

Abstract
Fat depots of different localization vary in their biological/metabolic function. We recently provided evidence for different regulation of lipolysis between perigonadal and mesenteric adipocytes; in particular insulin-induced suppression of lipolysis was significantly higher in perigonadal compared with mesenteric adipocytes in chow-fed mice. Moreover, insulin’s anti-lipolytic effect was maintained in mesenteric but lost in perigonadal adipocytes under high fat diet (HFD). Herein, we expanded our previous study and included inguinal (subcutaneous) adipocytes in our analysis. When compared with perigonadal adipocytes, inguinal adipocytes are equally sensitive to insulin’s anti-lipolytic effect under chow diet. However, they remain insulin-sensitive under HFD. Moreover, insulin-stimulated glucose incorporation was equally blunted in adipocytes of all three depots in HFD-fed mice. In conclusion, regulation of insulin sensitivity in murine adipocytes is diet-, depot- and function-dependent.
Keywords: adipocyte, fat depot, glucose incorporation, insulin resistance, insulin sensitivity, lipolysis
Fat depots of different localization vary in their biological/metabolic functions including lipid mobilization, glucose incorporation as well as adipokine and cytokine production.1,2 We recently showed that basal lipolysis is differently regulated in two different intra-abdominal (perigonadal and mesenteric) fat depots in mice.3 Whereas free fatty acid (FFA) and glycerol release were significantly higher in mesenteric compared with perigonadal adipocytes of standard chow-fed mice, basal lipolysis did not differ between these two depots in obese and glucose intolerant mice. Obesity was induced in male C57BL6J mice by feeding a high fat diet (HFD) for 8 weeks (58% of calories derived from fat compared with 12% in standard chow diet). As expected, HFD feeding impaired glucose tolerance as well as insulin sensitivity and induced hepatic steatosis.3 Of note, high fat feeding increased basal lipolysis in systemically drained perigonadal adipocytes (as shown before in ref. 4) but not in portally drained mesenteric adipocytes. Such finding was reflected and supported by the fact that a fat enriched diet led to an increase in systemic but no portal FFA levels.3 In contrast to similar lipolysis in mesenteric adipocytes of chow- and HFD-fed mice, cytokine release was clearly diet-regulated in portally drained mesenteric adipocytes. There was an increased release of the pro-inflammatory cytokines interleukin-6 (IL-6) and monocyte chemoattractant protein-1 (MCP-1) from mesenteric adipocytes of high fat diet compared with chow-fed mice,3 whereas secretion of the anti-inflammatory protein interleukin-10 (IL-10) tended to decrease. In the light of the “portal theory,” which proposed that the direct exposure of the liver to increasing amounts of free fatty acids and/or pro-inflammatory factors released from visceral fat into the portal vein promotes the development of hepatic insulin resistance and liver steatosis,5-7 our findings suggest a more important role for (pro-inflammatory) cytokines rather than FFAs in visceral obesity-associated (hepatic) insulin resistance in mice.
Remarkably, the ability of insulin to inhibit lipolysis was blunted in HFD-fed mice only in adipocytes isolated from the perigonadal fat depot but not in mesenteric adipocytes.3 To investigate whether such difference in insulin sensitivity is general or limited to certain function of insulin (i.e., inhibition of lipolysis vs. stimulation of glucose incorporation), insulin-stimulated glucose incorporation into isolated adipocytes was determined. Since most of the labeled glucose is incorporated into lipids such experiments are mainly a readout for lipid synthesis.8 Moreover, to have a broader overview regarding regulation of insulin sensitivity in different fat depots of chow and HFD-fed mice, we included subcutaneous (inguinal) besides perigonadal and mesenteric adipocytes into our analyses.
As shown for perigonadal and mesenteric adipocytes,3 diameter of inguinal adipocytes was significantly higher in HFD-fed compared with chow-fed mice (chow-fed 44.3 ± 0.9 µm vs. HFD 66.1 ± 2.3 µm, p < 0.001). Similarly, release of lactate dehydrogenase (LDH) from inguinal adipocytes was increased under HFD (chow-fed 2.3 ± 0.5 LDH/106 cells vs. HFD 8.2 ± 0.8 LDH/106 cells, p < 0.001). However, there was no significant difference in LDH release between adipocytes of all three depots for both diets [chow-fed: perigonadal 5.9 ± 1.1 LDH/106 cells vs. inguinal 2.3 ± 0.5 LDH/106 cells vs. mesenteric 5.4 ± 1.4 LDH/106 cells, p = 0.08 (ANOVA); HFD-fed: perigonadal 14.4 ± 2.9 LDH/106 cells vs. inguinal 8.2 ± 0.8 LDH/106 cells vs. mesenteric 10.2 ± 2.3 LDH/106 cells, p = 0.17 (ANOVA)] suggesting (1) comparable viability of adipocytes isolated from different depots and (2) increased vulnerability of adipocytes isolated from HFD-fed animals.
Basal FFA release was significantly lower in inguinal compared with mesenteric adipocytes both under chow and high fat diet (Fig. 1A). Of note, basal lipolysis was significantly increased in perigonadal adipocytes of obese compared with lean mice (Fig. 1A and shown in ref. 3), but not in inguinal and mesenteric adipocytes. In order to have a functional readout for stimulated lipolysis, we assessed the ability of isoproterenol to induce release of FFAs. Under either diet, the fold increase in FFA release upon isoproterenol stimulation was significantly higher in inguinal compared with mesenteric adipocytes [chow-fed: perigonadal 7.5 ± 1.2-fold vs. inguinal 11.7 ± 2.6-fold vs. mesenteric 2.6 ± 0.4-fold, p < 0.05 (ANOVA); HFD-fed: perigonadal 5.5 ± 1.0-fold vs. inguinal 10.3 ± 1.7-fold vs. mesenteric 5.1 ± 1.4-fold, p < 0.05 (ANOVA)]. In chow-fed mice, the anti-lipolytic effect of insulin was similar in perigonadal and inguinal adipocytes but clearly higher compared with mesenteric adipocytes (Fig. 1B). As reported previously, insulin’s ability to inhibit lipolysis was almost completely blunted in perigonadal adipocytes whereas it was maintained in mesenteric adipocytes.3 Similar to mesenteric adipocytes, the anti-lipolytic effect of insulin was conserved in inguinal adipocytes of HFD-fed mice (Fig. 1B).
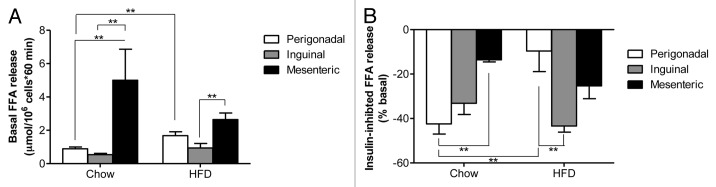
Figure 1. Basal and insulin-inhibited FFA release in adipocytes isolated from different fat depots of chow and HFD-fed mice. Basal (A) and insulin-inhibited (B) FFA release from isolated adipocytes of chow-fed (left bars) and HFD-fed (right bars) mice is depicted. Results are the means ± SEM of 3–9 independent experiments. **p < 0.01 (Student’s t-test, ANOVA).
Basal glucose incorporation was significantly higher in perigonadal compared with inguinal and mesenteric adipocytes of chow-fed mice whereas such difference was less marked in HFD-fed mice (Fig. 2A). Of note, basal glucose incorporation was significantly increased in all analyzed fat depots of HFD-fed mice when compared with chow-fed mice. In contrast to its effect on lipolysis, insulin’s ability to stimulated glucose incorporation was significantly higher in mesenteric compared with perigonadal adipocytes in chow-fed mice (Fig. 2B). However, this difference completely vanished in HFD-fed mice (Fig. 2B). Moreover, adipocytes of all analyzed depots showed significantly blunted insulin-stimulated glucose incorporation suggesting a similar degree of insulin resistance to glucose incorporation under HFD in all depots. GLUT4 protein levels were decreased to similar levels in all analyzed depots of obese mice (Fig. 2C). Hence, the HFD-induced reduction and convergence of GLUT4 protein content might be partially responsible for the observed reduction in insulin-stimulated glucose incorporation (Fig. 2B). Accordingly, reduction in GLUT4 was previously suggested to contribute to the development of insulin resistance in murine and human adipocytes.9,10 In contrast, GLUT4 levels do not explain observed differences in insulin-stimulated glucose incorporation in chow fed mice, as GLUT4 levels were highest in inguinal fat depots (Fig. 2C).
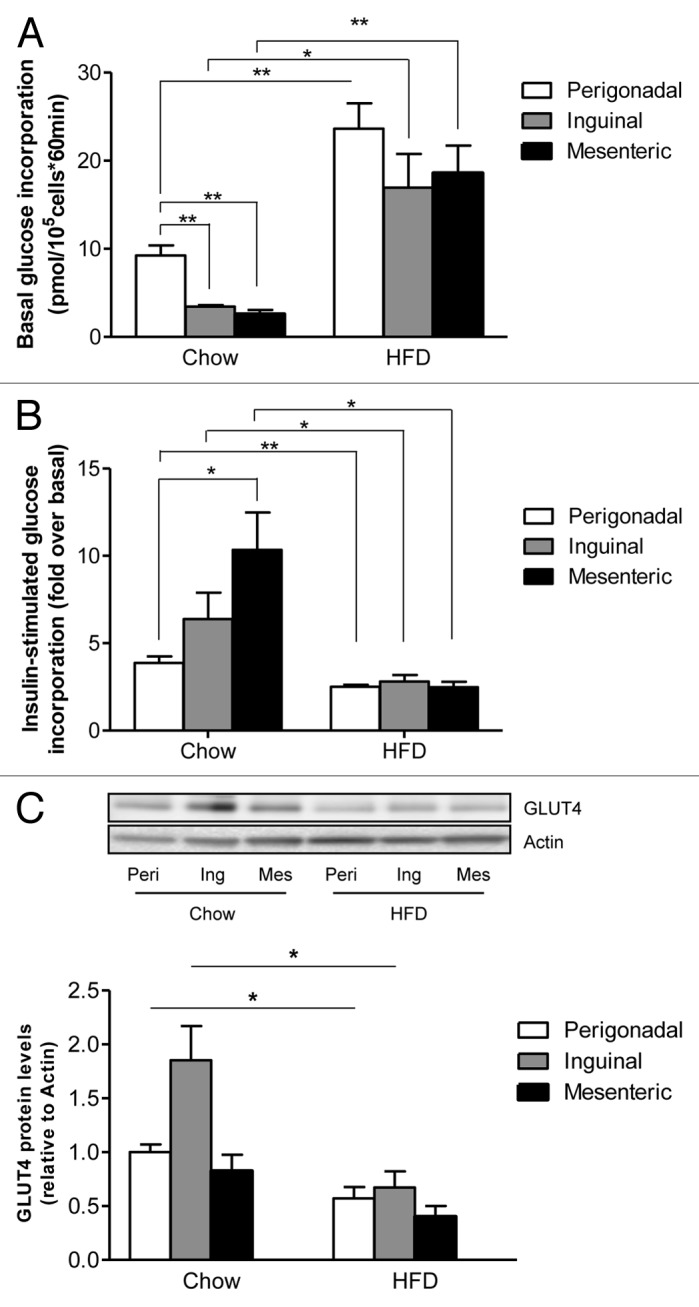
Figure 2. Basal and insulin-stimulated glucose incorporation in adipocytes isolated from different fat depots of chow and HFD-fed mice. Basal (A) and insulin-stimulated (B) 14C-D-glucose incorporation into isolated adipocytes of chow-fed (left bars) and HFD-fed (right bars) mice is presented. (C) GLUT4 protein levels were determined in different adipose depots of chow and HFD-fed mice and normalized to actin levels. Peri, perigonadal; Ing, inguinal; Mes, mesenteric. Results are the means ± SEM of 3–5 independent experiments. *p < 0.05, **p < 0.01 (Student’s t-test, ANOVA).
The aim of the present study was to expand our previous data comparing regulation of lipolysis in perigonadal and mesenteric fat depots of chow and HFD-fed mice.3 We found that under both chow and high fat diet basal lipolysis was significantly higher in mesenteric (visceral) adipocytes when compared with inguinal (subcutaneous) adipocytes. In addition, the anti-lipolytic effect of insulin was diminished in mesenteric adipocytes. Such finding may imply that increased release of FFAs from mesenteric fat depots plays an important role in the induction of obesity-associated hepatic insulin resistance and, hence, in the context of the portal theory. However, basal lipolysis in mesenteric adipocytes did not increase upon HFD. In accordance, FFA concentration in portal blood was similar in chow and high fat diet-fed animals even though the latter developed (hepatic) insulin resistance and steatosis.3 In contrast, we observed an increased release of pro-inflammatory cytokines such as IL-6 from mesenteric adipocytes of high fat- compared with standard diet-fed mice. Thus, the role of FFAs in the context of the portal theory needs to be further analyzed. In addition, the increased release of pro-inflammatory cytokines into the portal circulation may critically contribute to the portal theory. On the other hand, it is conceivable that elevated basal lipolysis in mesenteric adipocytes negatively affects the ability of insulin to inhibit FFA release.
In chow-fed mice, mesenteric adipocytes were most sensitive to insulin’s ability to stimulate glucose incorporation but least responsive to its ability to inhibit lipolysis. This fact suggests a higher relative metabolic turnover rate in mesenteric adipocytes postprandially. Moreover, these findings are in agreement with studies performed in human adipose tissue, which reported higher insulin-stimulated glucose uptake but decreased anti-lipolytic effect of insulin in visceral compared with subcutaneous fat.1
Whereas glucose incorporation into adipocytes of all three depots became equally resistant to insulin in HFD-fed mice (which might be partly explained by HFD-induced reduction of GLUT4 protein to similar levels in all three depots), insulin’s ability to suppress lipolysis was maintained in inguinal and mesenteric adipocytes whereas it was significantly blunted in perigonadal adipocytes. Thus, insulin sensitivity in murine adipocytes is regulated differently dependent on its localization and on its function; particularly insulin-stimulated glucose incorporation appears to be more prone to develop insulin resistance than lipolysis under a HFD challenge. In adipocytes, insulin-stimulated glucose uptake/incorporation is regulated via phosphorylation of Akt and translocation of GLUT4 to the plasma membrane, whereas lipolysis is inhibited via Akt-mediated phosphorylation/activation of phosphodiesterase 3B.11 Potentially, insulin signaling downstream of Akt may be differently affected/modulated in adipocytes of different fat pads thereby explaining the observed differences in the anti-lipolytic effect of insulin. Moreover, our data may also explain clinical observations showing maintained anti-lipolytic effect of insulin in patients with type 2 diabetes despite severe total body insulin resistance whereas patient with type 1 diabetes develop severe ketoacidosis as a consequence of unopposed lipolysis in the absence of insulin. Clearly, further studies are needed to unravel pathways involved in the regulation of insulin sensitivity to explain depot and function specific differences.
In conclusion, regulation of insulin sensitivity in murine adipocytes is depot-, diet- and function-dependent.
Materials and Methods
Animals
Six- to eight-week-old male C57BL6JOlaHsd mice were fed ad libitum with standard rodent diet or HFD (D12331, Research Diets) for eight weeks. HFD consisted of 58% of calories derived from fat, 28% from carbohydrate and 16% from protein. All protocols conformed to the Swiss animal protection laws and were approved by the Cantonal Veterinary Office in Zurich, Switzerland.
Viability assessment and cell size determination
Adipocytes were isolated and viability was determined with an LDH assay as described previously.12 Aliquots of isolated adipocytes were used to determine mean cell diameters. Photographs of isolated adipocytes were taken in the hematocytometer and images were analyzed using ImageJ software for quantification (National Institutes of Health). At least 100 adipocytes per four independent experiments were analyzed.
Glucose incorporation into isolated white adipocytes
Adipocyte isolation and glucose incorporation was performed as described previously.12-14 Briefly, adipocytes were incubated for 60 min with D-[U-14C]-glucose. In this setting, most of the labeled glucose is incorporated into lipids and therefore is a readout for lipid synthesis.8 Glucose incorporation was stopped by separating cells from the medium by centrifugation through phthalic acid dinonyl ester. Cells were then subjected to liquid scintillation counting. Cell number was determined with a hematocytometer under the light microscope.
Lipolysis assays
To assess lipolysis, isolated adipocytes were incubated in the absence or presence of 100 nM insulin or 1 μM isoproterenol (Sigma) for one hour. FFA levels were measured using the ACS-ACOD-MEHA method from Wako Chemicals GmbH.
Western blotting
Tissue samples were homogenized and western blotting was performed as described previously.13 The following primary antibodies were used: anti-GLUT4 (kind gift of Dr Amira Klip, The Hospital for Sick Children, Toronto, Canada), and anti-actin (Millipore). Membranes were exposed in an Image Reader and analyzed with Image Analyzer (FujiFilm). One of the perigonadal samples (chow-fed) was loaded on every gel and was used as a reference band for quantification; i.e., all bands on a membrane were normalized to the expression levels of this sample.
Data analysis
Statistical analyses were performed using unpaired Student’s t-test or ANOVA (Tukey’s multiple comparisons test).
Acknowledgments
This work was supported by research grants from the Swiss National Science Foundation 310030-124729 (to D.K.).
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/adipocyte/article/19910
References
- 1.Yang X, Smith U. Adipose tissue distribution and risk of metabolic disease: does thiazolidinedione-induced adipose tissue redistribution provide a clue to the answer? Diabetologia. 2007;50:1127–39. doi: 10.1007/s00125-007-0640-1. [DOI] [PubMed] [Google Scholar]
- 2.Yu R, Kim CS, Kwon BS, Kawada T. Mesenteric adipose tissue-derived monocyte chemoattractant protein-1 plays a crucial role in adipose tissue macrophage migration and activation in obese mice. Obesity (Silver Spring) 2006;14:1353–62. doi: 10.1038/oby.2006.153. [DOI] [PubMed] [Google Scholar]
- 3.Wueest S, Yang X, Liu J, Schoenle EJ, Konrad D. Inverse regulation of basal lipolysis in perigonadal and mesenteric fat depots in mice. Am J Physiol Endocrinol Metab. 2012;302:E153–60. doi: 10.1152/ajpendo.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaidhu MP, Anthony NM, Patel P, Hawke TJ, Ceddia RB. Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by high-fat diet: role of ATGL, HSL, and AMPK. Am J Physiol Cell Physiol. 2010;298:C961–71. doi: 10.1152/ajpcell.00547.2009. [DOI] [PubMed] [Google Scholar]
- 5.Kabir M, Catalano KJ, Ananthnarayan S, Kim SP, Van Citters GW, Dea MK, et al. Molecular evidence supporting the portal theory: a causative link between visceral adiposity and hepatic insulin resistance. Am J Physiol Endocrinol Metab. 2005;288:E454–61. doi: 10.1152/ajpendo.00203.2004. [DOI] [PubMed] [Google Scholar]
- 6.Rytka JM, Wueest S, Schoenle EJ, Konrad D. The portal theory supported by venous drainage-selective fat transplantation. Diabetes. 2011;60:56–63. doi: 10.2337/db10-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen MD. Role of body fat distribution and the metabolic complications of obesity. J Clin Endocrinol Metab. 2008;93(Suppl 1):S57–63. doi: 10.1210/jc.2008-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gliemann J, Gammeltoft S, Vinten J. Time course of insulin-receptor binding and insulin-induced lipogenesis in isolated rat fat cells. J Biol Chem. 1975;250:3368–74. [PubMed] [Google Scholar]
- 9.Garvey WT. Glucose transport and NIDDM. Diabetes Care. 1992;15:396–417. doi: 10.2337/diacare.15.3.396. [DOI] [PubMed] [Google Scholar]
- 10.Carvalho E, Jansson PA, Nagaev I, Wenthzel AM, Smith U. Insulin resistance with low cellular IRS-1 expression is also associated with low GLUT4 expression and impaired insulin-stimulated glucose transport. FASEB J. 2001;15:1101–3. [PubMed] [Google Scholar]
- 11.Zmuda-Trzebiatowska E, Manganiello V, Degerman E. Novel mechanisms of the regulation of protein kinase B in adipocytes; implications for protein kinase A, Epac, phosphodiesterases 3 and 4. Cell Signal. 2007;19:81–6. doi: 10.1016/j.cellsig.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 12.Wueest S, Rapold RA, Rytka JM, Schoenle EJ, Konrad D. Basal lipolysis, not the degree of insulin resistance, differentiates large from small isolated adipocytes in high-fat fed mice. Diabetologia. 2009;52:541–6. doi: 10.1007/s00125-008-1223-5. [DOI] [PubMed] [Google Scholar]
- 13.Wueest S, Rapold RA, Schumann DM, Rytka JM, Schildknecht A, Nov O, et al. Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest. 2010;120:191–202. doi: 10.1172/JCI38388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wueest S, Rapold RA, Schoenle EJ, Konrad D. Fas activation in adipocytes impairs insulin-stimulated glucose uptake by reducing Akt. FEBS Lett. 2010;584:4187–92. doi: 10.1016/j.febslet.2010.08.052. [DOI] [PubMed] [Google Scholar]