

Abstract
The cellular nutrient sensing apparatus detects nutritional depletion and transmits this information to downstream effectors that generate energy from alternate sources. Autophagy is a crucial catabolic pathway that turns over redundant cytoplasmic components in lysosomes to provide energy to the starved cell. Recent studies have described a role for hypothalamic autophagy in the control of food intake and energy balance. Activated autophagy in hypothalamic neurons during starvation mobilized neuron-intrinsic lipids to generate free fatty acids that increased AgRP levels. AgRP neuron-specific inhibition of autophagy decreased fasting-induced increases in AgRP levels and food intake. Deletion of autophagy in AgRP neurons led to constitutive increases in levels of proopiomelanocortin and its active processed product, α-melanocyte stimulating hormone that contributed to reduced adiposity in these rodents. The current manuscript discusses these new findings and raises additional questions that may help understand how hypothalamic autophagy controls food intake and energy balance. These studies may have implications for designing new therapies against obesity and insulin resistance.
Keywords: hypothalamus, AgRP, neuron, autophagy, food intake, energetic balance
Autophagy
Over millions of years organisms have evolved mechanisms and committed significant cellular resources to achieve one major objective—surveillance of the surroundings for food or predators. As part of this conserved mechanism, lack of nutrients initiate food-seeking behavior and/or activate processes that utilize internal fuel stores to maintain the energetic requirements of vital organs. Similarly, at the level of the cell, energy depletion is sensed by mechanisms that convey signals to catabolic pathways, which mobilize redundant cellular components for the provision of energy. A critical catabolic response to nutritional depletion is activation of macroautophagy (or autophagy).
Autophagy is an evolutionarily preserved mechanism that turns over cellular contents within acidic organelles called lysosomes, and in this way provides an alternate form of energy to the starved cell.1,2 Genetic studies in yeast have shown that autophagy requires greater than 30 autophagy gene (atg) products for its function.3 Briefly, autophagy entails the formation of a de novo limiting membrane that elongates to form a double-membraned vesicle, the autophagosome.1 Atg7 is a key E1-like ligase that activates two conjugation cascades, the LC3 and Atg5/Atg12 systems that are required for autophagosome formation (Fig. 1).1 The LC3 conjugation cascade involves lipidation of soluble cytosolic light chain 3 (LC3)-I to form the autophagosome-associated LC3-II, the specific autophagosome marker (Fig. 1).1 Once formed, autophagosomes sequester cytoplasmic contents that include damaged organelles and proteins, and then fuse with lysosomes (Fig. 1).1 Hydrolysis of engulfed cargo within lysosomes generates amino- and fatty acids that are released into the cytosol for essential biosynthetic functions (Fig. 1).1 Quite intriguingly, this “in-bulk” mechanism to eliminate cellular debris also displays significant selectivity for the degradation of organelles such as mitochondria and cellular lipid droplets via processes termed mitophagy and lipophagy, respectively.4,5

Figure 1. Autophagy and regulatory elements. Under basal fed conditions, nutrient and growth factor signals converge upon mTOR to inhibit autophagy. Nutrient deprivation activates AMPK, which activates autophagy by inhibiting mTOR and phosphorylating ULK1. Starvation-induced autophagy requires beclin to form a functional complex with vps34, vps30, vps15, which constitutes the active class III PI3K. The class III PI3K is a lipid kinase that contributes to the formation of the nucleation complex. Atg7 activates two independent conjugation systems, the LC3-II and Atg5–12 cascades that promote membrane elongation. The limiting membrane engulfs cytosolic cargo (mitochondria shown in cartoon) and seals to form an autophagosome, which then fuses with the lysosomes to enable cargo degradation. The products of lysosomal hydrolysis are released into the cytosol for biosynthetic functions. Atg, autophagy gene; LC3, light chain-3; PI3K, phosphoinositide 3-kinase; vps, vacuolar protein sorting.
Understandably, such a catabolic process will require tight regulatory control, and in fact an intricate signaling network integrates extrinsic and cellular cues to modulate autophagy.1 In essence, the functional significance of these signaling events is that autophagy is kept at low basal levels when nutrients and growth factors are abundant, whereas cellular energy depletion rapidly activates autophagy (Fig. 1). A crucial signaling axis conserved in yeast, flies and mammals is the nutrient sensor target of rapamycin (TOR), the best-characterized negative regulator of autophagy (Fig. 1).6 Available nutrients activate TOR that mediates growth, whereas starvation inhibits TOR, which releases its inhibition on autophagy.6 Depletion of nutrients activates a second important cellular energy sensor, AMP-activated protein kinase (AMPK) that activates autophagy by phosphorylating unc51-like kinase 1 (ULK1) (Fig. 1), a key autophagy activator and newly discovered AMPK substrate.7
Hypothalamic Control of Food Intake
The mediobasal hypothalamic (MBH) neurons play a pivotal role in control of food intake and energy balance.8 The localization of these neurons, which include the agouti-related peptide (AgRP) and the proopiomelanocortin (POMC) neurons, at distinct regions around the 3rd ventricle allows these neurons to monitor minute changes in levels of circulating nutrients and hormones.9 The AgRP neurons promote food intake in part through the release of AgRP, a physiological antagonist for the melanocortin receptors. AgRP neurons also provide inhibitory γ-aminobutyric acid (GABA)-ergic projections at POMC neurons.8 In contrast, the POMC neurons produce the POMC precursor that is cleaved to generate α-melanocyte stimulating hormone (MSH), which curtails food intake and promotes energy expenditure in the periphery.10
It is now well established that components of a number of signal transduction pathways, for instance, mTOR, AMPK, forkhead box O family transcription factor (FoxO), and class I phosphatidylinositol 3-kinase (PI3K), play important roles in the hypothalamic control of food intake.11-14 For example, studies in rodents have revealed that activation of hypothalamic mTOR reduces food intake,11 whereas activation of hypothalamic AMPK promotes food intake.12,13 Hypothalamic FoxO1 has also been shown to be a requirement for AgRP expression and food intake.15 Interestingly, a common denominator in all of these signaling cascades is in their ability to regulate autophagy. Besides the known roles for mTOR and AMPK in modulating autophagy in a number of cell types,6,7 FoxO-dependent activation of autophagy has been observed in neurons, and skeletal muscle.16,17 These observations suggest that the modulatory roles of at least some of these signaling events in control of food intake may have occurred, in part, via downstream effects on hypothalamic autophagy.
Metabolites, such as free fatty acids, have also been implicated in control of food intake,18 as well as in the activation of autophagy in non-neuronal tissues, for instance, hepatocytes.5 Cellular fatty acid oxidation is tightly regulated by carnitine palmitoyltransferase (CPT)-1-mediated influx of free fatty acids into the mitochondria. CPT-1-regulated hypothalamic fatty acid oxidation has been shown to drive food intake.19,20 Cellular energy depletion, as reflected by increases in AMP, activates AMPK that inhibits acetyl CoA carboxylase (ACC).20 Reduced ACC activity decreases cellular production of malonyl CoA, the allosteric inhibitor of CPT1 that, in turn, increases mitochondrial fatty acid availability and β-oxidation.20 Indeed, recent studies have suggested that neuronal free fatty acid availability and its oxidation may fulfill the bioenergetic needs in terms to rapid production of sufficient ATP molecules for neuronal activation and firing.18 While these studies have established links between hypothalamic signaling, fatty acid oxidation, and food intake, the mechanisms that generate neuronal free fatty acids during starvation have not been fully characterized.
Autophagy in AgRP Neurons Control Food Intake
Our studies in hepatocytes revealed a novel role for autophagy in the mobilization of cellular lipid droplets via lipophagy.5 Starvation typically increases circulating free fatty acids that are taken up by the liver and rapidly esterified in lipid droplets. We observed that activation of hepatocellular autophagy during starvation, and in response to acute fatty acid exposure, led to sequestration of cellular lipid droplets and their delivery to lysosomes wherein lipolysis liberated free fatty acids for β-oxidation.5 The effect of blocking liver-specific autophagy was one of increased hepatocellular lipid accumulation and reduced β-oxidation.5 Based on these observations in the liver, we hypothesized that hypothalamic autophagy is nutrient-responsive, and that the activation of hypothalamic autophagy during starvation mobilizes neuron-intrinsic lipids to generate free fatty acids that drive food intake.
Indeed, our studies demonstrate that hypothalamic autophagy is sensitive to nutritional depletion since fasting activated autophagy in the MBH, and in serum-deprived hypothalamic GT1-7 cells.21 Although, the brain is relatively resistant to starvation-induced activation of autophagy,22 our results reveal the unique characteristic of hypothalamic neurons in their ability to upregulate autophagy during starvation.21 In parallel to activation of autophagy, we observed that starvation increased hypothalamic fatty acid uptake in vivo and in hypothalamic cells.21 Indeed, the exposure of hypothalamic cells to serum from starved rodents dramatically increased cellular lipid accumulation,21 which indicated that the source of hypothalamic lipids during starvation was the periphery. Not surprisingly, the immediate fate of free fatty acids taken up by hypothalamic cells was triglyceride (TG) synthesis since exposure of starved cells to triacsin C, a TG synthesis inhibitor, remarkably blocked TG synthesis during starvation.21 These findings led us to ask the question whether fatty acid uptake was the mechanism for activation of starvation-induced hypothalamic autophagy. In agreement with our published findings in cultured hepatocytes,5 an acute exposure of hypothalamic GT1-7 cells to oleic and palmitic acid increased autophagy as determined by autophagy flux assays, and by measuring rates of long t1/2 protein degradation.21 Intriguingly, exposure of hypothalamic cells to fatty acids and starved rodent serum increased levels of phosphorylated AMPK and ULK1,21 suggesting the possibility that the hypothalamic AMPK/ULK1 axis forms part of a neuronal fatty acid sensing machinery that regulates autophagy during starvation.
To examine the physiological significance of activated hypothalamic autophagy, we examined whether autophagy served to mobilize hypothalamic lipids in this setting. Indeed, the activation of autophagy in cultured hypothalamic cells and in primary hypothalamic neurons increased interactions between neuronal lipids and lysosomes, indicating that hypothalamic autophagy functioned to turn over neuronal lipids during starvation.21 The functional relevance of these interactions was generation of neuron-intrinsic free fatty acids, since blocking lysosomal degradation or silencing atg5, a second autophagy gene, reduced hypothalamic free fatty acid levels.21 Inhibiting lysosomal lipolysis reduced fasting- and fatty acid-induced increases in orexigenic AgRP levels in hypothalamic cells, and in primary neurons cultured under basal conditions.21 These findings help establish a direct link between autophagy-derived neuronal free fatty acids and AgRP expression.
In parallel with our findings in the cell culture model, inhibiting AgRP neuron-selective autophagy in vivo decreased fasting-induced increases in hypothalamic AgRP levels and food intake.21 An exciting unexpected development was the finding of increased hypothalamic levels of the POMC precursor and its cleavage product α-MSH in AgRP neuronal autophagy deficient (KO) mice.21 These increases in α-MSH may have contributed to the reduced adiposity in the KO mice, at least in part, through increased locomotor activity and increased adipose triglyceride lipase levels observed in these rodents.21
In contrast to our present findings, a recent study that silenced atg7 within the MBH using acute siRNA injections has shown marked increases in adiposity and glucose intolerance.23 It is likely that adiposity in this model occurred from reduced autophagy in both MBH neuronal subsets, or possibly from inhibition of autophagy in additional cell types in the hypothalamus such as glial cells that have also been shown to be important for glucose homeostasis.24 Moreover, blocking autophagy nonspecifically in the hypothalamus may have obliterated the beneficial effects of AgRP neuron-selective deletion of autophagy.
Our observations allow us to present a new model for considering how activated hypothalamic autophagy during fasting serves to turn over neuronal lipids for the generation of free fatty acids, which upregulate AgRP levels and increase food intake specifically in response to starvation (Fig. 2).21
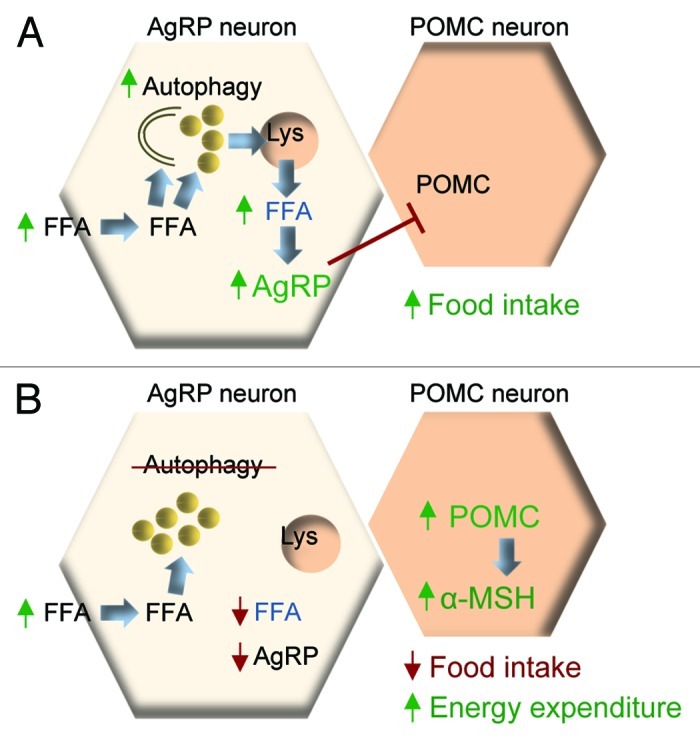
Figure 2. Autophagy in the control of food intake and energy balance. (A) During starvation, increased hypothalamic free fatty acid (FFA) uptake activates neuronal autophagy. These FFAs are rapidly esterified into triglycerides within lipid droplets (LD). Activated autophagy mobilizes LDs to generate neuron-intrinsic FFAs, which elevate AgRP levels and drive food intake. (B) Blocking autophagic mobilization of lipids during starvation reduces neuronal FFA availability, and AgRP expression. Blocking autophagy in AgRP neurons in vivo increases hypothalamic levels of POMC and α-MSH that contribute to reduced food intake, and increased energy expenditure.
What Remains Ahead?
While our findings reveal a new role for hypothalamic autophagy in control of food intake and energy balance,21 there are several new questions in place. For instance, what are the upstream signaling pathways that sense nutrients and hormones to modulate autophagy in the hypothalamus? How are autophagy-derived free fatty acids mechanistically linked to AgRP levels, for instance, do free fatty acids per se increase AgRP levels, or do free fatty acids form additional lipid species that may then regulate AgRP gene expression? Why is there a need for autophagy to generate neuron-intrinsic free fatty acids when starvation per se increases hypothalamic uptake of circulating free fatty acids? Is it possible that the neuron-intrinsic fatty acid pool is “physiologically distinct” from the adipose-derived circulating free fatty acids observed in the fasted state? Are there additional mechanisms, besides hypothalamic lipid metabolism, through which autophagy may control food intake and energy expenditure? Perhaps autophagy may directly contribute to key aspects of neuronal physiology, for example, regulation of synaptic plasticity, neuronal activation and/or neuropeptide secretion. In addition, hypothalamic autophagy may contribute to energy balance through its ability to “fine tune” the hypothalamic proteome.
The hypothalamic POMC neurons play crucial roles in regulating peripheral energy expenditure,8 which begs the next question—what is the role for autophagy in POMC neurons? Our recent work reveals that POMC neuron-selective deletion of autophagy leads to an early onset adiposity and altered glucose homeostasis,25 although in-depth studies will be required to unravel the precise function of autophagy in these neurons.
Autophagy is thought to decrease with age26 and advanced aging has been associated with reduced food intake.27 Thus, it is could be possible that chronic reduction of hypothalamic autophagy during aging may reduce food intake, as well as contribute to the metabolic dysregulation observed in the aged.28 Aging is typically associated with ectopic lipid accumulation in organs not suited for fat storage, and thus it is conceivable that chronic lipid build up in hypothalamic neurons as part of the aging process may negatively regulate autophagy. Indeed, reports reveal that high fat feeding and chronic lipid accumulation contribute to decreased autophagy in livers,5,29 which has been accredited to reduced autophagosome-lysosome fusion.30 To this end, it remains to be seen whether aged neurons display altered membrane lipid composition, which may affect these fusion events. Additionally, reduced autophagy/lysosomal function in aged hypothalamic neurons may result from accumulation of aggregates and inclusion bodies, which will affect energy metabolism, as recently demonstrated in rodents expressing aggregate-prone mutant huntingtin within the hypothalamus.31
A better understanding of how autophagy modulates neuronal physiology, as well as the detailed characterization of roles for autophagy in the hypothalamic control of energy balance will have implications for designing new strategies against obesity and insulin resistance.
Acknowledgments
This work was supported by the NIH NIDDK grant DK087776, and an Einstein Nathan Shock Basic Biology of Aging pilot grant to R.S. The author has no financial interests to declare.
Footnotes
Previously published online: www.landesbioscience.com/journals/adipocyte/article/18966
References
- 1.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–67. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 4.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neufeld TP. TOR-dependent control of autophagy: biting the hand that feeds. Curr Opin Cell Biol. 2010;22:157–68. doi: 10.1016/j.ceb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garfield AS, Lam DD, Marston OJ, Przydzial MJ, Heisler LK. Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol Metab. 2009;20:203–15. doi: 10.1016/j.tem.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Belgardt BF, Bruning JC. CNS leptin and insulin action in the control of energy homeostasis. Ann N Y Acad Sci. 2010;1212:97–113. doi: 10.1111/j.1749-6632.2010.05799.x. [DOI] [PubMed] [Google Scholar]
- 10.Mountjoy KG. Functions for pro-opiomelanocortin-derived peptides in obesity and diabetes. Biochem J. 2010;428:305–24. doi: 10.1042/BJ20091957. [DOI] [PubMed] [Google Scholar]
- 11.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–30. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 12.Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007;6:55–68. doi: 10.1016/j.cmet.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–74. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 14.Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115:951–8. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr., Xu AW, Barsh GS, et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–40. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- 16.Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011;25:310–22. doi: 10.1101/gad.1984311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao J, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy. 2008;4:378–80. doi: 10.4161/auto.5633. [DOI] [PubMed] [Google Scholar]
- 18.Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–51. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zammit VA, Arduini A. The AMPK-malonyl-CoA-CPT1 axis in the control of hypothalamic neuronal function. Cell Metab. 2008;8:175. doi: 10.1016/j.cmet.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 20.Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci. 2005;8:579–84. doi: 10.1038/nn1456. [DOI] [PubMed] [Google Scholar]
- 21.Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, et al. Autophagy in Hypothalamic AgRP Neurons Regulates Food Intake and Energy Balance. Cell Metab. 2011;14:173–83. doi: 10.1016/j.cmet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem. 2011;286:32324–32. doi: 10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chari M, Yang CS, Lam CK, Lee K, Mighiu P, Kokorovic A, et al. Glucose transporter-1 in the hypothalamic glial cells mediates glucose sensing to regulate glucose production in vivo. Diabetes. 2011;60:1901–6. doi: 10.2337/db11-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, Sahu S, et al. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012;13:258–65. doi: 10.1038/embor.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–12. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benelam B. Satiety and the anorexia of ageing. Br J Community Nurs. 2009;14:332–5. doi: 10.12968/bjcn.2009.14.8.43512. [DOI] [PubMed] [Google Scholar]
- 28.Bechtold M, Palmer J, Valtos J, Iasiello C, Sowers J. Metabolic syndrome in the elderly. Curr Diab Rep. 2006;6:64–71. doi: 10.1007/s11892-006-0054-3. [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–78. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–65. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hult S, Soylu R, Bjorklund T, Belgardt BF, Mauer J, Bruning JC, et al. Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab. 2011;13:428–39. doi: 10.1016/j.cmet.2011.02.013. [DOI] [PubMed] [Google Scholar]