

Abstract
Abnormal differentiation of the renal stem/progenitor pool into kidney tissue can lead to renal hypodysplasia (RHD), but the underlying causes of RHD are not well understood. In this multicenter study, we identified 20 Israeli pedigrees with isolated familial, nonsyndromic RHD and screened for mutations in candidate genes involved in kidney development, including PAX2, HNF1B, EYA1, SIX1, SIX2, SALL1, GDNF, WNT4, and WT1. In addition to previously reported RHD-causing genes, we found that two affected brothers were heterozygous for a missense variant in the WNT4 gene. Functional analysis of this variant revealed both antagonistic and agonistic canonical WNT stimuli, dependent on cell type. In HEK293 cells, WNT4 inhibited WNT3A induced canonical activation, and the WNT4 variant significantly enhanced this inhibition of the canonical WNT pathway. In contrast, in primary cultures of human fetal kidney cells, which maintain WNT activation and more closely represent WNT signaling in renal progenitors during nephrogenesis, this mutation caused significant loss of function, resulting in diminished canonical WNT/β-catenin signaling. In conclusion, heterozygous WNT4 variants are likely to play a causative role in renal hypodysplasia.
ESRD in children most commonly results from congenital anomalies of the kidney and urinary tract.1 The most common congenital anomalies of the kidney and urinary tract clinic-pathologic type is renal hypodysplasia (RHD).2 RHD can be diagnosed sporadically or with familial aggregation.1,3–6 For familial cases, the suggested mode of inheritance in most pedigrees is autosomal dominant with variable expression and reduced penetrance, estimated to range between 50% and 90%.7
The pathologic basis of RHD is the disturbance of normal nephrogenesis, possibly due to mutations in genes that direct the process.1,8 Most of the genes known to be involved are transcriptional factors and genes that encode for proteins involved in the mesenchymal to epithelial transition.2,8 To date, most forms of RHD have been found negative for abnormalities in recognized renal developmental genes,3,9,10 and it is highly likely that other, still unreported, genes will be identified, especially genes for which renal maldevelopment has been demonstrated in genetically modified models.11
We investigated the prevalence of mutations in 9 kidney developmental genes among a selected group of 20 families with isolated nonsyndromic familial RHD. We showed that mutations are present in a significant part of this group, and we outlined the possible role of mutated WNT4 as a cause for disturbed early kidney development leading to the RHD phenotype. Viewing RHD as a disease of the renal stem/progenitor cell pool, we hypothesized that human developing kidney cell–based systems would be extremely useful for disease modeling.
Our study group comprised 51 RHD-affected individuals and 91 unaffected family members from 20 unrelated families. The families’ clinical characteristics are shown in Supplemental Figure 1 and Supplemental Table 1. We identified 13 mutation-carrying participants from four unrelated families harboring mutations in three different genes: one family with a PAX2 mutation, two families with HNF1B mutations (for full genetic and clinical characterization see Supplemental Appendix), and one family (family 4) demonstrating a novel heterozygous missense variant in the human WNT4 gene. The latter family includes two affected brothers with severe left RHD who were found to harbor a novel missense WNT4 variant, c.t191c– p.M64T. This is the first description of an association between a WNT4 heterozygous variant and isolated human RHD. Genotype-phenotype correlation in this family revealed an autosomal dominant pattern of inheritance with incomplete penetrance, similar to other renal hypodysplasia-causing genes (Figure 1).
Figure 1.

Mutation analysis of the p.M64T, WNT4 variant shows high degree of conservation. (A) Sequence analysis reveals the WNT4 variant caused by heterozygous transition (c.t191c) (lower panel, blue arrow) resulting in amino acid substitution M64T. The wild-type (WT) sequence is given for comparison (upper panel). (B) WNT4 variant is found in patients 1, 3, 4, and 5. Whereas patients 3 and 4 have a clear isolated renal phenotype of severe left renal hypodysplasia, patient 1 has an isolated left renal cyst (sized 12.8×7 mm) in addition to normal BP and normal GFR. Patient 5, age 3 years, has normal pelvic and renal ultrasound results, normal BP, normal GFR, and low morning serum cortisol levels. Incomplete penetrance or variable expression can be considered in this family. Squares indicate male family members and circles female family members; black filled squares indicate that the patients are affected with RHD. Gray filled squares and circles indicate subtle clinical signs. (C) cDNA sequences of human WNT4 are compared with WNT4 orthologs in other species. (D) Conservation scale of WNT4 protein shows the amino acid methionine in position 64 (blue arrow) to be highly conserved (8 of 9), with “b” indicating a buried residue and “e” indicating an exposed residue. Note the high degree of conservation.
The WNT4 p.M64T variant affects a highly conserved methionin residue found in all living organisms for which the WNT4 sequence is known (Figure 1), suggesting possible functional importance. The variant was not found in a search of the single nucleotide polymorphism (SNPs) or mutation databases (dbSNP, 1000 genomes [http://browser.1000genomes.org/index.html]; Human Gene Mutation Database HGMD [http://www.hgmd.cf.ac.uk/ac/index.php]). Automatic computer prediction for possible effects of amino acid substitution on the structure and function of the WNT4 protein using PolyPhen software predictions (http://genetics.bwh.harvard.edu/pph)12 showed that this variant may be possibly damaging (position-specific independent count [PSIC] score, 1.79). Finally, screening of 280 ethnically matched control participants (560 alleles) for presence of the WNT4 variant was negative, suggesting that this is not a common polymorphism.
Further evidence supporting the causality of this rare WNT4 variant include that heterozygous mutation in this gene is known to cause renal malformation as part of the general Mayer–Rokitansky-like syndrome that was first described in an 18-year-old female presenting with primary amenorrhea and a single kidney.13 This is strikingly similar to the WNT4 knockout mouse model that showed defective mesenchymal-epithelial transition (MET), lack of nephron formation, and development of renal hypodysplasia,14,15 in addition to defective sexual differentiation exclusive to the female model. Supporting our findings, the male mouse model showed only isolated RHD, similar to both affected males described herein.15 The interdependence among other WNT proteins involved in nephrogenesis and different WNT pathways may explain why some of the phenotypic characteristics of this syndrome have incomplete penetrance.16 Importantly, our in vitro studies, which are described as follows, support the deleterious effect of this variant.
The best understood mechanism of WNT signaling is the canonical pathway that activates the nuclear functions of β-catenin, leading to changes in gene expression that influence proliferation and survival.17 Consequently, using human embryonic kidney cell line (HEK293) as a first platform, we assessed the consequence of the WNT4 variant by cell proliferation assay and TOPFlash reporter assay18 to study the canonical WNT signaling pathway. We found that the WNT4 p.M64T variant leads to significantly increased cell proliferation compared with the wild-type WNT4 (Figure 2C). Interestingly, analysis of Wnt/β-catenin target genes by real-time PCR after introduction of wild-type WNT4 or WNT4 M64T failed to show upregulation (Figure 2A). Moreover, activation of the canonical WNT pathway via the WNT3A ligand, a known activator of the canonical pathway in HEK293, showed that the wild-type WNT4 ligand is an inhibitor of the canonical pathway, in accordance with previous report19 (Figure 2B). Strikingly, in HEK293, the mutant WNT4 (p.M64T) led to significantly enhanced inhibition of the canonical WNT pathway compared with wild-type control, revealing a functional cell phenotype (Figure 2D).
Figure 2.

Functional analysis of the p.M64T,WNT4 variant in HEK293 shows enhanced inhibition of the canonical WNT pathway. (A) WNT4, wild-type and mutant, does not increase WNT canonical pathway target genes. Quantitative RT-PCR on HEK293 transfected cells with WNT4 wild-type (WT) and mutant (Mut) plasmids shows the mRNA expression levels of canonical WNT pathway target genes (MYC, CCND1, and AXIN). (B) WNT3A, but neither wild-type WNT4 nor mutant WNT4, activates the canonical WNT pathway. HEK293 cells are cotransfected on a 6-well plate, with TOPFlash reporter plasmid (4 µg) and either wild-type WNT4, mutant WNT4, or WNT3A expression plasmid (4 µg). pCMV-Renilla plasmid (0.4 µg) is used as the internal control. (C) Wild-type WNT4 and mutant variants increase cell proliferation compared with control. Mutant WNT4 leads to significantly increased proliferation compared with wild-type WNT4. *P<0.05. Results are presented as the mean absorbance at 492 nm using the MTS proliferation assay ± SEM of at least three replicates. (D) WNT4, wild-type and mutant, inhibit the canonical WNT pathway. WNT4 mutant compared with the wild-type leads to significantly enhanced inhibition of the canonical WNT pathway. Cells are cotransfected with TOPFlash reporter plasmid (500 ng), WNT3A expression plasmid (200 ng) to activate the canonical WNT pathway, and an increasing concentration of WNT4 wild-type or WNT4 mutant expression plasmid (200, 400, 800, and 1600 ng). pCMV-Renilla plasmid is used as an internal control (50 ng). Cell lysates are measured for luciferase activity 48 hours after transfection. Activities are expressed as fold activation of the relative luciferase activity. WNT3A and WNT4 does not activate the mutant TOPFlash reporter, FOPFlash, confirming assay specificity. Asterisks indicate a significant difference of the mutant WNT4 compared with dose-equivalent WNT4 wild-type transfection. *P<0.05; **P<0.01 (t test). Data are presented as the mean ± SEM from three separate experiments.
To further investigate the functional effect of the WNT4 variant on the canonical WNT pathway, we performed functional analysis experiments using a more physiologic disease model. We aimed for the original cells where the genetic insult first initiates its effect to produce RHD, hence, human developing kidney cells. Consequently we used primary cultures of human fetal kidneys (HFK-PC) obtained after curettage of elective abortions performed during the 18th week of gestation.
Cell characterization of low passage HFK-PC demonstrated that it comprises a significant portion of cells with renal epithelial stem/progenitor features. Expanded cells exhibit the morphology and clonogenic growth properties of renal epithelial stem/progenitors, express renal progenitor marker genes and proteins, and are devoid of hematopoietic and endothelial markers. Furthermore, HFK-PC cells exhibit basal canonical WNT activity and therefore are a reliable and relevant disease modeling platform (Figure 3).
Figure 3.
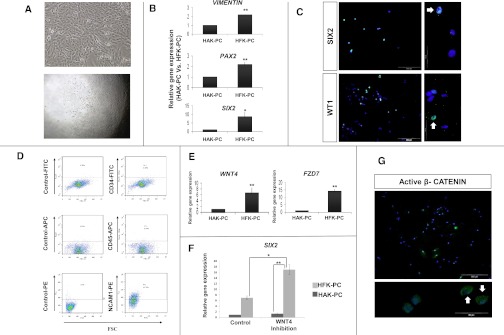
Human fetal kidney cells harbor renal epithelial stem/progenitor characteristics and exhibit basal canonical WNT activity. (A–D) HFK-PC cells express renal stem/progenitor markers. (A) Cellular appearance of cultured fetal kidney cells (upper panel) and clone formation by human fetal kidney cells, demonstrating their high clonogenic capacity. Culturing of 0.3 cells per well results in 5%–10% of single clone formation (lower panel). (B) Gene expression analysis of renal stem/progenitor genes. Quantitative RT-PCR (qat-PCR) analysis of Vimentin, PAX2, and SIX2, three representative genes expressed early during nephrogenesis. Normalization is performed against control GAPDH expression and real-time quantitative is calculated relative to the well differentiated HAK-PC cells. (C) Immunofluoresence staining for the expression of two representative early nephrogenesis transcriptional factors: WT1 and SIX2 (×20 and ×100). All nuclei are stained with DAPI (blue). The green florescence signal in the upper and lower panels corresponds to anti-SIX2 protein and anti-WT1 protein staining respectively. Both clearly show nuclear staining (representative stained nuclei are indicated with white arrows). (D) Flow cytometric analysis for CD34, CD45, and CD56/NCAM1 expression and corresponding isotype controls in HFK-PC cells. Results show negligible levels of CD34 (a well known marker of hematopoietic stem cells and vascular endothelial cells) and CD45 (leukocyte common antigen). Moreover, the HFK-PC contains 44% of CD56/NCAM1-positive cells. NCAM1 has been previously shown to be a stem/progenitor cell marker in the human fetal kidney.31 (E–G) HFK-PC cells harbor basal canonical WNT activity. (E) qRT-PCR analysis of WNT4 and frizzled 7 (FZD7), a representative receptor of the WNT signaling ligands. (F) Because WNT/β-catenin and SIX2 pathways have opposing actions (commitment and self-renewal of renal stem/progenitors, respectively), we sought to manipulate this balance so as to provide additional support that our HFK-PC cells contain a significant portion of cells with early renal stem/progenitor characteristics. Consequently, qRT-PCR analysis of SIX2, in HFK-PC after the addition of three different WNT pathway antagonists—dickkopf-related protein 1 (DKK1), secreted frizzled-related protein 1 (sFRP1), and Wnt inhibitory factor (WIF)—shows significant SIX2 increment compared with the control. Normalization is performed against control GAPDH expression and real-time quantitative is calculated relative to the well differentiated HAK-PC cells. Data are presented as the mean ± SEM from three separate experiments. (G) HFK-PC immunofluorescence staining for anti-active β-catenin (green) discloses its cytoplasmic and nuclear presence. All nuclei are stained with DAPI (blue). *P<0.05; **P<0.01 (t test). DAPI, 4',6-diamidino-2-phenylindole; NCAM-1, neural cell adhesion molecule 1.
Next, we assessed canonical pathway activation using the TOPFlash reporter assay; activation was only evident with wild-type WNT4. Neither WNT3A nor the WNT4 variant was found to activate this pathway (Figure 4A). Finally, we tested the consequence of the WNT4 variant using conditioned media experiments. We chose this type of experiment because WNT4 is a secreted glycol-protein that acts as a short-range ligand on the cell surface. HFK-PC cells were stimulated with conditioned medium from HEK293 cells transfected with wild-type WNT4, mutant, or empty vector (control) (Figure 4B). In contrast to wild-type WNT4, both mutant WNT4 and control conditioned media did not increase activated β-catenin levels nor it target gene levels in the treated HFK-PC cells (Figure 4, C and D).
Figure 4.

Functional analysis of the p.M64T, WNT4 variant in HFK-PC shows significant loss of function and diminished canonical WNT/b-catenin signaling. (A) Wild-type WNT4, but neither WNT3A nor mutant WNT4, activates the canonical WNT pathway in HFK-PC. HFK-PC cells are cotransfected on a 6-well plate, with TOPFlash reporter plasmid (4 µg) and wild-type WNT4, mutant WNT4, or WNT3A expression plasmid (4 µg). pCMV-Renilla plasmid (0.4 µg) is used as internal control. *P<0.05 (t test). Data are presented as the mean ± SEM from three separate experiments. (B) Illustration of the conditioned media experiments. HEK293 cells are transfected separately with wild-type WNT4, mutant WNT4, or empty vector (control). Conditioned media containing WNT proteins were applied on HFK-PC 24 hours after transfection. Cells are harvested for total RNA and total protein 6–24 hours later. (C) Results of a representative Western blot analysis of the HFK-PC cells after they are treated with wild-type WNT4, mutant WNT4, and control conditioned media, with the use of anti-active β-catenin antibody. Cbl (95 kD) is used as a loading control. Active β-catenin protein levels are adjusted to Cbl and are quantified compared with the control (empty vector). Three separate experiments yield a similar result in which wild-type WNT4 induces a significant increase in active β-catenin as opposed to the p.M64T Wnt4 variant, which results in a nonsignificant change. (D) Quantification by real-time RT-PCR of the expression of mRNA axin2 and ccnd1 in HFK-PC after treatment with wild-type WNT4, mutant WNT4, and control conditioned media.
WNT4 belongs to the WNT family, a large group of secreted glycoproteins encoded by 19 distinct genes involved in the WNT signaling pathway.17 In mammals, WNT4 function has mainly been studied during nephrogenesis because mice lacking WNT4 die shortly after birth as a result of kidney failure secondary to severe bilateral RHD.20 The mouse model has also shown that suboptimal WNT4 signaling results in RHD.14 During normal kidney development WNT4 plays an essential role in induction of mesenchymal progenitor cells to epithelialization (MET) and generation of pretubular aggregates, leading to renal vesicles and eventually tubule formation.21–23 Recapitulating normal development, WNT4 was also demonstrated to be upregulated in the proximal tubules during recovery from AKI24 and has a suggested role in the pathogenesis of renal fibrosis.25 Elegant studies have shown that WNT/β-catenin signaling is a necessary and sufficient trigger of the early stages of nephron differentiation.26,27 We linked the human WNT4 variant to defective WNT signaling by two independent model systems. In the first model, utilizing HEK293, WNT4 by itself does not trigger WNT/β-catenin signaling but rather inhibits a Wnt3A-induced signal, whereas the WNT4 variant induces significantly exaggerated inhibition. Although these data clearly show a differential response, they might not mimic renal development because the WNT3A ligand is not a physiologic participant in the process.28 Accordingly, in the second disease model utilizing early developing human kidney cells, design principles appear to more closely simulate renal development and WNT signaling due to the following findings. First, WNT3A fails to activate the canonical WNT signaling. Second, wild-type WNT4 is overexpressed. Third, a basal activation of canonical WNT pathway is present. Finally, wild-type WNT4 induces canonical activation on protein and target gene level, whereas the p.M64T WNT4 variant fails to do so. Clearly not all of the components required to study WNT signaling in human kidney development are present in HEK293.29 Given the fact that WNT4 does not stimulate canonical WNT signaling in HEK293, we hypothesized that the observed inhibition of the WNT3A-induced canonical WNT activation by WNT4 may be a result of competitive inhibition at the receptor level. In line with our findings and hypothesis, the ability of one WNT ligand to function in two distinct pathways based on receptor context was demonstrated with WNT5A.30
We therefore favor the mechanism delineated in the more physiologic HFK-PC model that is congruent with data generated in mouse models whereby loss-of-function mutation overall reduces WNT/β-catenin signaling, leading to lack of proper nephron differentiation and proliferation of undifferentiated cell types and resulting in a isolated phenotype of maldeveloped dysplastic kidney.
Our results clearly show that studying mutations in cell lines may pose problems in interpretation of mechanism, as opposed to human fetal kidney–derived primary cell models that can be further generalized to study mechanisms of other mutations affecting renal developmental diseases.
Concise Methods
Patients
A nationwide multicenter collaboration study was conducted, with six pediatric nephrology units in Israel enrolled in a prospective trial. The study comprised Israeli pedigrees identified using index pediatric patients with isolated, nonsyndromic, familial RHD. The diagnosis of isolated RHD was made by pediatric nephrologists and radiologists based on sonographic imaging studies. RHD was defined as familial when at least a second case of RHD was reported and verified in the proband's immediate family. Excluded from the study were patients with RHD associated with one of the following: posterior urethral valve, primary bladder abnormalities, previously identified or complex syndromes, and extrarenal major malformations.
The study was approved by the national and local Helsinki committees, and informed assent and/or consent for genetic screening was obtained from the patients and/or parents, as appropriate.
Genetic Analyses
Mutation analysis was carried out on DNA extracted according to standard methods from peripheral blood obtained from 20 probands. If mutations were detected, all additional family members were sequenced to look for segregation. Amplified products were initially screened using denaturing high-performance liquid chromatography (DHPLC). All 57 exons of nine candidate genes—PAX2, HNF1B, EYA1, SIX1, SIX2, SALL1, WNT4, GDNF, and WT1—were individually amplified using exon-flanking primers. DNA sequencing was conducted on the DHPLC-positive PCR products. After positive DHPLC products were sequenced, synonymous SNPs or variants previously reported in public SNP databases were excluded from further analysis.
DHPLC
Scanning for DNA mutations and variants using DHPLC involves subjecting PCR products to chromatography using an ion-pair reversed-phase cartridge (PCR primers are available upon request). PCR products are denaturated and allowed to re-anneal. Under conditions of partial denaturation with a linear acetonitrile gradient, heteroduplexes from PCR samples with internal sequence variation display reduced column retention time relative to their homoduplex counterparts. The elution profile for a heterozygous sample is typically quite distinct from that of either homozygous sequence, making identification of heterozygous mutations relatively straightforward.
Multiplex Ligation-Dependent Probe Amplification
Deletion screening of HNF1B was carried out as follows: All index patients were screened for gene deletions by the multiplex ligation-dependent probe amplification (MLPA) method using the SALSA MLPA kit P153EYA1 (MCR-Holland, Amsterdam, The Netherlands) in the conditions suggested by the manufacturer. Amplified samples were denatured and separated by capillary electrophoresis on an ABI 3130 sequencer (Applied Biosystems, Carlsbad, CA).
Expression Studies
Plasmids
The full-length WNT4 cDNA was cloned into pCMV6-AC-GFP (OG-RC-209205; OriGene Technologies Inc, Rockville, MD). The WNT4 variant c.t191c (p.M64T) was introduced to pCMV6-MYC-GFP -WNT4 by a QuikChange II XL Site-Directed Mutagenesis kit (200521; Agilent Technologies, Santa Clara, CA) using 5′-GCGGAACCTGGAAGTCACGGACTCGGTGCGCCGCG-3′ and 5′-CGCGGCGCACCGAGTCCGTGACTTCCAGGTTCCGC-3′ primers. The plasmids were verified by direct sequencing.
Cell Culture and Transfections
Renal HEK293 cells were grown in DMEM supplemented with 10% FCS, penicillin (100 U/ml), and streptomycin (100 µg/ml) at 37°C and 5% CO2. HEK293 cells were transfected using calcium phosphate with equal amounts of wild-type and mutant WNT4 DNA or with a combination of each with WNT3A. Expression of the encoded proteins was analyzed 2 days after transfection.
RT-PCR
Total RNA was extracted from transected/treated/primary cell cultures using the High Pure RNA Isolation Kit (Roche Diagnostics, Mannheim, Germany). Total cDNA was synthesized using the Reverse-iT 1st Strand Synthesis Kit (ABgene, Surrey, UK) and amplified using Taq polymerase, Q solution (QIAGEN, Valencia, CA), and intron-crossing specific primers.
Immunofluorescence
HFK-PC cells were fixed with 2% PFA 3% sucrose in PBS for 10 minutes and washed with PBS. Cells were blocked with 5% human serum and 1% BSA in PBS-Tween (0.05%) for 30 minutes followed by incubation with SIX2 (Norus), WT1 (Santa Cruz Biotechnology, Santa Cruz, CA), active β-catenin (Millipore, Billerica, MA) antibodies overnight in 4C. Cells were washed and then incubated with Alexa-488 conjugated anti-mouse or anti-rabbit IgG secondary antibody for 60 minutes. Mounting containing 4',6-diamidino-2-phenylindole (ProLonged Gold; Invitrogen, Grand Island, NY) was applied. The cells were analyzed by Olympus BX51 fluorescence microscope using Olympus DP72 camera and cellSens standard software.
Cell Proliferation Assay
The viability of transfected HEK293 cells carrying wt/mutant WNT4 was colorimetrically determined using a cell proliferation assay kit (Cell-Titter 96 AQueous One Solution Cell Proliferation Assay; Promega, Madison, WI) in a 96-well tissue culture plate, with 20 μl Cell-Titter 96 AQueous One Solution Reagent containing 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymeth-oxyphnyl)-2-(4-sufophnyl)-2H-terasolium (MTS) and phenazine ethosulfate added to each well. Cultured cells were incubated at 37°C in a humidified, 5% CO2 atmosphere for 4 hours. The absorbance of soluble formazan produced by cellular reduction of the MTS was measured at 492 nm using an ELISA plate reader.
Clonogenic Potential Evaluation
See the Supplemental Methods.
Luciferase Reporter Assay
To assay the canonical WNT4 pathway, we used the TOPFlash-TCF reporter plasmid, as previously described.18 Briefly, HEK293 cells were seeded at 1×105 cells per well in a 24-well plate 24 hours before transfection. Cells were transfected with the indicated vector, along with pTOPFlash/pFOPFlash and pCMV-Renilla plasmids. At 48 hours after transfection, the cells were harvested and subjected to Dual-Luciferase Reporter Assay System (E1910; Promega) according to manufacturer's instructions.
Establishment of Primary Cultures from Human Fetal Kidney and Human Adult Kidney
Normal human 18-week gestation kidneys (HFK) were obtained after curettage of elective abortions. Normal human adult kidney (HAK) samples were retrieved from borders of renal cell carcinoma tumors from partial nephrectomy patients. The tissues were handled within 1 hour after the procedure.31,32 All studies were approved by the local ethics committee and informed consent was provided by the patients involved in this research according to the Declaration of Helsinki.
Both HFK and HAK tissues were washed with cold Hanks’ balanced salt solution (Invitrogen, Carlsbad, CA) and minced into approximately 1-mm cubes using sterile surgical scalpels. The dissected tissue was then incubated for 2 hours at 37°C with Iscoves’s modified Dulbecco’s medium (IMDM; Invitrogen) supplemented with 0.1% collagenase IV (Invitrogen). The digested tissue was then forced through a 100-µm cell strainer to achieve a single cell suspension and after removal of the digesting medium, it was then resuspended in growth medium (IMDM containing 10% FBS [Invitrogen], 100 ng/ml epidermal growth factor, 100 ng/ml basic fibroblast growth factor, and 10 ng/ml stem cell factor [R&D Systems Inc, Minneapolis, MN]) and plated in flasks. HFK-PC cells (passages 0–1) were incubated and upon 90% confluence, cells were subjected to both luciferase reporter assay (TOPFlash) and conditioned media experiments. For conditioned media, HEK293 cells were transfected with 4 μg empty vector, wild-type Wnt4, or mutant WNT4, grown to near confluence. The media were then removed and used to replace the media from untransfected HFK-PC cells grown to near confluence in separate flasks. These cells were grown for 6–24 hours in the conditioned media and then harvested for protein and RNA extraction.
Protein Extraction and Western Blot Analyses
Cultured cells were washed twice in cold PBS and resuspended in lysis buffer (1% Triton-X100, 20 mM Tris-HCl, 120 mM NaCl) and a tablet of protease inhibitors (Complete Mini Protease Inhibitor Cocktail; Roche), incubated for 1 hour on ice and centrifuged at 16,100×g for 20 minutes. The cell supernatant was collected, concentration was determined using the Bradford method (Pierce), and the supernatant was then resuspended in loading buffer and denatured in 95°C for 5 minutes. SDS-PAGE analysis was carried out on Tris-glycine polyacrylamide gels and electrotransferred onto nitrocellulose membrane. For Western blot analysis, membranes were blocked with 5% skim milk, incubated with the relevant antibody, and then followed by incubation with a secondary, peroxidase-conjugated antibody (1:10,000, #115–035–146 and #115–035–144; Jackson ImmunoResearch, West Grove, PA). Peroxidase activity was detected by exposure of the membrane to chemiluminescence solution containing 150 mM Tris-HCl (pH 8.9), 0.22 mg/ml Luminol (Sigma, St. Louis, MO), 0.033 mg/ml paracoumaric acid (Sigma), and 0.015% H2O2. Bands were visualized and quantified using densitometer software.
FACS Analyses
See the Supplemental Methods.
Statistical Analyses
Functional in vitro experiments were interpreted using an unpaired, two-sided t test comparing effect and appropriate controls. Data are reported as mean and SEM.
Disclosures
None.
Acknowledgments
We thank all of the families for their generous participation in our study. We also thank Professor Elon Pras for providing 280 control DNA samples, as well as Dr. Rina Rosin-Arbesfeld and Dr. Michal Caspi for their generous support with the TOPFlash reporter assay. In addition, we thank Dr. Robert Kleta and Dr. Detlef Bockenhauer from University College London (London, UK) for their support and thoughtful comments. We also thank Sigal Koren and Dr. Dani Bercovich from MIGAL Galilee Technology Center (Kiryat Shmona, Israel) for technical assistance. This work was carried out in partial fulfillment of the PhD degree requirements for A.V.
This work was supported in part by the Israeli Society of Clinical Pediatrics, Bretler Foundation, Sackler School of Medicine, Tel Aviv University, Israel Scientific Foundation (ISF) 11/910 (B.D.), and the Israeli Ministry of Health.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2012010097/-/DCSupplemental.
References
- 1.Hildebrandt F: Genetic kidney diseases. Lancet 375: 1287–1295, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanna-Cherchi S, Caridi G, Weng PL, Scolari F, Perfumo F, Gharavi AG, Ghiggeri GM: Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr Nephrol 22: 1675–1684, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, Jankauskiené A, Mir S, Montini G, Peco-Antic A, Wühl E, Zurowska AM, Mehls O, Antignac C, Schaefer F, Salomon R: Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: Results of the ESCAPE study. J Am Soc Nephrol 17: 2864–2870, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Eccles MR, Schimmenti LA: Renal-coloboma syndrome: A multi-system developmental disorder caused by PAX2 mutations. Clin Genet 56: 1–9, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Bingham C, Bulman MP, Ellard S, Allen LI, Lipkin GW, Hoff WG, Woolf AS, Rizzoni G, Novelli G, Nicholls AJ, Hattersley AT: Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet 68: 219–224, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roodhooft AM, Birnholz JC, Holmes LB: Familial nature of congenital absence and severe dysgenesis of both kidneys. N Engl J Med 310: 1341–1345, 1984 [DOI] [PubMed] [Google Scholar]
- 7.McPherson E, Carey J, Kramer A, Hall JG, Pauli RM, Schimke RN, Tasin MH: Dominantly inherited renal adysplasia. Am J Med Genet 26: 863–872, 1987 [DOI] [PubMed] [Google Scholar]
- 8.Kerecuk L, Schreuder MF, Woolf AS: Renal tract malformations: Perspectives for nephrologists. Nat Clin Pract Nephrol 4: 312–325, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, Innis JW, Hoskins BE, Hoefele J, Otto EA, Hildebrandt F: Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int 81: 196–200, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas R, Sanna-Cherchi S, Warady BA, Furth SL, Kaskel FJ, Gharavi AG: HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 26: 897–903, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woolf AS: Renal hypoplasia and dysplasia: Starting to put the puzzle together. J Am Soc Nephrol 17: 2647–2649, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Ramensky V, Bork P, Sunyaev S: Human non-synonymous SNPs: Server and survey. Nucleic Acids Res 30: 3894–3900, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ: A WNT4 mutation associated with Müllerian-duct regression and virilization in a 46,XX woman. N Engl J Med 351: 792–798, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Iglesias DM, Hueber PA, Chu L, Campbell R, Patenaude AM, Dziarmaga AJ, Quinlan J, Mohamed O, Dufort D, Goodyer PR: Canonical WNT signaling during kidney development. Am J Physiol Renal Physiol 293: F494–F500, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Vainio S, Heikkilä M, Kispert A, Chin N, McMahon AP: Female development in mammals is regulated by Wnt-4 signalling. Nature 397: 405–409, 1999 [DOI] [PubMed] [Google Scholar]
- 16.Kispert A, Vainio S, McMahon AP: Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 125: 4225–4234, 1998 [DOI] [PubMed] [Google Scholar]
- 17.Moon RT, Kohn AD, De Ferrari GV, Kaykas A: WNT and beta-catenin signalling: Diseases and therapies. Nat Rev Genet 5: 691–701, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H: Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 275: 1784–1787, 1997 [DOI] [PubMed] [Google Scholar]
- 19.Bernard P, Fleming A, Lacombe A, Harley VR, Vilain E: Wnt4 inhibits beta-catenin/TCF signalling by redirecting beta-catenin to the cell membrane. Biol Cell 100: 167–177, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stark K, Vainio S, Vassileva G, McMahon AP: Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372: 679–683, 1994 [DOI] [PubMed] [Google Scholar]
- 21.Vainio SJ: Nephrogenesis regulated by Wnt signaling. J Nephrol 16: 279–285, 2003 [PubMed] [Google Scholar]
- 22.Vainio SJ, Uusitalo MS: A road to kidney tubules via the Wnt pathway. Pediatr Nephrol 15: 151–156, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Lyons JP, Miller RK, Zhou X, Weidinger G, Deroo T, Denayer T, Park JI, Ji H, Hong JY, Li A, Moon RT, Jones EA, Vleminckx K, Vize PD, McCrea PD: Requirement of Wnt/beta-catenin signaling in pronephric kidney development. Mech Dev 126: 142–159, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terada Y, Tanaka H, Okado T, Shimamura H, Inoshita S, Kuwahara M, Sasaki S: Expression and function of the developmental gene Wnt-4 during experimental acute renal failure in rats. J Am Soc Nephrol 14: 1223–1233, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Surendran K, McCaul SP, Simon TC: A role for Wnt-4 in renal fibrosis. Am J Physiol Renal Physiol 282: F431–F441, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Park JS, Valerius MT, McMahon AP: Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 134: 2533–2539, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Schmidt-Ott KM, Masckauchan TN, Chen X, Hirsh BJ, Sarkar A, Yang J, Paragas N, Wallace VA, Dufort D, Pavlidis P, Jagla B, Kitajewski J, Barasch J: beta-catenin/TCF/Lef controls a differentiation-associated transcriptional program in renal epithelial progenitors. Development 134: 3177–3190, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Schmidt-Ott KM, Barasch J: WNT/β-catenin signaling in nephron progenitors and their epithelial progeny. Kidney Int 74: 1004–1008, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw G, Morse S, Ararat M, Graham FL: Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 16: 869–871, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Mikels AJ, Nusse R: Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol 4: e115, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metsuyanim S, Harari-Steinberg O, Buzhor E, Omer D, Pode-Shakked N, Ben-Hur H, Halperin R, Schneider D, Dekel B: Expression of stem cell markers in the human fetal kidney. PLoS One 4: e6709, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buzhor E, Harari-Steinberg O, Omer D, Metsuyanim S, Jacob-Hirsch J, Noiman T, Dotan Z, Goldstein RS, Dekel B: Kidney spheroids recapitulate tubular organoids leading to enhanced tubulogenic potency of human kidney-derived cells. Tissue Eng Part A 17: 2305–2319, 2011 [DOI] [PubMed] [Google Scholar]