

Abstract
Although the prevalence of neurodegenerative diseases is increasing as a consequence of the growing aging population, the exact pathophysiological mechanisms leading to these diseases remains obscure. Multiple sclerosis (MS), an autoimmune disease of the central nervous system and the most frequent cause of disability among young people after traumatic brain injury, is characterized by inflammatory/demyelinating and neurodegenerative processes that occurr earlier in life. The ability to make an early diagnosis of MS with the support of conventional MRI techniques, provides the opportunity to study neurodegeneration and the underlying pathophysiological processes in earlier stages than in classical neurodegenerative diseases. This review summarizes mechanisms of neurodegeneration common to MS and to Alzheimer disease, Parkinson disease, and amiotrophic lateral sclerosis, and provides a brief overview of the neuroimaging studies employing MRI and PET techniques to investigate and monitor neurodegeneration in both MS and classical neurodegenerative diseases.
Keywords: MRI, multiple sclerosis, neurodegenerative diseases, microglia, iron
Introduction
Primary neurodegenerative diseases such as Alzheimer disease (AD), Parkinson disease (PD) and Amiotrophic Lateral Sclerosis (ALS) are characterized by a progressive neuroaxonal injury that becomes symptomatic in the late stages of the disease when 60–80% of neuron have already been lost.1 This makes it difficult to study the pathological mechanisms underlying the onset and progression of these conditions and limits the development of effective neuroprotective treatments. AD is the most common cause of acquired deterioration of cognitive functioning and emotional capacities leading to dementia. At histological examination, the disease is characterized by the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles.2 PD is characterized by asymmetrical bradikinesia, muscular rigidity, resting tremor and, in a later phase, by postural instability, bulbar dysfunction and impaired autonomic reflexes. The neuropathological hallmarks of the disease are the loss of cells in the Substantia Nigra (SN) pars compacta and the presence of neuronal Lewy inclusion bodies, resulting in a deficiency of dopamine in the striatum.1 ALS is a characterized by progressive degeneration of upper and lower motoneuron in corticospinal tract (CST), brain stem and spine3.
Multiple Sclerosis (MS) is a chronic inflammatory disease of the Central Nervous System (CNS) characterized, in the early phase, by massive leukocyte infiltration involving helper CD4+ T cells and extensive microglial activation that lead to axon demyelination and to neuroaxonal degeneration and brain atrophy that becomes more dominant as the disease progresses.4 First described by Charcot,5 axonal damage is a key feature of MS pathogenesis and correlates with permanent neurological deficits in MS patients.6 Since MS is most frequent between the second and third decades of life and it is diagnosed earlier than classical neurodegenerative diseases, it can provide a model for studying the pathogenesis of neurodegeneration. Magnetic resonance imaging (MRI) not only provides an useful tool for supporting clinical diagnosis, especially in MS, but provides advanced and sophisticated techniques to investigate disease pathology in-vivo and to monitor the pathological processes driving the progression of neurodegenerative diseases.7 In the present review, we will first summarize some of the pathological processes common to MS and classical neurodegenerative diseases such as iron accumulation, mitochondrial dysfunction and microglia activation; then we will discuss the main findings of imaging studies that have employed quantitative MRI and PET techniques to detect and monitor neurodegeneration in MS, AD, PD and ALS.
Iron deposition
Iron is essential for neuronal metabolism, mitochondrial energy production and myelination, but excessive levels of brain iron may lead to oxidative stress and neurodegeneration.8 Toxic iron products arise from the Fenton reaction when hydrogen peroxide reacts with free iron producing free radicals that lead to oxidative stress-induced lipid peroxidation, mitochondrial dysfunction and increase in intracellular free-calcium concentration, which in turn, causes cell death.9
MS
Iron deposition has been described in MS demyelinated plaques, myelinated white matter (WM), gray matter (GM) blood vessels as well as in the putamen and the thalamus and in macrophages and reactive microglia.10 Although brain iron deposition is associated with the development of brain atrophy, the exact underlying mechanism by which brain iron accumulates in MS is not fully understood and it is also not yet clear if iron deposition is a mere epiphenomenon or if it implicated in MS pathology.11
AD
Iron loading, in vitro, can regulate amyloid precursor protein (APP) expression, since mRNA shows the presence of iron responsive elements (IRE)12 and APP cleavage13 suggesting that iron causes APP misregulation and accumulation, particularly in cells that are associated with neuritic plaques in the cortex and hippocampus.12
PD
Iron accumulation in PD seems to be due to an overexpression of the divalent metal transporter 1 (DMT1) in the dopaminergic neurons within SN14 where iron deposition can trigger oxidative stress and alfa-synuclein deposition in Lewy bodies, the neuropathological hallmark of PD15.
ALS
Increased iron level has been detected in the ventral cervical spinal cord of ALS patients16 and abnormal iron homeostasis inducing excessive oxidative stress in the motor neurons has been postulated to contribute to disease pathogenesis. Moreover, the most frequently reported gene mutation in ALS, which accounts for 20% of familial ALS cases and of 2% of overall ALS cases, encodes the super oxide dismutase 1 (SOD1) that is involved in iron toxicity17.
Mitochondria dysfunction
The major mithocondrial function is the production of adenosine-5′-triphosphate (ATP), the main energy source of the cell, through the oxidative phosphorylation process. Any defect of oxidative phosphorylation complexes lead to a decreased ATP production as well as to electron leakage, with increased production of superoxide anion and other toxic reactive oxygen species (ROS) which oxidize a number of cellular macromolecules such as lipids, DNA and proteins. The resultant cellular damage determines the release of apoptotic inducing factors that lead to cell death. Alterations in mitochondria function and dynamics (shape, size, fission-fusion processes, distribution and movement) are supposed to be one of the causative factors of neurodegenerative disease. Moreover, mitochondrial DNA is more vulnerable to oxidative damage than nuclear DNA because of its close proximity to the site of ROS production and because of its limited repair mechanisms.18
MS
Neuroaxonal injury in MS can be caused by an imbalance between energy production and demand. The reorganization and upregulation of voltage-gated sodium channels on the axonal membrane subsequent to demyelination determines a switch from a saltatory to continuous nerve conduction with requires an increased amount of energy. At the same time mitochondrial energy production might be compromised by inherent defects in the electron transport chain. In active MS lesions mitochondrial density is increased in axons and astrocytes while complex IV electron transport chains activity is decreased.19 In MS chronic lesions intra axonal mitochondrial density and complex IV activity are both increased to supply the major metabolic demand of demyelinated axons. When the energy demand exceeds the ATP producing capacity of mitochondria the Na/K ATPase activity is impaired and that results in excessive sodium influx which drives the reverse action of the sodium calcium exchanger, leading to calcium influx and intracellular proteases activation which leads to axonal loss.20 In MS GM a decreased expression of genes involved in oxidative phosphorylation has been described with a consequent reduced activity of NADH-coenzyme Q oxidoreductase complex I and complex III).20
AD
A toxic effect on mitochondria can be exerted both by amyloid β and hyperphosphorilated tau, the two pathological hallmarks of AD. Amyloid β intracellular soluble aggregates decrease complex IV mRNAs expression.21 Furthermore, amyloid β peptides can alter mitochondrial dynamics inducing post-translational modifications in proteins involved in fusion and fission and disturbing the tracks along which mitochondria move, resulting in impaired delivery of functional mitochondria to axons and dendrites.22 Finally, intra-mitochondrial amyloid β alters calcium storage, increasing apoptotic pathways.23 Whether mitochondrial dysfunction can precede amyloid β plaques formation is still unclear but in transgenic APP mice, mitochondrial amyloid β accumulation was found to be increased before the formation of the plaques suggesting that mitochondrial dysfunction is an early event in AD.24
PD
The most common mitochondrial alteration associated with PD is complex I inhibition, that can be caused by glutathione depletion, one of the earliest oxidative signs detected in the course of the disease.25 Inhibition of complex I induces nigrostriatal degeneration and alfa synuclein aggregation26; on the other hand alfa synuclein is able, when localized in mitochondria, to inhibit complex I,27 leading to a vicious cycle. Mutations in genes encoding proteins related to mitochondrial function such as PINK 1, PARKIN, DJ-1, LRRK2, SNCA have been implicated in the pathogenesis of PD, however, whether mitochondrial dysfunction is the cause or the effect of protein aggregation in PD is still controversial.28
ALS
Mitochondrial damage and dysfunction have been reported in human ALS and SOD1 mutant transgenic mice.29 Mitochondrial abnormalities (swelling, deformed cristae, defects in respiratory chain activity, decrease in mtDNA copy number) are the earliest signs of disease developed by SOD1 mutant mice and axonal transport is disrupted both in humans and SOD1 mutant mice.30 Moreover, expressing mutant SOD1 confined to mitochondria is sufficient to produce loss of motor neurons and an ALS phenotype.31
Microglia activation
Microglia is a normally dormant population of brain tissue macrophages and represents the main form of immune defense in the CNS; while his acute activation conceives a beneficial inflammatory response that minimizes injury and promotes tissue repair, chronic neuroinflammation has been implicated as a causative factor in many neurodegenerative diseases.
MS
Focal inflammatory lesions occurring in MS are thought to be mediated by peripherally activated immune cells, but, in later stage, inflammation seems to be compartmentalized within the CNS, involving the resident macrophagic population and affecting diffusely the normal appearing brain tissue (NABT).32 Thus, neurological decline in the progressive stage of MS results from axon loss that is dependent on this compartmentalized inflammation and chronic activation of the innate immune system.33
AD
Amyloid β deposition in the brain is associated with a local inflammatory response with reactive gliosis and consequent activation and proliferation of astrocytes and microglial cells. Although the release of inflammatory cytokines increases oxidative stress leading to cellular damage, the inflammatory response may also play a beneficial role in AD pathogenesis. In particular, complement activation, operated by amyloid β fibrillar aggregates through the classic and alternative pathway, may help clearing amyloid β deposits switching microglial cells from a resting state to the active, phagocytic state.34
PD
Activated microglia was first reported in SN of PD post mortem specimens35 and, more recently, it has been described also in the putamen, hippocampus, cingulate and temporal cortex of PD patients.36 Dopaminergic neurons show a prominent vulnerability to microglia-derived cytokines37 and alfa-synuclein itself can induce microglial activation.38 Microglial transition from the resting to the activated state takes place before other signs of cellular or tissue injury can be seen; therefore, microglial activation is an early, albeit nonspecific, indicator of neuronal damage39.
ALS
Several studies supported the idea that inflammatory mechanisms are important mediators of cell death and survival in ALS. Accumulation of large numbers of activated microglia and astrocytes, as well as small numbers of T cells, mostly adhering to postcapillary venules, has been detected in the brainstem and spinal cord of ALS cases40 and in mouse models41 of the disease where microglial infiltration increases with disease progression.42 Activated microglia cells have been shown to secrete the ligand for the apoptotic receptor Fas, triggering motor neuronal cell death.43 According to other studies however, motoneural injury preceded microglial activation suggesting that it was a secondary response.44
Imaging techniques to detect and monitor neurodegeneration
Conventional MRI (cMRI) is currently regarded as the most sensitive non-invasive tool for supporting MS diagnosis and monitor treatment response in the contest of clinical trials of antinflammatory agents. Despite these advantages, cMRI lacks pathological specificity and sensitivity to the presence of gray matter (GM) lesions and to the microscopic injury occurring in normal-appearing WM and GM. In the last few years new MRI techniques, that provide better correlation with pathological features such as de- and remyelination, neuroaxonal injury and astrogliosis, have been developed.45 We will briefly summarize the principles of more advanced neuroimaging techniques such as measures of brain atrophy; diffusion tensor imaging (DTI), functional MRI (fMRI), and 11C-PK11195 PET; then we will briefly review the findings of the application of these techniques to the study of MS and, finally, we will discuss the similarity of findings of the application of the same techniques to the study of classical neurodegenerative diseases.
Brain atrophy
Brain atrophy reflects loss of brain parenchyma, especially myelin, neurons and axons. Several algorithms are available to perform its quantification and are classified in segmentation-based and registration-based techniques. Segmentation-based techniques are based on tissue classification (cerebrospinal fluid -CSF-, GM, and WM) in a semiautomated or automated way and provide brain volume measures at a given time. They can be used for cross-sectional studies by including normalization for head size.46 Registration-based methods have been developed to improve longitudinal evaluations. They include brain edge motion analysis for structural image evaluation, using normalization, of atrophy (SIENA) (http://www.fmrib.ox.ac.uk/fsl), voxel-based statistical analysis for voxel-based morphometry (VBM), statistical parametric mapping, and local Jacobian determinant analysis after nonlinear matching between coregistered images.46
Diffusion tensor imaging (DTI)
Diffusion MRI allows the assessment of brain tissue microscopic properties.47 Cellular structures in the CNS restrict water random molecular motion and, therefore, pathologic processes that modify tissue integrity can result in an increased diffusion coefficient. Commonly measured DTI-derived metrics are mean diffusivity (MD) that reflects cellular integrity, fractional anisotropy (FA) that reflects fibers integrity and directionality, radial diffusivity (RD) that provides a specific measure of myelin integrity, and axial diffusivity (AD) that correlates with axons injury. In addition, DTI allows the segmentation and visualization of the major WM tracts. Fiber tracking uses each voxel’s primary eigenvector of the diffusion tensor, that express the direction in which water diffusion is highest, which typically is parallel to white matter fiber fascicles, to follow an axonal tract in 3D from voxel to voxel through the brain, thus allowing the delineation of specific cerebral WM tracts48 (See example in Figure 1) . A voxel based analysis of diffusion tensor imaging (DTI) data called tract based spatial statistic (TBSS) permits the assessment of the whole brain, and, unlike the histogram approach, also yields information about the location of damage49 (See example in Figure 2).

Figure 1. Overall brain tractography from a 62 y-old male Parkinson patient (A) and a 61 y-old male healthy control (B). The color code represents fiber direction: blue-inferior to superior, red - left to right, green- anterior to posterior. The 3D view from the superior plane shows a diffuse reduction of white matter fibers in the PD patient compared with the control.
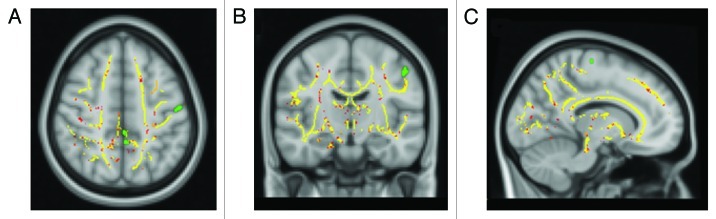
Figure 2. TBSS results and SPM-VBM analysis of cortical brain volume from 15 RR-MS patients and 15 healthy controls displayed on the Montreal Neurological Institute template. Clusters of reduced FA in patients compared with healthy controls are indicated in red. Areas of significant reduction of gray matter volume in patients compared with healthy controls are indicated in green.
Functional MRI (fMRI)
fMRI measures the level of blood oxygenation, by detecting the signal generated by changes in the local magnetic field secondary to the increased amount of oxygenated hemoglobin in the venous system. fMRI has been used in research settings to examine the neural correlates of known motor, visual, sensory and neuropsychological deficits.45 Resting state (rs) functional MRI measures low-frequency (< 0.1 Hz) BOLD (blood oxygenation level-dependent) fluctuations caused by spontaneous neuronal activity within and between different regions throughout the entire human brain. At the neural level, rs-fMRI is thought to be based on very infrequent bursts of spiking cortical activity that slowly diffuse through several brain networks.50
11C-PK11195 PET
Positron emission tomography (PET) is a functional imaging technique that detects gamma rays emitted by a positron-emitting radionuclide (tracer), which is introduced into the body on a biologically active molecule. Three-dimensional images of tracer concentration within the body are then constructed by computer analysis. Many radiotracers are used in PET to image the tissue concentration of many types of molecules of interest. Among them, the selective radioligand 11C-PK11195 express the concentration of translocator protein (TSPO),51 a protein that is upregulated in activated microglia.52 In absence of invading peripheral blood cells, the increased 11C-PK11195 binding indicates the transition of microglia from a resting to an activated state.51 Regional increased 11C-PK11195 binding may also indicate active tissue response to disconnection in retrograde, anterograde or trans-synaptic projection areas.53 Such remote activation of microglia may potentially be responsible for secondary progressive brain pathology extending beyond the initially affected neural system.54
Relationship between WM and GM degeneration
Although MS has traditionally been considered a disease of the white matter, there is an increasing body of evidence that gray matter is involved by the pathological process since the early stage of the disease and has a significant impact on physical and cognitive disability.55 Conversely, although classic neurodegenerative diseases are thought to be characterized by a selective and localized damage of GM, advanced quantitative MRI techniques can demonstrate a diffuse involvement of WM.56
Very little is known about the dynamics of GM and WM damage in MS and in the classical neurodegenerative diseases. In MS, it has been suggested that the retrograde degeneration of axons transected in WM lesions leads to a secondary neuronal loss in the cortex of MS patients. This is in part supported by MRI studies showing a certain degree of correlation between WM injury and cortical atrophy.57-59 However, a number of MRI studies55 have demonstrated that GM pathology (both cortical lesions and atrophy) is already evident at the earliest stages of the disease when the WM lesion load is quite modest60 suggesting that GM could be the primary target of the disease process, resulting in secondary axonal degeneration and demyelination of WM.61
MRI studies combining measures of Brain atrophy and DTI
Abnormal brain DTI metrics have been reported in all the MS clinical phenotypes, clinical isolated syndrome (CIS), relapsing-remitting MS (RR-MS), primary-progressive MS (PP-MS), secondary- progressive MS (SP-MS) demonstrating that demyelination and axonal loss occur in lesions and to less extent in NAWM since the early stage of the disease62 and become more prominent as the disease progresses.63
MRI studies using DTI and measure of atrophy to investigate the relationship between the damage occurring in the NAWM and in the GM revealed that while NAWM decreased volume and FA values, GM volume was relatively preserved in patients with CIS.57 However, at 1 y follow-up the same patients reported no significant changes in global and local WM FA, while longitudinal volumetric analysis revealed a reduction in global GM volume involving several cortical and subcortical regions, especially in patients that converted from CIS to clinically definite MS.58 The absence of GM atrophy in the early phase of MS suggests that GM pathology develops as a result of WM damage.57,58 Another study investigating the relationship between WM and GM applying DTI and measures of brain volume in patients with PP-MS showed a significant association between the right sensory motor region with the adjacent CST, the left and right thalamus with the corresponding thalamic radiations and the left insula with the adjacent WM59 suggesting a link between the pathological processes occurring in the NAWM and in the GM in these patients. However, in the same study, there were other WM tracts and areas of GM atrophy that did not correlate with each other, suggesting that the pathological processes occurring in the NAWM and in the GM could, to some extent, develop independently from each other.
Similarly, MRI studies using DTI and measure of atrophy in patients with AD and mild cognitive impairment (MCI) reported an increase of MD in all major WM tracts and significant and anatomically congruent correlations between WM changes and regional GM atrophy suggesting that WM damage was secondary to Wallerian degeneration of axons whose cell bodies were injured in the cortex.64 However, since no correlation was found between most WM tracts and corresponding regional cortical atrophy in patients with aMCI64 it has been speculated that WM damage is not the mere consequence of neuronal loss but might be affected by pathological mechanisms independent from those affecting GM regions.
In patients with ALS, voxel-based analysis of DTI data showed changes of diffusion parameters not only in the cortico-spinal tracts but also in frontal, temporal and parietal lobes, supporting the view that ALS is a multisystem degenerative disease where abnormalities of extra-motor areas play an important role in the disease physiopathology.65
Finally, in DTI studies of de novo patients with >PD, FA was found to be decreased in the SN, with greater reduction in the caudal part, suggesting that DTI could be a noninvasive early biomarker of neurodegeneration for PD.66 Moreover, voxel-based analysis of brain volumetric data acquired in patients with PD and depression showed tissue loss in several WM regions within the cortical-limbic network suggesting an increased vulnerability of this neural circuit in PD that could partially explain the high prevalence of depression in PD.67
Functional MRI: fMRI
Brain plasticity might have a role in limiting the clinical consequences of MS-related damage, especially at the beginning of the disease and in clinically stable patients.68 Task-related fMRI studies of MS patients have demonstrated differences of brain activation between patients and healthy controls in terms of extent of activations and number of regions recruited during the performance of a given task. Resting state (rs) fMRI studies of MS patients have showed alteration of the default mode network (DMN) that is the most consistently and commonly reported resting-state network highly relevant for human cognition.50
fMRI during the performance of simple motor task in MS patients demonstrated a correlations between measures of functional connectivity and structural damage to some of the major WM bundles, suggesting an adaptive role of functional connectivity changes in limiting the clinical consequences of structural damage in patients with RR-MS.69
A similar study conducted in patients with ALS demonstrated an higher connectivity in patients without structural damage of CST assessed by DTI metrics, and a less widespread increase in connectivity in ALS patients with CST damage, suggesting that functional brain changes do occur in ALS with mild disability and that these changes might have a role in compensating for limited structural damage and might exhaust with increasing burden of disease pathology.70
The better compensatory abilities found in MS patients can depend not only on the different underlying pathogenetic mechanism, but also on the age at disease onset that is significantly lower in patients with MS than in patients with ALS.
11C-PK11195 PET
11C-PK11195 PET studies in EAE and in MS post mortem brains demonstrated that 11C-PK11195 uptake overlapped with the distribution pattern of activated microglia51; the binding was increased not only in areas of focal pathology identified by T1-W and T2-W sequences71 and in gadolinium-enhanced lesions72 but also in NAWM and NAGM51 suggesting a subtle inflammatory process microglia mediated, that occur independently of relapses.
In AD patients, significant 11C-PK11195 binding was observed in the temporal, parietal and occipital cortices73; it was inversely correlated with the patient Mini-Mental State Examination (MMSE) scores, which was compatible with a role of microglia in neuronal damage. Elevated levels of activated microglia were also described in patients with MCI74 suggesting that microglial activation could be an early event in the AD pathogenesis that begins at the MCI stage.
11C-PK11195 uptake was detected in the midbrain and putamen of PD patients where it correlated positively with the motor severity of Parkinsonism75 but also in the basal ganglia, pons, frontal and temporal cortical regions.76 The increased microglial activation remained unchanged over time, while the patients deteriorated clinically, suggesting that microglia is activated early in PD and remain activated for longer periods possibly driving the progression of the disease.76
In ALS patients significant increased binding of 11C-PK11195 was measured in the motor cortex, with strong correlation with clinical upper motor neuron (UMN) score and in the frontal lobe, in agreement with the subtle cognitive deficits related to executive function, identified in up to 50% of ALS patients suggesting the clinical relevance of microglial activation.54
Conclusion
Imaging techniques provide practical tools to detect and monitor in vivo neurodegeneration in MS and classical neurodegenerative diseases and more sophisticated tools to investigate the underlying pathological mechanisms. The MRI and PET studies performed to date support the concept that GM and WM damage coexist in both MS and classic neurodegenerative diseases. While in MS GM damage is in part secondary to neurodegeneration of WM, in classical neurodegenerative diseases WM damage is in part secondary to GM degeneration. However, an increasing number of studies suggest that GM and WM might be independent and concurrent targets of pathological mechanisms in both MS and classical neurodegenerative diseases. Unlike AD, PD and ALS, MS is diagnosed early in life when most of the neuroaxonal is still preserved and, therefore, provides the unique opportunity to study the dynamics of neurodegeneration and may serve as a model for better understanding the pathogenetic mechanisms leading neuroaxonal loss and, eventually, for developing neuroprotective strategies in that might benefit classical neurodegenerative diseases.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/22650
References
- 1.Gibb WR. Neuropathology of Parkinson’s disease and related syndromes. Neurol Clin. 1992;10:361–76. [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 4.Lassmann H, van Horssen J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011;585:3715–23. doi: 10.1016/j.febslet.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Charcot M. Histologie de la sclerose en plaques. Gazette Hopitaux. 1868;141:554–8. [Google Scholar]
- 6.Stadelmann C. Multiple sclerosis as a neurodegenerative disease: pathology, mechanisms and therapeutic implications. Curr Opin Neurol. 2011;24:224–9. doi: 10.1097/WCO.0b013e328346056f. [DOI] [PubMed] [Google Scholar]
- 7.Poloni G, Minagar A, Haacke EM, Zivadinov R. Recent developments in imaging of multiple sclerosis. Neurologist. 2011;17:185–204. doi: 10.1097/NRL.0b013e31821a2643. [DOI] [PubMed] [Google Scholar]
- 8.Oshiro S, Morioka MS, Kikuchi M. Dysregulation of iron metabolism in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Adv Pharmacol Sci. 2011;2011:378278. doi: 10.1155/2011/378278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salvador GA. Iron in neuronal function and dysfunction. Biofactors. 2010;36:103–10. doi: 10.1002/biof.80. [DOI] [PubMed] [Google Scholar]
- 10.LeVine SM. Iron deposits in multiple sclerosis and Alzheimer’s disease brains. Brain Res. 1997;760:298–303. doi: 10.1016/S0006-8993(97)00470-8. [DOI] [PubMed] [Google Scholar]
- 11.Khalil M, Teunissen C, Langkammer C. Iron and neurodegeneration in multiple sclerosis. Mult Scler Int. 2011;2011:606807. doi: 10.1155/2011/606807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–73. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 13.Bodovitz S, Falduto MT, Frail DE, Klein WL. Iron levels modulate alpha-secretase cleavage of amyloid precursor protein. J Neurochem. 1995;64:307–15. doi: 10.1046/j.1471-4159.1995.64010307.x. [DOI] [PubMed] [Google Scholar]
- 14.Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci U S A. 2008;105:18578–83. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerlach M, Riederer P, Double KL. Neuromelanin-bound ferric iron as an experimental model of dopaminergic neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord. 2008;14(Suppl 2):S185–8. doi: 10.1016/j.parkreldis.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 16.Kasarskis EJ, Tandon L, Lovell MA, Ehmann WD. Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study. J Neurol Sci. 1995;130:203–8. doi: 10.1016/0022-510X(95)00037-3. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida M, Takahashi Y, Koike A, Fukuda Y, Goto J, Tsuji S. A mutation database for amyotrophic lateral sclerosis. Hum Mutat. 2010;31:1003–10. doi: 10.1002/humu.21306. [DOI] [PubMed] [Google Scholar]
- 18.Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27:1129–36. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131:1722–35. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478–89. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- 21.Hong WK, Han EH, Kim DG, Ahn JY, Park JS, Han BG. Amyloid-beta-peptide reduces the expression level of mitochondrial cytochrome oxidase subunits. Neurochem Res. 2007;32:1483–8. doi: 10.1007/s11064-007-9336-7. [DOI] [PubMed] [Google Scholar]
- 22.Silva DF, Esteves AR, Arduino DM, Oliveira CR, Cardoso SM. Amyloid-β-induced mitochondrial dysfunction impairs the autophagic lysosomal pathway in a tubulin dependent pathway. J Alzheimers Dis. 2011;26:565–81. doi: 10.3233/JAD-2011-110423. [DOI] [PubMed] [Google Scholar]
- 23.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–9. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 25.Danielson SR, Held JM, Oo M, Riley R, Gibson BW, Andersen JK. Quantitative mapping of reversible mitochondrial Complex I cysteine oxidation in a Parkinson disease mouse model. J Biol Chem. 2011;286:7601–8. doi: 10.1074/jbc.M110.190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chinta SJ, Andersen JK. Nitrosylation and nitration of mitochondrial complex I in Parkinson’s disease. Free Radic Res. 2011;45:53–8. doi: 10.3109/10715762.2010.509398. [DOI] [PubMed] [Google Scholar]
- 27.Chinta SJ, Mallajosyula JK, Rane A, Andersen JK. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett. 2010;486:235–9. doi: 10.1016/j.neulet.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–70. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 30.Magrané J, Sahawneh MA, Przedborski S, Estévez AG, Manfredi G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J Neurosci. 2012;32:229–42. doi: 10.1523/JNEUROSCI.1233-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Igoudjil A, Magrané J, Fischer LR, Kim HJ, Hervias I, Dumont M, et al. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J Neurosci. 2011;31:15826–37. doi: 10.1523/JNEUROSCI.1965-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–12. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- 33.Weiner HL. A shift from adaptive to innate immunity: a potential mechanism of disease progression in multiple sclerosis. J Neurol. 2008;255(Suppl 1):3–11. doi: 10.1007/s00415-008-1002-8. [DOI] [PubMed] [Google Scholar]
- 34.Golde TE. Inflammation takes on Alzheimer disease. Nat Med. 2002;8:936–8. doi: 10.1038/nm0902-936. [DOI] [PubMed] [Google Scholar]
- 35.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–91. doi: 10.1212/WNL.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 36.Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003;106:518–26. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- 37.De Lella Ezcurra AL, Chertoff M, Ferrari C, Graciarena M, Pitossi F. Chronic expression of low levels of tumor necrosis factor-alpha in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiol Dis. 2010;37:630–40. doi: 10.1016/j.nbd.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–42. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 39.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–8. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 40.Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140:691–707. [PMC free article] [PubMed] [Google Scholar]
- 41.Troost D, Van den Oord JJ, Vianney de Jong JM. Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 1990;16:401–10. doi: 10.1111/j.1365-2990.1990.tb01276.x. [DOI] [PubMed] [Google Scholar]
- 42.Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–9. doi: 10.1212/WNL.57.7.1282. [DOI] [PubMed] [Google Scholar]
- 43.Ciesielski-Treska J, Ulrich G, Chasserot-Golaz S, Zwiller J, Revel MO, Aunis D, et al. Mechanisms underlying neuronal death induced by chromogranin A-activated microglia. J Biol Chem. 2001;276:13113–20. doi: 10.1074/jbc.M009711200. [DOI] [PubMed] [Google Scholar]
- 44.Mariotti R, Bentivoglio M. Activation and response to axotomy of microglia in the facial motor nuclei of G93A superoxide dismutase transgenic mice. Neurosci Lett. 2000;285:87–90. doi: 10.1016/S0304-3940(00)01034-X. [DOI] [PubMed] [Google Scholar]
- 45.Filippi M, Rocca MA, De Stefano N, Enzinger C, Fisher E, Horsfield MA, et al. Magnetic resonance techniques in multiple sclerosis: the present and the future. Arch Neurol. 2011;68:1514–20. doi: 10.1001/archneurol.2011.914. [DOI] [PubMed] [Google Scholar]
- 46.Durand-Dubief F, Belaroussi B, Armspach JP, Dufour M, Roggerone S, Vukusic S, et al. Reliability of Longitudinal Brain Volume Loss Measurements between 2 Sites in Patients with Multiple Sclerosis: Comparison of 7 Quantification Techniques. AJNR Am J Neuroradiol. 2012 doi: 10.3174/ajnr.A3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmierer K, Wheeler-Kingshott CA, Boulby PA, Scaravilli F, Altmann DR, Barker GJ, et al. Diffusion tensor imaging of post mortem multiple sclerosis brain. Neuroimage. 2007;35:467–77. doi: 10.1016/j.neuroimage.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mori S, van Zijl PC. Diffusion weighting by the trace of the diffusion tensor within a single scan. Magn Reson Med. 1995;33:41–52. doi: 10.1002/mrm.1910330107. [DOI] [PubMed] [Google Scholar]
- 49.Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, et al. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage. 2006;31:1487–505. doi: 10.1016/j.neuroimage.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 50.Bonavita S, Gallo A, Sacco R, Corte MD, Bisecco A, Docimo R, et al. Distributed changes in default-mode resting-state connectivity in multiple sclerosis. Mult Scler. 2011;17:411–22. doi: 10.1177/1352458510394609. [DOI] [PubMed] [Google Scholar]
- 51.Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123:2321–37. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- 52.Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35:306–28. doi: 10.1111/j.1365-2990.2008.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banati RB. Brain plasticity and microglia: is transsynaptic glial activation in the thalamus after limb denervation linked to cortical plasticity and central sensitisation? J Physiol Paris. 2002;96:289–99. doi: 10.1016/S0928-4257(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 54.Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–9. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 55.Popescu BF, Lucchinetti CF. Meningeal and cortical grey matter pathology in multiple sclerosis. BMC Neurol. 2012;12:11. doi: 10.1186/1471-2377-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xekardaki A, Giannakopoulos P, Haller S. White matter changes in bipolar disorder, Alzheimer disease, and mild cognitive impairment: New insights from DTI. J Aging Res. 2011;2011:286564. doi: 10.4061/2011/286564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raz E, Cercignani M, Sbardella E, Totaro P, Pozzilli C, Bozzali M, et al. Clinically isolated syndrome suggestive of multiple sclerosis: voxelwise regional investigation of white and gray matter. Radiology. 2010;254:227–34. doi: 10.1148/radiol.2541090817. [DOI] [PubMed] [Google Scholar]
- 58.Raz E, Cercignani M, Sbardella E, Totaro P, Pozzilli C, Bozzali M, et al. Gray- and white-matter changes 1 year after first clinical episode of multiple sclerosis: MR imaging. Radiology. 2010;257:448–54. doi: 10.1148/radiol.10100626. [DOI] [PubMed] [Google Scholar]
- 59.Bodini B, Khaleeli Z, Cercignani M, Miller DH, Thompson AJ, Ciccarelli O. Exploring the relationship between white matter and gray matter damage in early primary progressive multiple sclerosis: an in vivo study with TBSS and VBM. Hum Brain Mapp. 2009;30:2852–61. doi: 10.1002/hbm.20713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Calabrese M, Atzori M, Bernardi V, Morra A, Romualdi C, Rinaldi L, et al. Cortical atrophy is relevant in multiple sclerosis at clinical onset. J Neurol. 2007;254:1212–20. doi: 10.1007/s00415-006-0503-6. [DOI] [PubMed] [Google Scholar]
- 61.Tsunoda I, Kuang LQ, Libbey JE, Fujinami RS. Axonal injury heralds virus-induced demyelination. Am J Pathol. 2003;162:1259–69. doi: 10.1016/S0002-9440(10)63922-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Inglese M, Bester M. Diffusion imaging in multiple sclerosis: research and clinical implications. NMR Biomed. 2010;23:865–72. doi: 10.1002/nbm.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Preziosa P, Rocca MA, Mesaros S, Pagani E, Stosic-Opincal T, Kacar K, et al. Intrinsic damage to the major white matter tracts in patients with different clinical phenotypes of multiple sclerosis: a voxelwise diffusion-tensor MR study. Radiology. 2011;260:541–50. doi: 10.1148/radiol.11110315. [DOI] [PubMed] [Google Scholar]
- 64.Agosta F, Pievani M, Sala S, Geroldi C, Galluzzi S, Frisoni GB, et al. White matter damage in Alzheimer disease and its relationship to gray matter atrophy. Radiology. 2011;258:853–63. doi: 10.1148/radiol.10101284. [DOI] [PubMed] [Google Scholar]
- 65.Sage CA, Peeters RR, Görner A, Robberecht W, Sunaert S. Quantitative diffusion tensor imaging in amyotrophic lateral sclerosis. Neuroimage. 2007;34:486–99. doi: 10.1016/j.neuroimage.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 66.Vaillancourt DE, Spraker MB, Prodoehl J, Abraham I, Corcos DM, Zhou XJ, et al. High-resolution diffusion tensor imaging in the substantia nigra of de novo Parkinson disease. Neurology. 2009;72:1378–84. doi: 10.1212/01.wnl.0000340982.01727.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kostić VS, Agosta F, Petrović I, Galantucci S, Spica V, Jecmenica-Lukic M, et al. Regional patterns of brain tissue loss associated with depression in Parkinson disease. Neurology. 2010;75:857–63. doi: 10.1212/WNL.0b013e3181f11c1d. [DOI] [PubMed] [Google Scholar]
- 68.Filippi M, Rocca MA. Cortical reorganisation in patients with MS. J Neurol Neurosurg Psychiatry. 2004;75:1087–9. doi: 10.1136/jnnp.2004.036020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rocca MA, Pagani E, Absinta M, Valsasina P, Falini A, Scotti G, et al. Altered functional and structural connectivities in patients with MS: a 3-T study. Neurology. 2007;69:2136–45. doi: 10.1212/01.wnl.0000295504.92020.ca. [DOI] [PubMed] [Google Scholar]
- 70.Agosta F, Pagani E, Rocca MA, Caputo D, Perini M, Salvi F, et al. Voxel-based morphometry study of brain volumetry and diffusivity in amyotrophic lateral sclerosis patients with mild disability. Hum Brain Mapp. 2007;28:1430–8. doi: 10.1002/hbm.20364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vowinckel E, Reutens D, Becher B, Verge G, Evans A, Owens T, et al. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci Res. 1997;50:345–53. doi: 10.1002/(SICI)1097-4547(19971015)50:2<345::AID-JNR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 72.Debruyne JC, Versijpt J, Van Laere KJ, De Vos F, Keppens J, Strijckmans K, et al. PET visualization of microglia in multiple sclerosis patients using [11C]PK11195. Eur J Neurol. 2003;10:257–64. doi: 10.1046/j.1468-1331.2003.00571.x. [DOI] [PubMed] [Google Scholar]
- 73.Yokokura M, Mori N, Yagi S, Yoshikawa E, Kikuchi M, Yoshihara Y, et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2011;38:343–51. doi: 10.1007/s00259-010-1612-0. [DOI] [PubMed] [Google Scholar]
- 74.Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, et al. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72:56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bartels AL, Willemsen AT, Doorduin J, de Vries EF, Dierckx RA, Leenders KL. [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat Disord. 2010;16:57–9. doi: 10.1016/j.parkreldis.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 76.Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404–12. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]