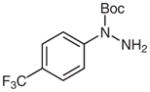 1d |
 2d |
81 |
| 3 |
 1e |
 2e |
54 |
| 4 |
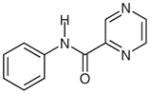
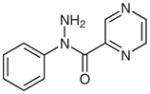 1f |
 2f |
47 |
| 5 |
 1g |
 2g |
68b |
| 6 |
 1h |
 2h |
51b |
| 7 |
 1i |
 2i |
37 |
| 8 |
 1j |
– | – |
| 9 |
 1k |
 2k |
36 |
| 10 |
 1l |
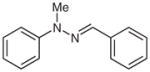 2l |
88 |
| 11 |
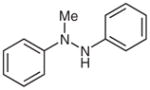 1m |
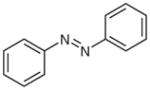 2m |
58 |
Reactions were performed on 0.3–0.5 mmol scale in 3–5 mL solvent with 2 mol% 3a. A household 13 W light bulb was used as the light source. Reaction time ranged from 4 to 28 h.
Due to the instability of these hydrazine derivatives, the crude products were subjected to the cleavage reaction directly after Boc-deprotection. The yields given refer to overall yields for the two steps.
Secondary aromatic amines are of great interest in many areas of chemistry because of the prevalence of these compounds in pharmaceuticals, materials, etc. Their preparation through nucleophilic amination has been recently popularized by the Goldberg reaction17 and Buchwald–Hartwig amination.18 On the other hand, synthesis of the secondary aromatic amines through electrophilic amination has received much less attention. Knochel recently reported an electrophilic amination protocol for the synthesis of diarylamines that involves addition of nucleophilic aryl donors to arylazo tosylates followed by cleavage of N–N bonds (Scheme 2).7a Excess allyl iodide (3 equiv) and zinc (10 equiv) were required for the cleavage of the N–N bonds. Separately, Mäeorg reported that a number of alkyl and aromatic nucleophiles were efficiently added to N-tert-butoxycarbonylaryldiazenes to generate Boc-protected hydrazines.19 Although N-tert-butoxycarbonylaryldiazenes are potentially a synthon of ‘ArNH(+)’, the author did not pursue the further conversion of the addition product, Boc-protected hydrazines, to secondary aromatic amines. We felt that, coupled with Mäeorg’s method, our photochemistry could be developed into an efficient electrophilic amination protocol that is complementary to Knochel’s. As shown in Table 4, we were able to add a wide variety of nucleophiles, including aryl boronic acids [catalyzed by Cu(OAc)2], alkyl lithiums, aryl lithiums, aryl Grignard reagents, and lithium amide enolates, to N-tert-butoxycarbonylaryldiazenes. The Boc group of the hydrazines was easily removed by treatment with trifluoroacetic acid (TFA). Some of the deprotected hydrazines were not stable to silica gel chromatography. For those, the deprotected products were directly subjected to the cleavage reactions. In general, the cleavage step worked as well as those in our substrate scope studies (see above) with one exception where the ruthenium catalyst (3a) was found to decompose with a substrate containing an iodide group (entry 9).
Scheme 2.
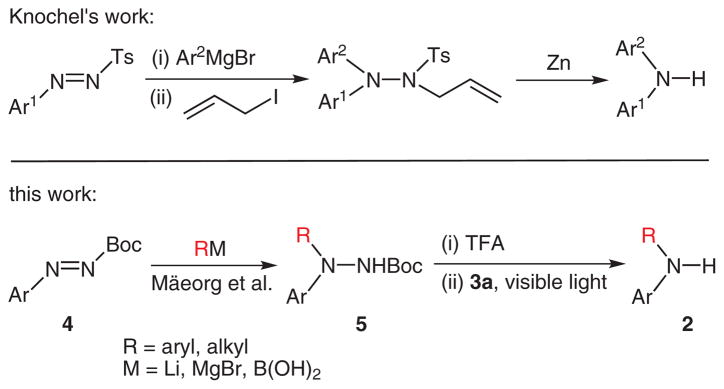
Electrophilic amination via aryl azo compounds
Table 4.
Synthesis of Secondary Aromatic Aminesa
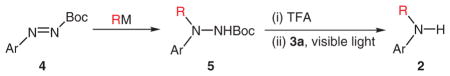
| ||||
|---|---|---|---|---|
| Entry | 4 | RM | 5 (Yield %) | 2 (Yield %) |
| 1 |
 4n |
|
 5n (95) |
 2n (91)b |
| 2 |
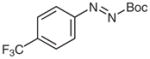 4o |
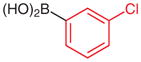
|
 5o (95) |
 2o (87)b |
| 3 |
 4p |
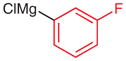
|
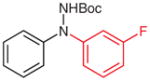 5p (96) |
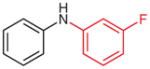 2p (84)b |
| 4 |
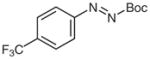 4q |
PhMgBr |
 5q (96) |
 2q (85)b |
| 5 |
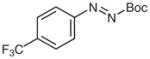 4r |
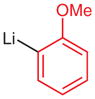
|
 5r (79) |
 2r (65)b |
| 6 |
 4s |
|
 5s (73) |
 2s (55) |
| 7 |
 4t |
|
 5t (50) |
 2t (72)b |
| 8 |
 4u |
BuMgCl |
 5u (92) |
 2u (70)b |
| 9 |
 4v |
BuMgCl |
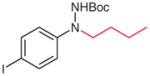 5v (95) |
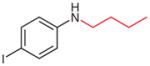 2v (21)b,c |
| 10 |
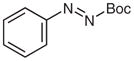 4w |
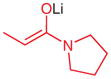
|
 5w (71) |
 2w (51)b |
Reactions were performed on 0.3–0.5 mmol scale in 3–5 mL of MeCN–MeOH (1:1 v/v) with 2 mol% 3a. A household 13 W light bulb was used as the light source. Reaction time ranged from 15 to 48 h.
Due to the instability of these hydrazine derivatives, the crude products were subjected to the cleavage reaction directly after Boc-deprotection. The yields refer to overall yields for the two steps.
Another 2 mol% of 3a was added after 10 h; 43% of hydrazine was recovered.
We thought it likely that several competing reaction pathways would operate under the photocatalytic conditions (Scheme 3). Similar to tertiary amines, hydrazine or hydrazide 1 would reductively quench a photoexcited state [Ru(bpyrz)3]2+*, which is generated by exposure of [Ru(bpyrz)3](PF6)2·2H2O (3a) to visible light. Deprotonation of the resulting nitrogen radical cation 6 would produce radical 7, which could then react with O2 to form radical 8. Depending on R1, two different pathways are proposed to diverge from 8. When R1 is H, a rearrangement of 8 to 10 via 9 is proposed. Similar rearrangements have been observed with nitroso oxides.20 Fragmentation of 10 would provide nitrous acid and amine radical 10, which could be reduced by ruthenium(I) to afford amine 2. On the other hand, when R1 is non-hydrogen, radical 8 would be reduced by ruthenium(I) to yield 12. Elimination of HOO− would generate diazenium 13, which could undergo two different isomerization processes (cf. Table 3). When R1 is a benzyl group, isomerization of 13 to hydrazone 2l would be favorable. In contrast, isomerization of 13 to 16 would be favorable when R1 is a phenyl group. Conversion of 16 into 17 followed by oxidation in situ would provide azobenzene 2m.
Scheme 3.

Proposed pathways
To support the hypothesis that hydrazine or hydrazide 1 would be initially oxidized by a photoexcited state [Ru(bpyrz)3]2+*, we performed two series of fluorescence quenching experiments (Stern–Volmer studies21) using substrates 1a and 1b (Figure 1). The ratio of the fluorescene intensity without a quencher (Io) versus that with a quencher (I) was shown to be first order dependent on the concentration of 1a or 1b, indicating that both act as reductive quenchers of the excited state of [Ru(bpyrz)3]2+. Hydrazine 1b is expected to be a stronger reducing agent than hydrazide 1a because the latter compound is substituted with a benzoyl group instead of a methyl group. The slope of the line for hydrazine 1b was larger than that for hydrazide 1a, which was consistent with this expectation. Furthermore, protonation of hydrazines such as 1b would remove one of the two lone pairs on the two nitrogen atoms, rendering 1b an ineffective quencher of [Ru(bpyrz)3]2+*. Indeed, the HCl salt of hydrazine 1b was found to be inert under the optimized conditions.
Figure 1.
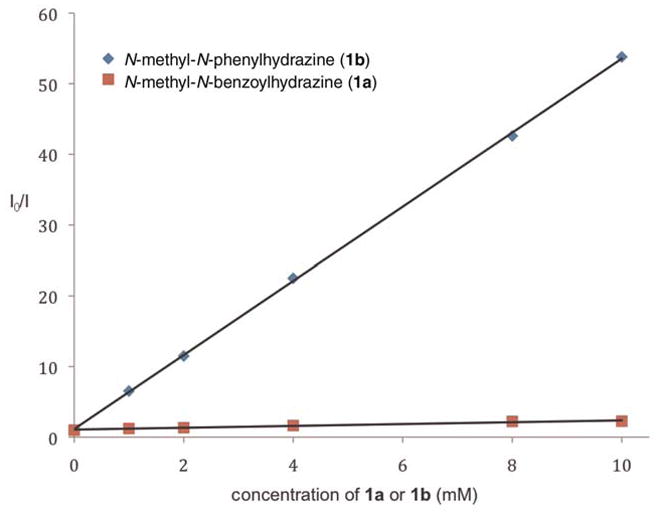
Fluorescence quenching of [Ru(bpyrz)3](PF6)2·2H2O (3a)by hydrazide 1a and hydrazines 1b
In conclusion, we have developed a photocatalytic method for cleavage of the N–N bonds of hydrazines and hydrazides. The method, using 2 mol% ruthenium(II) catalyst, visible light, and air, is operationally very simple. The utility of this method has been demonstrated by the synthesis of secondary aromatic amines through electrophilic amination. Furthermore, this study has shed some light on the fate of nitrogen radical cations generated by quenching the photoexcited state [Ru(bpyrz)3]2+*.
All reactions were carried out under a nitrogen atmosphere unless otherwise stated. Anhydrous solvents were purchased and used as received except THF, CH2Cl2, Et2O, and toluene, which were rigorously purged with argon for 2 h and then further purified by passing through two packed columns of neutral alumina (for THF and Et2O) or through neutral alumina and copper(II) oxide (for toluene and CH2Cl2) under argon from a solvent purification system. All new compounds were characterized by 1H NMR, 13C NMR, and IR spectroscopy (thin film on NaCl plates), in addition to elemental analysis and/or high resolution mass spectroscopy. Melting points are uncorrected. For starting materials and intermediates, copies of the 1H and 13C NMR spectra are given in the Supporting Information. Copies of the 1H and 13C NMR spectra for all the final products are given in the Supporting Information. NMR spectra were recorded in deuterated solvents. All 1H NMR experiments are reported in δ units in parts per million (ppm) and were measured relative to the signals for residual chloroform (δ = 7.26 ppm), DMSO-d6 (δ = 2.50 ppm), CD3CN (δ = 1.96 ppm), or CD3OD (δ = 3.34 ppm). All 13C NMR spectra (obtained with 1H decoupling) are reported in ppm relative to CDCl3 (δ = 77.00 ppm), DMSO-d6 (δ = 39.51 ppm), CD3CN (δ = 118.26 ppm), or CD3OD (δ = 49.86 ppm). The following abbreviations are used to designate multiplicities of NMR signals: s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet, quint = quintet, and dd (doublet of doublet). Coupling constants (J values) are reported in hertz (Hz).
Deprotection of N-tert-Butoxycarbonyl N′,N′-Disubstituted Hydrazines and Hydrazides; General Procedure A
To a solution of a Boc-hydrazine or hydrazide derivative (0.3 mmol) in CH2Cl2 (5 mL) was added TFA (0.5 mL) at 0 °C. The flask was placed in a fridge until no starting material was observed by TLC. Sat. aq Na2CO3 (10 mL) was added and the resulting mixture was stirred at 0 °C for 5 min, then the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic solution was washed with H2O (1 × 5 mL), brine (1 × 5 mL) and dried over anhydrous MgSO4. The solvents were removed under reduced pressure to afford the crude N′,N′-disubstituted hydrazine or hydrazide, which was purified by silica gel column chromatography. If the hydrazine or hydrazide was not stable on silica gel, it was then used directly in the next step.
Ruthenium(II)-Catalyzed Photolytic Cleavage of N–N Bonds; General Procedure B
A screw-capped, disposable test tube with a stir bar was charged with [Ru(bpyrz)3](PF6)2·2H2O (2 mol%), substituted hydrazine or hydrazide (0.5 mmol), and solvent (5 mL). A 20-gauge 11/2″ needle was pierced through the Teflon cap of the tube so that the reaction system was exposed to air. The reddish solution was stirred at r.t. under the irradiation of a 13 W GE compact fluorescent light bulb. The reaction was monitored by TLC. When the reaction was complete, the mixture was filtered through a short silica pad and eluted with Et2O (20 mL) and EtOAc (5 mL). The solution was concentrated under reduced pressure and the residue was purified by silica gel chromatography to afford the product. A photograph of the setup is shown in the Supporting Information.
N-Benzoylaniline (2a)22
Following general procedure B, 1a (106 mg, 0.5 mmol) in MeCN (2.5 mL) and MeOH (2.5 mL) was subjected to the reaction for 12 h. Purification on silica gel column chromatography (hexanes–Et2O, 5:1) gave the product.
Yield: 70 mg (71%); white crystals; mp 165–166 °C (Lit.22 165–167 °C).
1H NMR (400 MHz, DMSO-d6): δ = 10.25 (s, 1 H), 7.94–7.97 (m, 2 H), 7.79 (d, J = 8.0 Hz, 2 H), 7.51–7.61 (m, 3 H), 7.35 (t, J = 7.8 Hz, 2 H), 7.10 (t, J = 7.5 Hz, 1 H).
13C NMR (100 MHz, DMSO-d6): δ = 165.6, 139.2, 135.0, 131.6, 128.6, 128.4, 127.7, 123.7, 120.4.
N-Acetyl-N-methylaniline (2b)23
Following general procedure B, N-methyl phenylhydrazine (35 μL, 0.30 mmol) in MeCN (2 mL) and MeOH (1 mL) was subjected to the reaction for 24 h. The crude product was dissolved in EtOAc (5 mL), and AcCl (47 μL, 0.66 mol) was added. The solution was heated at 50 °C for 20 min, and then diluted with Et2O (60 mL). After washing with sat. aq NaHCO3 (1 × 5 mL), H2O (1 × 5 mL), and brine (1 × 5 mL), the solvent was removed in vacuo. The residue was purified by silica gel column chromatography (hexanes–acetone, 4:1) to give the product.
Yield: 37 mg (83%); white crystals; mp 108–109 °C (Lit.24 106–108 °C). The 1H and 13C NMR data are consistent with those reported.23
N-tert-Butoxycarbonylaniline (2c)25
Based on general procedure B, 1c (103 mg, 0.5 mmol) was subjected to the reaction in MeCN (2.5 mL) and MeOH (2.5 mL) for 24 h. After purification by silica gel column chromatography (hexanes–EtOAc, 10:1), the product was obtained.
Yield: 79 mg (82%); white crystals; mp 140–143 °C (Lit.26 134–136 °C). The 1H and 13C NMR data are consistent with those reported.25
N-tert-Butoxycarbonyl-4-trifluoromethylaniline (2d)27
Based on general procedure B, compound 1d (122 mg, 0.44 mmol) in MeCN (2.5 mL) and MeOH (2.5 mL) was subjected to the reaction for 28 h. Purification by silica gel column chromatography (hexanes–EtOAc, 20:1) gave the desired product.
Yield: 93 mg (81%); white crystals; mp 120–123 °C (Lit.27 121–122 °C). The 1H and 13C NMR data are consistent with those reported.27
N-Styrylacetylaniline (2e)28
Following the general procedure B, 1e (101 mg, 0.4 mmol) in MeCN (2.0 mL) and MeOH (2.0 mL) was subjected to the reaction for 10 h. Purification by silica gel column chromatography (hexanes–EtOAc, 6:1) gave the product.
Yield: 51 mg (54%); white crystals; mp 95–97 °C.
1H NMR (400 MHz, CDCl3): δ = 7.49–7.52 (m, 2 H), 7.41–7.44 (m, 2 H), 7.27–7.37 (m, 5 H), 7.11 (t, J = 7.4 Hz, 1 H), 6.65 (d, J = 15.9 Hz, 1 H), 6.39 (dt, J = 15.8, 7.3 Hz, 1 H), 3.34 (dd, J = 7.3, 1.2 Hz, 1 H).
13C NMR (100 MHz, CDCl3): δ = 168.8, 137.6, 136.4, 135.4, 129.0, 128.7, 128.0, 126.4, 124.5, 121.8, 119.9, 41.9.
(N-Phenylcarboxamido)pyrazine (2f)29
Following general procedure B, compound 1f (62 mg, 0.29 mmol) in MeCN (1.5 mL) and MeOH (1.5 mL) was irradiated for 28 h. Purification by silica gel column chromatography (hexanes–EtOAc, 2:1) gave the product.
Yield: 27 mg (47%); white crystals; mp 128–130 °C (Lit.30 123–125 °C). The 1H and 13C NMR data are consistent with those reported.29
N-Benzylaniline (2g)31
Based on general procedure B, crude 1g in MeCN (2.6 mL) and MeOH (1.3 mL) was subjected to the reaction for 24 h. After purified by silica gel column chromatography (hexanes–CH2Cl2, 2:1) the product was obtained as a yellow oil (51 mg, 68%). The 1H and 13C NMR data are consistent with those reported.31
N-Benzyl-2-chloroaniline (2h)32
Following general procedure B, crude 1h was subjected to the reaction in MeCN (3 mL) and MeOH (1.5 mL) for 15 h. After purification by silica gel column chromatography (hexanes–CH2Cl2, 10:1→8:1), the desire product (43 mg, 51%) was obtained as a colorless oil. The 1H and 13C NMR data are consistent with those reported.32
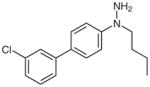
N-Butyl-(3′-chlorobiphenyl-4-yl)amine (2i)
Following general procedure B, compound 1i (87 mg, 0.32 mmol) in MeCN (2.0 mL) and MeOH (1.0 mL) was subjected to the reaction for 15 h. Purification by silica gel column chromatography (hexanes–EtOAc, 50:1) gave the product.
Yield: 31 mg (37%, two steps); colorless oil.
IR (neat): 3416, 3063, 3030, 2962, 2931, 2869, 1615, 1594, 1530, 1478, 1330, 824, 782 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.53 (t, J = 1.9 Hz, 1 H), 7.40–7.44 (m, 3 H), 7.31 (t, J = 7.8 Hz, 1 H), 7.21–7.24 (m, 1 H), 6.65–6.69 (m, 2 H), 3.75 (br s, 1 H), 3.17 (t, J = 7.1 Hz, 2 H), 1.60–1.68 (m, 2 H), 1.46 (sext, J = 7.4 Hz, 2 H), 0.99 (t, J = 7.4 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 148.4, 143.1, 134.5, 129.8, 128.2, 127.9, 126.2, 125.8, 124.2, 112.8, 43.5, 31.6, 20.3, 13.9.
Anal. Calcd for C16H18ClN: C, 73.98; H, 6.98; N, 5.39. Found: C, 73.99; H, 7.12; N, 5.51.
2-Methyl-1-methylene-2-phenylhydrazine (2k)33
Following general procedure B, compound 1k (68 mg, 0.5 mmol) in MeCN (2.5 mL) and MeOH (2.5 mL) was subjected to the cleavage reaction for 15 h. After purification by silica gel column chromatography (hexanes–CH2Cl2, 2:1), 2k was isolated as the major product.
Yield: 24 mg (36%); colorless oil. The 1H and 13C NMR data are consistent with those reported.33
1-Benzylidene-2-methyl-2-phenylhydrazine (2l)34
Following general procedure B, compound 1l (105 mg, 0.49 mmol) in MeCN (5 mL) was subjected to the cleavage reaction for 36 h. After purification by silica gel chromatography (hexanes–EtOAc, 30:1), 2l was obtained.
Yield: 91 mg (88%); white crystals; mp 107–109 °C (Lit.35 106–107 °C). The 1H and 13C NMR data are consistent with those reported.34
Azobenzene (2m)
Following general procedure B, 1m (67 mg, 0.34 mmol) in MeCN (1.5 mL) and MeOH (1.5 mL) was subjected to the reaction for 2 d. After purification by silica gel column chromatography (hexanes–EtOAc, 100:1), the product was obtained.
Yield: 36 mg (58%); yellow crystals.
1H NMR (400 MHz, CDCl3): δ = 7.94–7.98 (m, 4 H), 7.48–7.57 (m, 6H).
13C NMR (100 MHz, CDCl3): δ = 152.6, 131.0, 129.1, 122.8. NMR spectra are identical to those from an authentic sample.
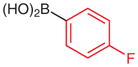
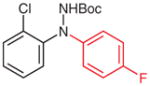
N-tert-Butoxycarbonyl-N′-(2-chlorophenyl)-N′-(4-fluorophenyl)hydrazine (5n)
Based on the literature procedure,19b a solution of 4n (300 mg, 1.25 mmol), 4-fluorophenylboronic acid (280 mg, 2.0 mmol), and Cu(OAc)2·H2O (12.5 mg, 0.0625 mmol) in dried MeOH (5 mL) was heated at reflux for 5 min in an atmosphere of N2. The solution was cooled to r.t. and 100 mg of silica gel was added. After stirring for 5 min, the solvent was removed and the residue was subjected to silica gel column chromatography (hexanes–EtOAc, 25:1) to give the pure product.
Yield: 398 mg (95%); white crystals; mp 107–109 °C.
IR (neat): 3307, 3067, 2983, 2935, 1729, 1708, 1509, 1481, 1370, 1230, 1160, 831 cm−1.
1H NMR (400 MHz, CD3OD): δ = 7.55 (d, J = 7.6 Hz, 1 H), 7.47 (dd, J = 8.0, 1.5 Hz, 1 H), 7.35 (t, J = 7.6 Hz, 1 H), 7.25 (t, J = 7.6 Hz, 1 H), 6.93 (t, J = 8.7 Hz, 2 H), 6.65–6.68 (m, 2 H), 1.48/1.37 (rotamers, 2 × s, 9 H).
13C NMR (100 MHz, CDCl3): δ = 157.2 (d, J = 240.0 Hz), 154.7, 143.4, 141.7, 131.9, 130.7, 130.2, 128.1, 127.9, 115.36 (d, J = 22.5 Hz), 114.7, 81.3, 28.1.
Anal. Calcd for C17H18ClFN2O2: C, 60.63; H, 5.39; N, 8.32. Found: C, 60.48; H, 5.29; N, 8.24.
N-(2-Chlorophenyl)-N-(4-fluorophenyl)hydrazine (1n)
Compound 5n (110 mg, 0.33 mmol) was subjected to general procedure A to give crude 1n as a reddish oil.
1H NMR (400 MHz, CDCl3): δ = 7.48–7.50 (m, 1 H), 7.28–7.35 (m, 2H), 7.20–7.24 (m, 1 H), 6.87–6.95 (m, 4 H), 4.23 (s, 2 H).
13C NMR (100 MHz, CDCl3): δ = 156.9 (d, J = 236.2 Hz), 146.1 (d, J = 1.5 Hz), 146.0, 131.9, 131.1, 128.1 (d, J = 22.2 Hz), 127.5, 115.7 (d, J = 7.5 Hz), 115.2, 115.0.
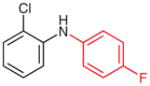
N-(2-Chlorophenyl)-4-fluoroaniline (2n)36
The crude compound 1n in MeCN (3.0 mL) and MeOH (1.5 mL) was subjected to general procedure B. Purification by silica gel column chromatography (hexanes–CH2Cl2, 6:1) gave the product.
Yield: 66 mg (91%); yellow oil.
1H NMR (400 MHz, CDCl3): δ = 7.35 (dd, J = 7.9, 1.4 Hz, 1 H), 7.01–7.17 (m, 6 H), 6.77–6.81 (m, 1 H), 6.01 (br s, 1 H).
13C NMR (100 MHz, CDCl3): δ = 159.0 (d, J = 240.7 Hz), 141.0, 137.3 (d, J = 2.5 Hz), 129.7, 127.5, 123.2 (d, J = 8.0 Hz), 120.8, 120.0, 116.1 (d, J = 22.4 Hz), 114.5.
N-tert-Butoxycarbonyl-N′-(4-trifluoromethylphenyl)-N′-(3-chlorophenyl)hydrazine (5o)
This compound was prepared according to the literature procedure.19b A mixture of 4o (300 mg, 1.09 mmol), 3-chlorophenylboronic acid (274 mg, 1.75 mmol), and Cu(OAc)2·H2O (11 mg, 0.055 mmol) in anhydrous MeOH (5 mL) was heated at reflux for 2 h. The solvent was removed with a rotary evaporator and the residue was purified by silica gel column chromatography (hexanes–EtOAc, 5:1) to give the product.
Yield: 400 mg (95%); white crystals; mp 119–121 °C.
IR (neat): 3284, 3072, 2986, 2936, 1710, 1619, 1597, 1524, 1480, 1372, 1329, 1251, 1163, 1117, 1075, 838 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.52 (d, J = 8.6 Hz, 2 H), 7.26 (t, J = 8.2 Hz, 1 H), 7.22–7.24 (m, 1 H), 7.13 (d, J = 8.6 Hz, 2 H), 7.09–7.11 (m, 2 H), 6.92/6.69 (rotamers, 2 × br s, 1 H), 1.50/1.32 (rotamers, 2 × br s, 9 H).
13C NMR (100 MHz, CDCl3): δ = 154.5, 148.6, 146.1, 135.0, 130.3, 126.4, 124.3 (q, J = 270.5 Hz), 124.5/124.3, 123.9 (q, J = 31.7 Hz), 121.4, 119.4/119.1, 116.9, 82.4/82.1, 28.2/28.0.
HRMS (ESI): m/z [M − H]− calcd for C18H18ClF3N2O2: 385.0925; found: 385.0928.
N′-(4-Trifluoromethylphenyl)-N′-(3-chlorophenyl)hydrazine (1o)
According to general procedure A, 5o (120 mg, 0.31 mmol) afforded crude 1o after 4 h at r.t.
Yield: 85 mg; yellow oil.
IR (neat): 3359, 3076, 2928, 1620, 1592, 1517, 1478, 1327, 1166, 1117, 1073, 839, 785 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.50 (d, J = 8.6 Hz, 2 H), 7.31 (t, J = 1.9 Hz, 1 H), 7.28–7.24 (m, 3 H), 7.18–7.15 (m, 1 H), 7.08–7.05 (m, 1 H), 4.20 (s, 2 H).
13C NMR (100 MHz, CDCl3): δ = 150.9, 148.8, 135.0, 130.3, 126.2 (q, J = 3.8 Hz), 124.5 (q, J = 269.4 Hz), 123.7, 122.6 (q, J = 32.5 Hz), 121.3, 119.3, 117.0.
N-(3-Chlorophenyl)-4-trifluoromethylaniline (2o)
According to general procedure B, crude 1o obtained above (ca. 0.31 mmol) in MeCN (1.5 mL) and MeOH (1.5 mL) was subjected to the cleavage reaction for 15 h to give 2o.
Yield: 73 mg (87%, 2 steps); off-white crystals; mp 60–63 °C.
IR (neat): 3414, 1597, 1584, 1532, 1327, 1117, 1070, 844, 769 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.51 (d, J = 8.6 Hz, 2 H), 7.23 (t, J = 8.1 Hz, 1 H), 7.13 (t, J = 2.1 Hz, 1 H), 7.08 (d, J = 8.6 Hz, 2 H), 7.00 (dd, J = 8.1, 2.1 Hz, 2 H), 5.92 (br s, 1 H).
13C NMR (100 MHz, CDCl3): δ = 145.6, 142.7, 135.1, 130.5, 126.8 (q, J = 3.7 Hz), 124.4 (q, J = 269.2 Hz), 122.6 (q, J = 32.6), 122.4, 118.9, 117.2, 116.3.
Anal. Calcd for C13H9ClF3N: C, 57.47; H, 3.34; N, 5.16. Found: C, 57.71; H, 3.23; N, 5.11.
N-tert-Butoxycarbonyl-N′-(3-fluorophenyl)-N′-phenylhydrazine (5p)
Isopropylmagnesium chloride (2 M in THF, 1.1 mL, 2.2 mmol) was added dropwise to a solution of 1-fluoro-3-iodobenzene (235 μL, 2 mmol) in THF (5 mL) at −20 °C. After 30 min, the solution was cooled to −95 °C and a solution of 4p (412 mg, 2 mmol) in THF (2 mL) was added. The reaction was quenched with sat. aq NH4Cl (5 mL) after 5 min. The mixture was extracted with EtOAc (3 × 20 mL), and the combined organic layers were washed with H2O (1 × 5 mL), brine (1 × 5 mL) and dried over anhydrous MgSO4. After being concentrated in vacuo, the residue was purified by silica gel column chromatography (hexanes–EtOAc, 20:1) to give 5p.
Yield: 581 mg (96%); white crystals; mp 123–125 °C.
IR (neat): 3289, 3071, 3040, 3006, 2983, 2936, 1711, 1613, 1594, 1493, 1374, 1252, 1164, 759 cm−1.
1H NMR (400 MHz, DMSO-d6): δ = 9.87/9.46 (rotamers, 2 × br s, 1 H), 7.36 (t, J = 7.8 Hz, 2 H), 7.27 (q, J = 7.8 Hz, 1 H), 7.16–7.20 (m, 2 H), 7.07–7.12 (m, 1 H), 6.63–6.77 (m, 3 H), 1.42/1.21 (rotamers, 2 × s, 9 H).
13C NMR (100 MHz, DMSO-d6): δ = 162.8 (d, J = 240.3 Hz), 155.1, 148.5 (d, J = 10.1 Hz), 145.1, 130.5 (d, J = 9.8 Hz), 129.2, 123.8, 121.1, 112.3, 107.2 (d, J = 21.0 Hz), 103.0 (d, J = 25.9 Hz), 79.8, 28.4.
Anal. Calcd for C17H19FN2O2: C, 67.53; H, 6.33; N, 9.27. Found: C, 67.42; H, 6.31; N, 9.18.
N-(3-Fluorophenyl)-N-phenylhydrazine (1p)
This crude compound was obtained from compound 5p (100 mg, 0.33 mol) following general procedure A.
1H NMR (400 MHz, CDCl3): δ = 7.34–7.39 (m, 2 H), 7.27–7.30 (m, 2 H), 7.15–7.21 (m, 1 H), 7.09–7.13 (m, 1 H), 6.90–6.95 (m, 2 H), 6.57–6.62 (m, 1 H), 4.18 (br s, 2 H).
13C NMR (100 MHz, CDCl3): δ = 163.5 (d, J = 242.0 Hz), 151.0 (d, J = 10.0 Hz), 148.4, 129.8 (d, J = 9.9 Hz), 129.4, 123.8, 121.8, 112.6 (d, J = 2.5 Hz), 107.0 (d, J = 21.4 Hz), 104.3 (d, J = 25.6 Hz).
N-(3-Fluorophenyl)aniline (2p)37
Following general procedure B, after 18 h, crude 1p in MeCN (2.0 mL) and MeOH (1.0 mL) gave the desired product after silica gel column chromatography (hexanes–Et2O, 8:1).
Yield: 52 mg (84%); yellow oil. The 1H and 13C NMR data are consistent with those reported.37
N-tert-Butoxycarbonyl-N′-(4-trifluoromethylphenyl)-N′-phenylhydrazine (5q)
According to the literature procedure,19a a solution of phenylmagnesium bromide (3 M in THF, 317 μL, 0.95 mmol) was added drop-wise to a solution of 4o (200 mg, 0.73 mmol) in anhydrous THF (5 mL) at −100 °C. After 15 min, the reaction was quenched by addition of sat. aq NH4Cl (5 mL). The mixture was extracted with Et2O (3 × 20 mL) and the combined organic layers were washed with H2O (1 × 5 mL), brine (1 × 5 mL), and dried over anhydrous MgSO4. After the solvents were removed by rotary evaporation, the residue was purified by silica gel column chromatography (hexanes–EtOAc, 10:1) to give the desired product.
Yield: 246 mg (96%); white crystals; mp 112–115 °C.
IR (neat): 3289, 2988, 2938, 1711, 1622, 1327, 1165, 1117, 1071, 836 cm−1.
1H NMR (400 MHz, DMSO-d6): δ = 9.97/9.55 (rotamers, 2 × br s, 1H), 7.57 (d, J = 8.6, 2 H), 7.41 (t, J = 7.7 Hz, 2 H), 7.26–7.29 (m, 2 H), 7.16–7.21 (m, 1 H), 7.00 (d, J = 8.6 Hz, 2 H), 1.43/1.22 (rotamers, 2 × s, 9 H).
13C NMR (100 MHz, CDCl3): δ = 156.2/155.0, 149.4, 144.6, 129.3, 126.1, 125.1/124.8, 124.5 (q, J = 269.2 Hz), 122.8/122.3, 122.3 (q, J = 32.5 Hz), 115.4/115.2, 82.0/81.5, 28.1/27.9.
Anal. Calcd for C18H19F3N2O2: C, 61.36; H, 5.44; N, 7.95. Found: C, 61.39; H, 5.36; N, 7.99.
N-(4-Trifluoromethylphenyl)aniline (2q)38
According to general procedure A, 5q (120 mg, 0.34 mmol) was subjected to the deprotection reaction for 1 h. The crude hydrazine 1q in MeCN (2.0 mL) and MeOH (1.0 mL) was subjected to the cleavage reaction for 4 h following general procedure B. After purification by silica gel column chromatography (hexanes–EtOAc, 40:1), compound 2q was obtained.
Yield: 69 mg (85%, two steps); yellow oil.
IR (neat): 3351, 3224, 3066, 3042, 2931, 2858, 1620, 1597, 1517, 1493, 1327, 1280, 1164, 1114, 1070, 839, 702.
The 1H and 13C NMR data are consistent with those reported.38
Anal. Calcd for C13H10F3N: C, 65.82; H, 4.25; N, 5.90. Found: C, 65.82; H, 4.27; N, 5.84.
N-tert-Butoxycarbonyl-N′-(2-methoxyphenyl)-N′-(4-trifluoromethylphenyl)hydrazine (5r)
To a solution of anisole (218 μL, 2 mmol) and TMEDA (362 μL, 2.4 mmol) in THF (10 mL), was added n-BuLi (1.6 M in THF, 1.5 mL, 2.4 mmol) at 0 °C. The solution was stirred at r.t. for 2 h, then 1.8 mL of the above solution was added to a solution of 4o (137 mg, 0.5 mmol) in THF (5 mL) at −78 °C. The reaction was quenched with sat. aq NH4Cl (5 mL) after 20 min. Purification by silica gel column chromatography (hexanes–Et2O, 4:1) afforded the desired product.
Yield: 151 mg (79%); yellow oil.
IR (neat): 3371, 3306, 3008, 2981, 2940, 2845, 1734, 1620, 1525, 1501, 1469, 1373, 1329, 1248, 1162, 1117, 1067, 834, 760 cm−1.
1H NMR (400 MHz, DMSO-d6): δ = 9.75/9.27 (2 × br s, 1 H), 7.49 (d, J = 8.7 Hz, 2 H), 7.42 (d, J = 6.8 Hz, 1 H), 7.31–7.36 (m, 1 H), 7.14–7.16 (m, 1 H), 7.03 (t, J = 7.6 Hz, 1 H), 6.61 (d, J = 7.8 Hz, 2H), 3.72 (s, 3 H), 1.42 (br s, 9 H).
13C NMR (100 MHz, DMSO-d6): δ = 155.2, 154.6, 150.7, 131.8, 128.6, 127.8, 126.0 (q, J = 3.6 Hz), 125.1 (q, J = 270.2 Hz), 121.1, 118.3 (q, J = 32.0 Hz), 112.8, 112.1, 79.8, 55.5, 28.1.
Anal. Calcd for C19H21F3N2O3: C, 59.68; H, 5.54; N, 7.33. Found: C, 59.74; H, 5.48; N, 7.31.
N-(2-Methoxyphenyl)-N-(4-trifluoromethylphenyl)hydrazine (1r)
Following general procedure A, compound 5r (135 mg, 0.35 mmol) afforded crude 1r.
1H NMR (400 MHz, CDCl3): δ = 7.37–7.40 (m, 2 H), 7.31–7.35 (m, 1 H), 7.26–7.29 (m, 1 H), 6.97–7.07 (m,4 H), 4.29 (br s, 2 H), 3.83 (s, 3 H).
13C NMR (100 MHz, CDCl3): δ = 155.3, 153.0, 135.1, 128.6, 128.5, 125.9 (q, J = 3.8 Hz), 125.0 (q, J = 270.5 Hz), 121.3, 119.4 (q, J = 32.2 Hz), 112.8, 112.4, 55.5.
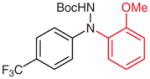
N-(2-Methoxyphenyl)-(4-trifluoromethyl)aniline (2r)
Following general procedure B, crude 1r in MeCN (2 mL) and MeOH (1 mL) was subjected to the cleavage reaction for 12 h. Another 2 mol% of the catalyst 3a was added and the reaction was complete after another 12 h. Purification by silica gel column chromatography (hexanes–Et2O, 10:1) afforded the desired product.
Yield: 61 mg (65%, two steps); pink crystals; mp 60–62 °C.
IR (neat): 3415, 3077, 3009, 2970, 2946, 2845, 1622, 1602, 1533, 1465, 1323, 1246, 1115, 1070, 1029, 750 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.50 (d, J = 8.7 Hz, 2 H), 7.36–7.39 (m, 1 H), 7.14 (d, J = 8.7 Hz, 2 H), 6.92–7.02 (m, 3 H), 3.90 (s, 3H).
13C NMR (100 MHz, CDCl3): δ = 149.3, 146.3, 130.8, 126.6 (q, J = 3.8 Hz), 124.6 (q, J = 270.6 Hz), 122.0, 121.7 (q, J = 32.7 Hz), 120.7, 117.2, 116.0, 110.8, 55.6.
Anal. Calcd for C14H12F3NO: C, 62.92; H, 4.53; N, 5.24. Found: C, 63.20; H, 4.31; N, 5.33.
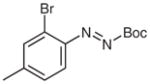
N-tert-Butoxycarbonyl-N′-(2-bromo-4-methylphenyl)-N′-(pyridn-3-yl)hydrazine (5s)
According to the literature procedure,19a i-PrMgCl·LiCl (1.3 M in THF, 1.5 mL, 2.0 mmol) was added dropwise to a solution of 3-bro-mopyridine (193 μL, 2.0 mmol) in THF (2 mL) at 0 °C. The solution was stirred at r.t. for 30 min, then cooled to −100 °C and a solution of 5i (598 mg, 2.0 mmol) in THF (5 mL) was added. The reaction was quenched after 20 min with sat. aq NH4Cl (5 mL). The mixture was extracted with Et2O (3 × 20 mL) and the combined organic layers were washed with H2O (1 × 5 mL), brine (1 × 5 mL), and dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes–EtOAc, 5:1→1:1) to give 5s.
Yield: 277 mg (73%); white crystals; mp 58–61 °C.
IR (neat): 3159, 2978, 2933, 1732, 1581, 1491, 1369, 1242, 1164 cm−1.
1H NMR (400 MHz, DMSO-d6): δ = 9.91/9.49 (rotamers, 2 × br s, 1 H), 8.02 (d, J = 4.0 Hz, 1 H), 7.84 (br s, 1 H), 7.55–7.57 (m, 1 H), 7.49 (d, J = 7.7 Hz, 1 H), 7.31 (d, J = 7.7 Hz, 1 H), 7.22 (dd, J = 8.5, 4.7 Hz, 1 H), 6.83 (d, J = 7.0 Hz, 1 H), 2.33 (s, 3 H), 1.42 (br s, 9 H).
13C NMR (100 MHz, CDCl3): δ = 154.6, 143.4, 140.9, 140.1, 139.2, 135.2, 134.2, 131.0, 129.7, 123.2, 122.6, 119.3, 81.6, 28.2, 20.8.
HRMS (ESI): m/z [M + H]+ calcd for C17H21BrN3O2: 378.0812; found: 378.0812.
N-(2-Bromo-4-methylphenyl)-N-(pyridin-3-yl)hydrazine (1s)
Compound 5s (78 mg, 0.21 mmol) was subjected to general procedure A. Purification by silica gel column chromatography (CH2Cl2–Et2O, 2:1) gave 1s.
Yield: 24 mg (42%); colorless oil.
IR (neat): 3338, 3310, 3167, 3037, 2959, 2926, 2856, 1584, 1488, 1428, 1319, 1249, 1044, 800, 710 cm−1.
1H NMR (400 MHz, CDCl3): δ = 8.24 (d, J = 2.8 Hz, 1 H), 8.05 (dd, J = 4.6, 1.3 Hz, 1 H), 7.52–7.53 (m, 1 H), 7.17–7.22 (m, 3 H), 7.08 (ddd, J = 8.4, 4.6, 0.7 Hz, 1 H), 4.19 (s, 2 H), 2.38 (s, 3 H).
13C NMR (100 MHz, CDCl3): δ = 145.7, 143.2, 139.8, 139.4, 136.0, 134.8, 130.0, 128.7, 123.1, 122.5, 119.9, 20.8.
HRMS (ESI): m/z [M + H]+ calcd for C12H13BrN3: 278.0288; found: 278.0287.
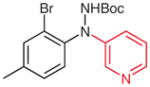
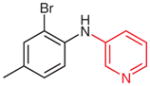
N-(Pyridn-3-yl)-2-bromo-4-methylaniline (2s)
Following general procedure B, 1s (58 mg, 0.21 mmol) in MeCN (2.0 mL) and MeOH (1.0 mL) was subjected to the cleavage reaction for 6 h. After purification by silica gel column chromatography (hexanes–acetone, 4:1) the product was obtained.
Yield: 30 mg (55%); yellow oil.
IR (neat): 3393, 3235, 3032, 2923, 1587, 1517, 1485, 1314, 806, 710 cm−1.
1H NMR (400 MHz, CDCl3): δ = 8.42 (s, 1 H), 8.22 (d, J = 4.3 Hz, 1 H), 7.38–7.41 (m, 2 H), 7.20 (dd, J = 8.2, 4.6 Hz, 1 H), 7.14 (d, J = 8.2 Hz, 1 H), 7.01–7.04 (m, 1 H), 5.90 (s, 1 H), 2.29 (s, 3 H).
13C NMR (100 MHz, CDCl3): δ = 142.7, 141.0, 139.1, 137.5, 133.5, 132.6, 128.9, 124.8, 123.7, 117.4, 113.8, 20.3.
HRMS (EI): m/z [M]+ calcd for C12H11BrN2: 262.0106; found: 262.0101.
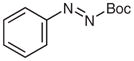
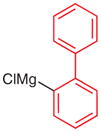
N-tert-Butoxycarbonyl-N′-(2-byphenyl)-N′-phenylhydrazine (5t)
To a solution of 2-bromobiphenyl (259 μL, 1.5 mmol) in THF (5 mL) was added i-PrMgCl·LiCl (1.3 M in THF, 1.3 mL, 1.65 mmol) at 0 °C. The solution was stirred at r.t. for 15 h (GC analysis showed incomplete halogen exchange) and cooled to −78 °C. A solution of 4p (309 mg, 1.5 mmol) in THF (2 mL) was added and the reaction was quenched with sat. aq NH4Cl (5 mL) after 30 min. The mixture was extracted with Et2O (3 × 20 mL), and the combined organic layers were washed with H2O (1 × 5 mL), brine (1 × 5 mL) and then dried over anhydrous MgSO4. After being concentrated in vacuo, the residue was purified by silica gel column chromatography (hexanes–CH2Cl2, 3:1→1:1) to give 5t.
Yield: 270 mg (50%); colorless oil.
IR (neat): 3373, 3320, 3065, 2982, 2934, 1744, 1726, 1706, 1599, 1500, 1477, 1162, 750, 705 cm−1.
1H NMR (400 MHz, DMSO-d6): δ = 9.71/9.08 (2 × s, 1 H), 7.50–7.68 (m, 3 H), 7.41–7.47 (m, 1 H), 7.29–7.33 (m, 2 H), 7.18 (t, J = 7.3 Hz, 2 H), 7.07–7.12 (m, 1 H), 6.91–6.99 (m, 2 H), 1.45/1.31 (2 × s, 9 H).
13C NMR (100 MHz, DMSO-d6): δ = 155.6, 146.9, 142.6, 139.4, 136.7, 130.9, 128.8, 128.1, 127.8, 126.7, 126.1, 125.1, 119.3, 114.5, 79.4, 28.2.
Anal. Calcd for C23H24N2O2: C, 76.64; H, 6.71; N, 7.77. Found: C, 76.50; H, 6.61; N, 7.78.
N-(2-Biphenyl)-N-phenylhydrazine (1t)
Following general procedure A, 5t (180 mg, 0.5 mmol) was treated at 0 °C for 3 h to give crude 1t (126 mg).
1H NMR (400 MHz, CDCl3): δ = 7.47–7.51 (m, 3 H), 7.32–7.41 (m, 6 H), 7.23–7.28 (m, 2 H), 7.05–7.08 (m, 2 H), 6.81–6.86 (m, 1 H), 3.63 (s, 2 H).
13C NMR (100 MHz, CDCl3): δ = 150.3, 146.4, 139.5, 139.1, 131.7, 128.7 (2 × C), 128.6, 128.3, 127.4, 126.9, 126.6, 118.2, 113.8.
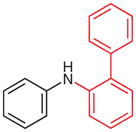
N-(2-Biphenyl)aniline (2t)39
Following general procedure A, crude 1t in MeCN (3 mL) and MeOH (1.5 mL) was subjected to the cleavage reaction for 24 h. After purification by silica gel column chromatography (hexanes–Et2O, 5:1), 2t was obtained.
Yield: 88 mg (72%, two steps); as yellow oil.
IR (neat): 3409, 3053, 3029, 1584, 1593, 1516, 1503, 1474, 1438, 1314, 747, 705 cm−1.
The 1H and 13C NMR data are consistent with those reported.39
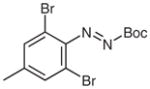
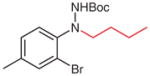
N-tert-Butoxycarbonyl-N′-(2-bromo-4-methylphenyl)-N′-butylhydrazine (5u)
According to the literature procedure,19a n-butylmagnesium bromide (2 M in THF, 0.35 mL, 0.67 mmol) was added to a solution of 4s (200 mg, 0.67 mmol) in THF (5 mL) at −100 °C. The color of the reactant turned from red to yellow during the addition. The reaction was quenched with sat. aq NH4Cl (5 mL) after 15 min, the mixture was extracted with Et2O (3 × 20 mL), and the combined organic layers were washed with H2O (1 × 5 mL), brine (1 × 5 mL) and then dried over MgSO4. After being concentrated in vacuo, the residue was purified by silica gel column chromatography to give the product.
Yield: 220 mg (92%); yellow oil.
IR (neat): 3247, 2965, 2933, 2874, 1701, 1493, 1371, 1169 cm−1.
1H NMR (400 MHz, DMSO-d6): δ = 8.78/8.40 (rotamers, 2 × s, 1H), 7.36 (s, 1 H), 7.24 (d, J = 8.0 Hz, 1 H), 7.11 (d, J = 8.0 Hz, 1 H), 2.96–3.13 (m, 2 H), 2.23/2.17 (rotamers, 2 × s, 3 H), 1.47–1.55 (m, 2 H), 1.26–1.44 (m, 11 H), 0.87 (t, J = 7.4 Hz, 3 H).
13C NMR (100 MHz, DMSO-d6): δ = 154.4, 147.3, 134.6, 133.4, 128.6, 122.1, 116.9, 78.5, 55.7, 29.2, 28.1, 19.8, 19.5, 14.1.
Anal. Calcd for C16H25BrN2O2: C, 53.79; H, 7.05; N, 7.84. Found: C, 54.05; H, 7.14; N, 7.82.
N-Butyl-N-(2-bromo-4-methylphenyl)hydrazine (1u)
Following general procedure A, 5u (120 mg, 0.34 mmol) afforded the crude product (85 mg) as a yellow oil that was used directly in the next step.
1H NMR (400 MHz, CDCl3): δ = 7.37–7.39 (m, 1 H), 7.18 (d, J = 8.1 Hz, 1 H), 7.06–7.10 (m, 1 H), 3.56 (br s, 2 H), 3.01–3.05 (m, 2 H), 2.28 (s, 3 H), 1.60–1.69 (m, 2 H), 1.40 (quin, J = 7.4 Hz, 2 H), 0.94 (t, J = 7.4 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 149.5, 134.9, 133.9, 128.6, 120.7, 118.9, 59.6, 29.5, 20.3, 20.1, 14.0.
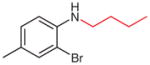
N-Butyl-2-bromo-4-methylaniline (2u)40
Based on general procedure B, compound 1u (crude, 85 mg, ca. 0.33 mol) in MeCN (2.0 mL) and MeOH (1.0 mL) was subjected to the cleavage reaction for 15 h and then purified by silica gel column chromatography (hexanes–EtOAc, 50:1) to give the desired product.
Yield: 57 mg (70%).
1H NMR (400 MHz, CDCl3): δ = 7.24–7.25 (m, 1 H), 6.97–7.00 (m, 1H), 6.55 (d, J = 8.3 Hz, 1 H), 4.10 (br s, 1 H), 3.13 (q, J = 6.7 Hz, 2 H), 2.22 (s, 3 H), 1.61–1.68 (m, 2 H), 1.45 (quin, J = 7.4 Hz, 2 H), 0.97 (t, J = 7.4 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 142.9, 132.7, 128.9, 126.9, 111.2, 109.5, 43.8, 31.4, 20.3, 20.0, 13.9.
MS (CI): m/z [M + H]+ calcd for C11H17BrN2: 242; found: 242.
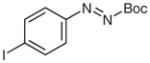
N-tert-Butoxycarbonyl-N′-butyl-N′-(4-iodophenyl)hydrazine (5v)
According to the literature procedure,19a n-butylmagnesium chloride (2.0 M in THF, 375 μL, 0.75 mmol) was added dropwise to a solution of 4v (250 mg, 0.75 mmol) in THF (10 mL) at −100 °C. The solution color turned from orange to green. After 5 min, the reaction was quenched with sat. aq NH4Cl (5 mL), the mixture was extracted with Et2O (3 × 20 mL), and the combined organic layers were washed with H2O (1 × 5 mL), brine (1 × 5 mL), and dried over anhydrous MgSO4. After being concentrated in vacuo, the residue was purified by silica gel column chromatography (hexanes–EtOAc, 100:5) to give the product.
Yield: 280 mg (95%); yellow crystals; mp 72–75 °C.
IR (neat): 3299, 2965, 2934, 2874, 1709, 1590, 1489, 1368, 1251, 1163, 813 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.45–7.49 (m, 2 H), 6.56–6.60 (m, 2 H), 6.42/6.23 (rotamers, 2 × br s, 1 H), 3.34–3.48 (m, 2 H), 1.56–1.64 (m, 2 H), 1.32–1.48 (m, 11 H), 0.95 (t, J = 7.3 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 154.6, 149.0, 137.8, 114.8, 81.1, 80.5, 52.1, 28.6, 28.3, 20.2, 14.0.
Anal. Calcd for C15H23IN2O2: C, 46.16; H, 5.94; N, 7.18. Found: C, 46.36; H, 6.03; N, 7.25.
N-Butyl-N-(4-iodophenyl)hydrazine (1v)
Based on general procedure A, 5v (170 mg, 0.436 mmol) gave the crude product (115 mg, 91%) as a yellow oil.
1H NMR (400 MHz, CDCl3): δ = 7.45–7.49 (m, 2 H), 6.73–6.77 (m, 2 H), 3.54 (br s, 2 H), 3.56 (t, J = 7.5 Hz, 2 H), 1.57–1.64 (m, 2 H), 1.38 (sext, J = 7.5 Hz, 2 H), 0.97 (t, J = 7.4 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 151.1, 137.5, 115.2, 78.8, 55.1, 27.6, 20.1, 14.0.
N-Butyl-4-iodoaniline (2v)41
The crude product 1v (ca. 0.4 mmol) was subjected to the cleavage reaction in MeCN (4.0 mL) following general procedure B. After 10 h, another 2 mmol% of catalyst was added. After another 10 h, the reaction mixture was loaded onto a silica gel column (hexanes–CH2Cl2, 3:1→2:1) to give the desired product.
Yield: 23 mg (21%); yellow oil.
1H NMR (400 MHz, CDCl3): δ = 7.39–7.42 (m, 2 H), 6.36–6.40 (m, 2 H), 3.63 (br s, 1 H), 3.07 (t, J = 7.1 Hz, 2 H), 1.55–1.62 (m, 2 H), 1.42 (sext, J = 7.2 Hz, 2 H), 0.96 (t, J = 7.3 Hz, 3 H).
13C NMR (100 MHz, CD3CN): δ = 149.8, 138.5, 115.6, 76.5, 43.8, 31.9, 20.9, 14.1.
1-(N-tert-Butoxycarbonylamino-N-phenyl-DL-alanyl)pyrrolidine (5w)
Freshly prepared LDA in THF (0.26 M, 1.83 mmol) was added to 1-propionylpyrrolidine (234 mg, 1.83 mmol) in THF (5 mL) at −20 °C. After 1 h, the solution was cooled to −78 °C and a solution of 4p (170 mg, 0.825 mmol) in THF (2 mL) was added. The reaction was quenched with sat. aq NH4Cl (5 mL) after 30 min, the mixture was extracted with EtOAc (3 × 20 mL) and the combined organic layers were washed with H2O (1 × 5 mL), brine (1 × 5 mL) and then dried over anhydrous MgSO4. After being concentrated in vacuo, the residue was purified by silica gel column chromatography (hexanes–EtOAc, 3:2) to give 5w.
Yield: 195 mg (71%); colorless oil.
IR (neat): 3472, 3362, 3258, 3065, 2981, 2934, 2881, 1738, 1637, 1456, 1234, 1168, 759 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.72 (br s, 1 H), 7.21–7.31 (m, 2H), 6.79–6.89 (m, 3 H), 4.67 (q, J = 6.9 Hz, 1 H), 3.33–3.57 (m, 4H), 1.98–2.06 (m, 2 H), 1.76–1.88 (m, 2 H), 1.43–1.54 (m, 11 H).
13C NMR (100 MHz, CDCl3): δ = 173.1, 156.2, 149.6, 129.1, 120.5, 114.2, 79.9, 56.7, 46.1, 45.6, 28.3, 26.2, 23.9, 14.7.
Anal. Calcd for C18H27N3O3: C, 64.84; H, 8.16; N, 12.6. Found: C, 64.82; H, 8.26; N, 12.33.
1-(N-Amino-N-phenyl-DL-alanyl)pyrrolidine (1w)
Following general procedure A, 5w (120 mg, 0.36 mmol) afforded crude 1w (78 mg).
1H NMR (400 MHz, CDCl3): δ = 7.23–7.28 (m, 2 H), 7.03–7.06 (m, 2 H), 6.79–6.83 (m, 1 H), 4.60 (q, J = 6.8 Hz, 1 H), 3.73 (br s, 2 H), 3.40–3.55 (m, 4 H), 1.77–1.95 (m, 4 H), 1.39 (d, J = 6.8 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 170.8, 150.8, 129.1, 118.9, 114.0, 57.4, 46.2, 45.9, 26.2, 24.0, 11.7.
1-(N-Phenyl-DL-alanyl)pyrrolidine (2w)42
Following general procedure B, crude 1w in MeCN (2 mL) and MeOH (2 mL) was subjected to the cleavage reaction for 24 h. After purification by silica gel column chromatography (hexanes–EtOAc, 1:1), the product 2w was obtained.
Yield: 40 mg (51%, two steps); white crystals; mp 111–112 °C (Lit.42 113–115 °C).
IR (neat): 3320, 3116, 3050, 2976, 2923, 2872, 2854, 1643, 1427, 1643, 1332, 756, 693 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.14–7.19 (m, 2 H), 6.68–6.73 (m, 1 H), 6.56–6.63 (m, 2 H), 4.63 (br s, 1 H), 4.25 (q, J = 6.6 Hz, 1 H), 3.44–3.63 (m, 4 H), 1.97–2.03 (m, 2 H), 1.84–1.91 (m, 2 H), 1.38 (d, J = 6.6 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 172.0, 146.7, 129.3, 117.7, 113.5, 50.5, 46.2, 46.0, 26.1, 24.1, 18.0.
MS (CI): m/z (%) = 219 (100) [M + H]+.
Acknowledgments
We thank the University of Arkansas, the Arkansas Bioscience Institute, and the NIH NCRR COBRE grant (P20 RR15569) for generous support of this research. Core facilities were funded by the NIH NCRR COBRE grant (P20 RR15569) and the Arkansas Bioscience Institute. We also thank Prof. Bill Durham for insightful suggestions and technique assistance in early screening experiments.
Footnotes
Supporting Information for this article is available online at http://www.thieme-connect.com/ejournals/toc/synthesis.
References
- 1.For reviews, see: Zeitler K. Angew Chem Int Ed. 2009;48:9785. doi: 10.1002/anie.200904056.Ravelli D, Dondi D, Fagnoni M, Albini A. Chem Soc Rev. 2009;38:1999. doi: 10.1039/b714786b.Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n.
- 2.For photophysical and redox properties of Ru(II), see: Campagana S, Puntoriero F, Nastasi F, Bergamini G, Balzani V. Top Curr Chem. 2007;280:117.Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, von Zelewsky A. Coord Chem Rev. 1988;84:85.Kalyanasundaram K, Grätzel M. Coord Chem Rev. 1998;177:347.Kalyanasundaram K. Coord Chem Rev. 1982;46:159.Balzani V, Bergamini G, Marchioni F, Ceroni P. Coord Chem Rev. 2006;250:1254.
- 3.For photophysical and redox properties of Ir(III), see: Lowry MS, Bernhard S. Chem Eur J. 2006;12:7970. doi: 10.1002/chem.200600618.Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top Curr Chem. 2007;281:143.
- 4.For early applications of Ru(II) as a photocatalyst in organic synthesis, see: DeLaive PJ, Giannotti C, Whitten DG. J Am Chem Soc. 1978;100:7413.DeLaive PJ, Sullivan BP, Meyer TJ, Whitten DG. J Am Chem Soc. 1979;101:4007.DeLaive PJ, Foreman TK, Giannotti C, Whitten DG. J Am Chem Soc. 1980;102:5627.Montserrat K, Foreman TK, Gratzel M, Whitten DG. J Am Chem Soc. 1981;103:6667.Pac C, Ihama M, Yasuda M, Miyauchi Y, Sakurai H. J Am Chem Soc. 1981;103:6495.Cano-Yelo H, Deronzier A. J Chem Soc, Perkin Trans. 1984;2:1093.Ishitani O, Yanagida S, Takamuku S, Pac C. J Org Chem. 1987;52:2790.Zen JM, Liou SL, Kumar AS, Hsia MS. Angew Chem Int Ed. 2003;42:577. doi: 10.1002/anie.200390166.Okada K, Okamoto K, Morita N, Okubo K, Oda M. J Am Chem Soc. 1991;113:9401.
- 5.Recent applications of Ru(II) and Ir(III) as photocatalysts in organic synthesis. Stephenson’s group: Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756. doi: 10.1021/ja9033582.Tucker JW, Narayanam JMR, Krabbe SW, Stephenson CRJ. Org Lett. 2010;12:368. doi: 10.1021/ol902703k.Tucker JW, Nguyen JD, Narayanam JMR, Krabbe SW, Stephenson CRJ. Chem Commun. 2010;46:4985. doi: 10.1039/c0cc00981d.Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org Lett. 2010;12:3104. doi: 10.1021/ol101146f.Condie AG, González-Gómez JC, Stephenson CRJ. J Am Chem Soc. 2010;132:1464. doi: 10.1021/ja909145y.MacMillan’s group: Nicewicz D, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976.Nagib DA, Scott ME, MacMillan DWC. J Am Chem Soc. 2009;131:10875. doi: 10.1021/ja9053338.Shih HW, Vander Wal MN, Grange RL, MacMillan DWC. J Am Chem Soc. 2010;132:13600. doi: 10.1021/ja106593m.Yoon’s group: Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886. doi: 10.1021/ja805387f.Du J, Yoon TP. J Am Chem Soc. 2009;131:14604. doi: 10.1021/ja903732v.Yoon TP, Ischay MA, Du J. Nature Chem. 2010;2:527. doi: 10.1038/nchem.687.Ischay MA, Lu Z, Yoon TP. J Am Chem Soc. 2010;132:8572. doi: 10.1021/ja103934y.Other groups: Koike T, Akita M. Chem Lett. 2009;38:166.Hasegawa E, Takizawa S, Seida T, Yamaguchi A, Yamaguchi N, Chiba N, Takahashi T, Ikeda H, Akiyama K. Tetrahedron. 2006;62:6581.Inagaki A, Edure S, Yatsuda S, Akita M. Chem Commun. 2005:5468. doi: 10.1039/b508013d.
- 6.For dyes, see: Hedstrand DM, Kruizinga WH, Kellogg RM. Tetrahedron Lett. 1978:1255.Neumann M, Füldner S, König B, Zeitler K. Angew Chem Int Ed. 2011;50:951. doi: 10.1002/anie.201002992.
- 7.(a) Sapountzis I, Knochel P. Angew Chem Int Ed. 2004;43:897. doi: 10.1002/anie.200353241. [DOI] [PubMed] [Google Scholar]; (b) Liu X, Barry M, Tsou HR. Tetrahedron Lett. 2007;48:8409. [Google Scholar]; (c) Enders D, Noll S, Raabe G, Runsink J. Synthesis. 2008:1288. [Google Scholar]
- 8.For selected reductive examples, see: Alonso F, Candela P, Gómez C, Yus M. Adv Synth Catal. 2003;345:275.Seino H, Masumori T, Hidai M, Mizobe Y. Organometallics. 2003;22:3424.Sinha P, Kofink CC, Knochel P. Org Lett. 2006;8:3841. doi: 10.1021/ol061303m.Enders D, Funabiki K. Org Lett. 2001;3:1575. doi: 10.1021/ol015869g.
- 9.For selected oxidative examples, see: Tang Q, Zhang C, Luo M. J Am Chem Soc. 2008;130:5840. doi: 10.1021/ja711153b.Lim Y, Pang S, Paik S, Cho C. Bull Korean Chem Soc. 2003;24:543.Lee K, Lim Y, Cho C. Tetrahedron Lett. 2002;43:7463.Prata JV, Clemente DTS, Prabhakar S, Lobo AM, Mourata I, Branco PS. J Chem Soc, Perkin Trans. 2002;1:513.Fernández R, Ferrete A, Llera JM, Magriz A, Martín-Zamora E, Díaz E, Lassaletta JM. Chem Eur J. 2004;10:737. doi: 10.1002/chem.200305501.
- 10.(a) Magnus P, Garizi N, Seibert KA, Ornholt A. Org Lett. 2009;11:5646. doi: 10.1021/ol902313v. [DOI] [PubMed] [Google Scholar]; (b) Winter AH, Thomas SI, Kung AC, Falvey DE. Org Lett. 2004;6:4671. doi: 10.1021/ol048250y. [DOI] [PubMed] [Google Scholar]
- 11.Lebrun S, Couture A, Deniau E, Grandclaudon P. Synlett. 2009:2621. [Google Scholar]
- 12.(a) Rillema DP, Allen G, Meyer TJ, Conrad D. Inorg Chem. 1983;22:1617. [Google Scholar]; (b) Crutchley RJ, Lever ABP. J Am Chem Soc. 1980;102:7128. [Google Scholar]
- 13.The abbreviation bpyrz stands for 2,2′-bipyrazine.
- 14.The acetylation was performed because of the volatility of N-methylaniline.
- 15.Beeston RF, Aldridge WS, Treadway JA, Fitzgerald MC, DeGraff BA, Stitzel SE. Inorg Chem. 1998;37:4368. doi: 10.1021/ic971322f. [DOI] [PubMed] [Google Scholar]
- 16.(a) Neckers DC. J Photochem Photobiol, A. 1989;47:1. [Google Scholar]; (b) Alberti MN, Vougioukalakis GC, Orfanopoulos M. J Org Chem. 2009;74:7274. doi: 10.1021/jo9012942. [DOI] [PubMed] [Google Scholar]
- 17.For selected recent reviews, see: Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054. doi: 10.1021/cr8002505.Ma D, Cai Q. Acc Chem Res. 2008;41:1450. doi: 10.1021/ar8000298.Monnier F, Taillefer M. Angew Chem Int Ed. 2009;48:6954. doi: 10.1002/anie.200804497.Surry DS, Buchwald SL. Chem Sci. 2010;1:13. doi: 10.1039/C0SC00107D.
- 18.For selected recent reviews, see: Hartwig JF. Acc Chem Res. 2008;41:1534. doi: 10.1021/ar800098p.Surry DS, Buchwald SL. Chem Sci. 2011;2:27. doi: 10.1039/C0SC00331J.
- 19.(a) Tšubrik O, Sillard R, Mäeorg U. Synthesis. 2006:843. [Google Scholar]; (b) Kisseljova K, Tšubrik O, Sillard R, Mäeorg S, Mäeorg U. Org Lett. 2006;8:43. doi: 10.1021/ol052403f. [DOI] [PubMed] [Google Scholar]
- 20.(a) Ishikawa S, Tsuji S, Sawaki Y. J Am Chem Soc. 1991;113:4282. [Google Scholar]; (b) Harder T, Wessig P, Bendig J, Stösser R. J Am Chem Soc. 1999;121:6580. [Google Scholar]
- 21.Lakowicz JR, editor. Principles of Fluorescence Spectroscopy. Vol. 8. Springer; Berlin: 2006. [Google Scholar]
- 22.Hu Z, Ye W, Zou H, Yu Y. Synth Commun. 2010;40:222. [Google Scholar]
- 23.Tobisu M, Nakamura R, Kita Y, Chatani N. J Am Chem Soc. 2009;131:3174. doi: 10.1021/ja810142v. [DOI] [PubMed] [Google Scholar]
- 24.Lusch MJ, Woller KR, Keller AM, Turk MC. Synthesis. 2005:551. [Google Scholar]
- 25.Moraczewski AL, Banaszynski LA, From AM, White CE, Smith BD. J Org Chem. 1998;63:7258. doi: 10.1021/jo980644d. [DOI] [PubMed] [Google Scholar]
- 26.Bailey WJ, Griffith JR. J Org Chem. 1978;43:2690. [Google Scholar]
- 27.Swenson RE, Sowin TJ, Zhang HQ. J Org Chem. 2002;67:9182. doi: 10.1021/jo0203387. [DOI] [PubMed] [Google Scholar]
- 28.Lewis FD, Wagner-Brennan JM, Miller AM. Can J Chem. 1999;77:595. [Google Scholar]
- 29.Takács A, Jakab B, Petz A, Kollár L. Tetrahedron. 2007;63:10372. [Google Scholar]
- 30.McKenzie WL, Foye WO. J Med Chem. 1972;15:570. doi: 10.1021/jm00275a040. [DOI] [PubMed] [Google Scholar]
- 31.Milburn RR, Snieckus V. Angew Chem Int Ed. 2004;43:892. doi: 10.1002/anie.200352634. [DOI] [PubMed] [Google Scholar]
- 32.Bedford RB, Cazin CSJ. Chem Commun. 2002:2310. doi: 10.1039/b207712b. [DOI] [PubMed] [Google Scholar]
- 33.Kamitori Y, Hojo M, Msuda R, Yoshida T, Ohara S, Yamada K, Yoshikawa N. J Org Chem. 1988;53:519. [Google Scholar]
- 34.Mino T, Shirae Y, Sakamoto M, Fujita T. J Org Chem. 2005;70:2191. doi: 10.1021/jo048107i. [DOI] [PubMed] [Google Scholar]
- 35.Aylward JB. J Chem Soc C. 1970:1494. [Google Scholar]
- 36.Thu-Cuc ST, Buu-Hoi NP, Xuong ND. J Heterocycl Chem. 1964;1:28. [Google Scholar]
- 37.Desmarets C, Schneider R, Fort Y. J Org Chem. 2002;67:3029. doi: 10.1021/jo016352l. [DOI] [PubMed] [Google Scholar]
- 38.Yu Y, Srogl J, Liebeskind LS. Org Lett. 2004;6:2631. doi: 10.1021/ol048982q. [DOI] [PubMed] [Google Scholar]
- 39.Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald S. J Am Chem Soc. 2003;125:6653. doi: 10.1021/ja035483w. [DOI] [PubMed] [Google Scholar]
- 40.Commercially available. CAS number: 1019542-99-4.
- 41.Okano K, Tokuyama H, Fukuyama T. Org Lett. 2003;5:4987. doi: 10.1021/ol035942y. [DOI] [PubMed] [Google Scholar]
- 42.Wright WB, Jr, Brabander HJ, Hardy HA., Jr J Org Chem. 1961;26:476. [Google Scholar]