

Abstract
The molecular mechanism of cyst formation and expansion in autosomal recessive polycystic kidney disease (ARPKD) is poorly understood, but impaired mechanosensitivity to tubular flow and dysfunctional calcium signaling are important contributors. The activity of the mechanosensitive Ca2+-permeable TRPV4 channel underlies flow-dependent Ca2+ signaling in murine collecting duct (CD) cells, suggesting that this channel may contribute to cystogenesis in ARPKD. Here, we developed a method to isolate CD-derived cysts and studied TRPV4 function in these cysts laid open as monolayers and in nondilated split-open CDs in a rat model of ARPKD. In freshly isolated CD-derived cyst monolayers, we observed markedly impaired TRPV4 activity, abnormal subcellular localization of the channel, disrupted TRPV4 glycosylation, decreased basal [Ca2+]i, and loss of flow-mediated [Ca2+]i signaling. In contrast, nondilated CDs of these rats exhibited functional TRPV4 with largely preserved mechanosensitive properties. Long-term systemic augmentation of TRPV4 activity with a selective TRPV4 activator significantly attenuated the renal manifestations of ARPKD in a time-dependent manner. At the cellular level, selective activation of TRPV4 restored mechanosensitive Ca2+ signaling as well as the function and subcellular distribution of TRPV4. In conclusion, the functional status of TRPV4, which underlies mechanosensitive Ca2+ signaling in CD cells, inversely correlates with renal cystogenesis in ARPKD. Augmenting TRPV4 activity may have therapeutic potential in ARPKD.
Polycystic kidney disease (PKD) is a cohort of monogenic disorders that result in development and subsequent growth of renal cysts filled with fluid.1–5 Cyst enlargement compromises function of surrounding nephrons and progresses to ESRD.1,6 In the more common form of PKD, autosomal dominant PKD (ADPKD), which is caused by mutations of polycystin 1 (PC1) and polycystin 2 (PC2), renal cysts are formed along the full length of the nephron with prevalence to the collecting duct (CD).1,7 In the rarer and more severe autosomal recessive PKD (ARPKD), renal cyst formation is virtually restricted to the CD.1,2,5,8 Mutations of the PKHD1 gene encoding fibrocystin underlie the genetic basis of the disease.6,8,9 Although the exact function of the protein is unknown, fibrocystin was shown to be expressed in primary cilia where it can interact and form complexes with PC2, possibly participating in mechanotransduction.10–12
It is accepted that the CD cells elevate [Ca2+]i in response to mechanical stress arising from variations in tubular flow or tubular composition.13–23 Impaired mechanosensitive [Ca2+]i responses, reported for both cultured ADPKD24 and ARPKD25,26 cells, point to a possible fundamental role of disrupted [Ca2+]i signaling in cystogenesis. The central cilia and cilia-associated PC1 and PC2 were proposed to mediate flow-induced cellular responses.19,27 However, homomeric PC2 channels are not mechanosensitive and fail to increase [Ca2+]i in response to flow and hypotonicity.28,29 Furthermore, intercalated cells, which lack primary cilia, respond to flow changes with comparable increases in [Ca2+]i as observed in principal cells, which have primary cilia.16,30 Therefore, additional mechanisms conferring mechanosensitivity to the CD cells need to be considered.
Transient receptor potential (TRP) channels are known to participate in cellular responses to a variety of environmental stimuli, including thermosensation, chemosensation, and mechanical forces (reviewed in Song and Yuan31). Several TRP channels, including TRPC3, TRPC6, and TRPV4, can be detected in the native CD cells and CD-originated cultured lines.19,32–34 Among these channels, TRPV4 has routinely been shown to be activated by mechanical stimuli.34–38 Indeed, we documented that endogenous TRPV4 in M-1 CD cells is stimulated by increases in flow, a response that is abolished by TRPV4 small interfering RNA knockdown.34,38 We further demonstrated a lack of flow-mediated [Ca2+]i elevations in CD from TRPV4−/− mice.30 Consistently, flow-mediated Ca2+-dependent K+ secretion in the CD is disrupted in TRPV4 knockout animals.39 TRPV4 directly interacts with PC2 to form mechanosensitive heteromeric complexes.28,29 The fact that PC2 interacts with both PC140 and fibrocystin10–12 suggests that TRPV4 could be an essential part of this mechanotransducing sensory complex.
Current PKD management is directed toward pharmacologic interference with abnormal signaling pathways causing exaggerated cell proliferation, dedifferentiation, apoptosis, and cyst growth.41 Specifically, PKD is associated with elevated circulating vasopressin levels, increased basal cellular cAMP levels, and strong upregulation of cAMP-dependent fluid secretion and proliferation.42,43 V2 antagonism greatly diminishes disease progression in rodent models of both ADPKD and ARPKD.42,44 Elevated cAMP levels might be directly related to the reduced [Ca2+]i, possibly due to impaired ability to sense changes in flow.42,44,45 This raises the possibility that manipulation with the mechanosensitivity in the CD along the TRPV4 axis modulates [Ca2+]i signalization and, in turn, renal cystogenesis.
In this study, we developed a new approach to isolate native CD-derived cyst monolayers and nondilated CDs from a rat model of ARPKD to thoroughly investigate how functional TRPV4 status determines the development and growth of renal cysts. We found that the disease leads to disruption of mechanosensitive [Ca2+]i signaling and impaired TRPV4 activity specifically in CD cysts but not in nondilated CDs. Long-term pharmacologic potentiation of TRPV4 activity gradually restores mechanosensitivity in cyst cells and greatly blunts renal ARPKD progression. From a global prospective, this study establishes a temporal link between disruption of TRPV4-based mechanosensitivity in the CD and cystogenesis. This also suggests pharmacologic potential of targeting TRPV4 activity as a treatment strategy in retarding development of ARPKD.
Results
Accumulating evidence points to impaired flow-dependent Ca2+ signalization in cultured ARPKD cells.25,26 To assess mechanosensitive properties of native cyst cells, we developed and implemented mechanical isolation of CD-derived cyst monolayers from 2-month-old ARPKD PCK453 rats. This novel technique enables real-time monitoring changes in [Ca2+]i at the single cell level in native tissue in response to mechanical and pharmacologic inputs (Figure 1). CD origin of the isolated cyst fragments was confirmed using staining with AQP2, as discussed in the Concise Methods. We used freshly isolated split-opened CDs from age-matched Sprague-Dawley (S/D) rats as controls. An abrupt 10× elevation in flow (producing shear stress of approximately 3 dyn/cm2, as discussed in the Concise Methods) on the apical surface caused a rapid and sustained increase (at least for 10 minutes) (Supplemental Figure 1) in [Ca2+]i in control CD cells, whereas it had a minimal effect on cyst cells (Figure 2A). Importantly, basal [Ca2+]i levels are prominently decreased in ARPKD cells.
Figure 1.

Ca2+ imaging in individual cells of normal CDs and CD-derived cyst monolayers of ARPKD. The top row shows representative micrographs of a typical kidney slice (left) from a control Sprague-Dawley (S/D) rat and respective split-opened CD after loading with Fura-2 taken with bright-field illumination (middle) and 380 nm excitation (right). The bottom row shows representative micrographs of a kidney slice (left) from a PCK453 rat and respective freshly isolated CD-derived cyst monolayer after loading with Fura-2 taken with bright-field illumination (middle) and 380 nm excitation (right).
Figure 2.

Cyst cells in ARPKD have disrupted flow sensitivity, decreased [Ca2+]i, and impaired TRPV4 activity. (A) The average time course of [Ca2+]i changes in individual CD cells (black) and CD-derived cyst cells (gray) in response to an abrupt 10× elevation in flow (for 90 seconds) from apical side (shown with a bar on top). (B) The average time course of [Ca2+]i changes in individual cells within split-opened area of CDs from Sprague-Dawley (S/D) rats in response to elevated flow (gray bars) in the control conditions and in the presence of TRPV4 inhibition with 4 μM of HC067047 (black bar). All other conditions are the same as in A. (C) The left side shows a summary graph of the magnitudes of flow-induced Ca2+ responses in CD cells from S/D rats (black bars) in the control, after pretreatment with 4 μM of HC067047 and 2 μM of Ruthenium red (RuR), and in CD-derived cyst cells from PCK453 rats (gray bar). The right side shows the summary graph of the basal [Ca2+]i in CD cells from S/D rats (black bars) in the control and after pretreatment with 4 μM of HC067047 and in CD-derived cyst cells from PCK453 rats (gray bar). *Significant decrease versus S/D rats. (D) The average time course of [Ca2+]i changes in individual CD cells from S/D rats (black) and cyst cells within CD-derived cyst monolayers (gray) in response to 3-minute application of TRPV4 activator GSK1016790A (30 nM) followed by RuR (2 μM).
We demonstrated that genetic deletion of the Ca2+-permeable TRPV4 channel abolished flow-mediated [Ca2+]i elevations in CD cells of mice.30 To probe whether TRPV4 is also critical for mechanosensitive properties in rat CD cells, we took advantage of a potent and selective TRPV4 antagonist, HC067047. Acute treatment with HC067047 (4 μM) nearly abolished elevations in [Ca2+]i to the mechanical stimulation (Figure 2B). Pretreatment with a less selective TRPV4 antagonist, Ruthenium red (2 μM) yielded similar results (Figure 2C). Inhibition of TRPV4 with HC067047 also significantly decreased basal [Ca2+]i levels (Figure 2B). These results suggest a key role of TRPV4 in mediating flow-dependent Ca2+ responses and shaping basal [Ca2+]i in rat CD cells. Importantly, pharmacologic inhibition of TRPV4 recapitulates the state of compromised [Ca2+]i signalization observed in cyst cells (Figure 2, A and C).
Lack of flow-mediated Ca2+ responses and diminished basal [Ca2+]i levels in ARPKD cyst cells indicate TRPV4 dysfunction. Indeed, stimulation of TRPV4 with a highly selective activator, GSK1016790A (30 nM), caused [Ca2+]i elevations in control CDs that were four times greater than in cyst cells (Figure 2D). GSK1016790A had no effect when TRPV4 was inhibited with Ruthenium red (Supplemental Figure 2). We concluded that functional activity of TRPV4 is drastically diminished in ARPKD cyst cells contributing to the impaired flow-mediated Ca2+ signalization.
TRPV4 dysfunction in cyst monolayers may result from decreased levels of the protein. Figure 3A shows a representative Western blot monitoring TRPV4 expression in whole kidney homogenates from S/D and PCK453 rats. As expected, TRPV4 antibodies recognized both nonglycosylated (lower) and glycosylated (upper) bands around 95 kD in S/D rats. Renal TRPV4 levels were modestly but significantly reduced in PCK453 rats. Strikingly, the glycosylated form of TRPV4 was virtually absent in these animals (Figure 3). We concluded that post-translational modification of TRPV4 is compromised during ARPKD.
Figure 3.

Renal TRPV4 levels are modestly decreased and TRPV4 glycosylation is disrupted in ARPKD rats. (A) Representative Western blot from whole kidney lysates of Sprague-Dawley (S/D) and PCK453 rats simultaneously probed with anti-TRPV4 and anti-actin antibodies. Samples are run in triplicate. (B) Summary graph comparing total renal TRPV4 expression (left), glycosylated (middle), and nonglycosylated (right) forms of TRPV4 in S/D and PCK453 rats from Western blots similar to that shown in A. Intensities of TRPV4-reporting bands were normalized to the intensities of the respective actin bands. *Significant decrease versus S/D rats. g, glycosylated form of TRPV4.
We next probed subcellular TRPV4 distribution in control split-opened CDs from S/D rats and CD-derived cyst monolayers from PCK453 rats with immunofluorescence. TRPV4 is distributed in subapical cytosolic compartments and at the apical plasma membrane, with stronger expression in AQP2-positive cells (Figure 4A; see Supplemental Figure 3 for a lower magnification). Surprisingly, we detected a striking shift of the TRPV4-reporting fluorescent signal toward the apical membrane in CD-derived cyst monolayers (Figure 4B; see Supplemental Figure 3 for a lower magnification). We also observed AQP2 targeting to the apical side, which is consistent with reported upregulation of cAMP levels in ARPKD cells.42,46 For quantitative estimation, we utilized line-scan analysis (Figure 4C) of TRPV4-reporting fluorescent signal distribution along the z-axis in cross-sections of three-dimensional stacks similar to that shown in Figure 4, A and B. Cyst cells clearly exhibit a leftward narrowing of the bell-shape distribution of TRPV4 along the z-axis toward the apical plasma membrane. As summarized in Figure 4D, the half-width was significantly reduced from 2.63±0.12 μm in the control to 1.89±0.03 μm in CD and cyst cells, respectively.
Figure 4.

Subcellular TRPV4 distribution is shifted toward the apical membrane in cyst cells. Representative confocal plane micrographs (axes are shown) and corresponding cross-sections (pointed by arrows) showing three-dimensional stacks of TRPV4 localization (anti-TRPV4, pseudocolor green), AQP2 localization (anti-AQP2, pseudocolor red), and the combined image for a split-opened CD from a wild-type Sprague-Dawley (S/D) rat (A) and a CD-derived cyst monolayer from a PCK453 rat (B). Nuclear DAPI staining is shown by pseudocolor blue. (C) The distribution of averaged relative fluorescent signals representing TRPV4 localization along a line on z-axis in CD and cyst cells. For each individual cell, the fluorescent signals are normalized to their corresponding maximal value. The position of the apical and basolateral sides is shown with arrows at the top. (D) The summary graph of half-width means for distributions of fluorescent signals shown in C. #Significant decrease versus control CDs. a, apical side; b, basolateral side; DAPI, 4',6-diamidino-2-phenylindole.
To exclude the possibility that isolation of CD-derived cysts might potentially cause subcellular TRPV4 redistribution, we monitored TRPV4 localization in kidney sections from S/D and PCK453 rats (Figure 5). TRPV4 was preferentially localized to the AQP2-positive distal nephrons and cysts (respective top rows in Figure 5). Higher-resolution images of the same respective kidney sections revealed that TRPV4- and AQP2-reporting fluorescent signals are restricted to the apical/subapical domain of cyst cells (Figure 5B, middle row). This is consistent with the results obtained in freshly isolated cyst fragments (Figure 4B). In contrast, we detected diffuse subcellular distribution of TRPV4 and AQP2 in nondilated CDs (Figure 5B, bottom row). These expression patterns are clearly different from observed in cyst cells and are reminiscent of those observed in control CD cells from S/D rats (Figure 5A).
Figure 5.

Subcellular TRPV4 localization is different in cysts and nondilated CDs in PKC453 rats. (A) Immunofluorescence of a kidney section from Sprague-Dawley (S/D) rat. A representative low-magnification 10-μm-thick kidney section (top row) shows staining patterns for TRPV4 (pseudocolor green), AQP2 (pseudocolor red), and the combined image. The area defined by a rectangle is shown below with a higher magnification. (B) Immunofluorescence of kidney section from a PCK453 rat. A representative low-magnification 10-μm-thick kidney section (top row) shows staining patterns for TRPV4 (pseudocolor green), AQP2 (pseudocolor red), and the combined image. Areas defined by rectangles and containing a cyst (middle) and a nondilated CD (bottom) are shown below with a higher magnification.
Different subcellular TRPV4 distribution in nondilated CD cells and cyst cells might indicate altered TRPV4 functionality. Thus, we assessed mechanosensitive Ca2+ signalization and functional TRPV4 status in nondilated CDs from PCK453 rats. We detected prominent [Ca2+]i response to elevations in luminal flow and unchanged basal [Ca2+]i values (Figure 6, A and B). Activation of TRPV4 with GSK1016790A also produced a similar rise in [Ca2+]i as in control CD cells from Sprague-Dawley rats (Figure 6C and Supplemental Figure 4). Consistently, subcellular TRPV4 and AQP2 distribution in freshly isolated nondilated CDs was not different from that in control CDs as visualized with immunofluorescence (Figure 6D). Therefore, the results in Figure 6 strongly argue for unchanged mechanosensitivity in nondilated CDs.
Figure 6.
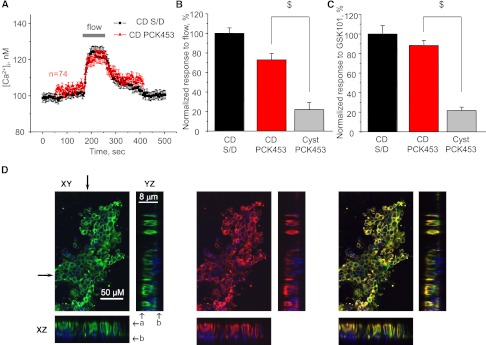
Mechanosensitivity and TRPV4 function are intact in nondilated CDs of PCK453 rats. (A) The average time course of [Ca2+]i changes in individual cells from nondilated CDs (red) in response to an abrupt 10× elevation in flow (for 90 seconds) from the apical side (shown with a bar on top). The average time course of [Ca2+]i changes in CD cells from Sprague-Dawley (S/D) rats (black) in response to elevated flow is reproduced from Figure 2A. (B) Summary graph of relative responses to elevated flow in nondilated CDs and CD-derived cysts. The mean amplitude of flow-mediated Ca2+ response in control CD cells from S/D rats was used for normalization. (C) Summary graph of relative responses to application of the selective TRPV4 activator GSK1016790A (30 nM) in nondilated CDs and CD-derived cysts. The responses were normalized to the mean value for CD cells from S/D rats. $Significant decrease versus CD PCK453. (D) Representative confocal plane micrographs and corresponding cross-sections (shown by arrows) of three-dimensional stacks of TRPV4 localization (pseudocolor green), AQP2 localization (pseudocolor red), and the combined image for a split-opened nondilated CD from PCK453 rat. a, apical side; b, basolateral side.
Substantial experimental evidence supports a critical role of paracrine ATP release in facilitating [Ca2+]i responses in the CD.21 Development of ARPKD results in a virtually closed environment inside renal cysts, creating favorable conditions for amplification of the purinergic signal.14,47,48 Thus, we next characterized the functional status of purinergic cascade in CD-derived cyst monolayers and nondilated CDs from PCK453 rats. Exogenous ATP (10 μM) induced a transient [Ca2+]i peak followed by a sustained plateau (Figure 7). The magnitude of the initial peak was indeed markedly increased in cyst cells compared with the control CD cells from S/D rats (Figure 7). However, the plateau phase was nearly absent in ARPKD cells. In nondilated CDs from PCK453 rats, ATP produced Ca2+ response undistinguishable from that observed in control CDs (Figure 7A). We recently identified a dominant role of TRPV4 in generating ATP-induced Ca2+ plateau in murine CD cells.18 The absence of the plateau phase in cyst cells supports the conception of TRPV4 dysfunction in ARPKD. These results suggest malfunctioning of purinergic signaling in CD-derived cysts but not in nondilated CDs.
Figure 7.
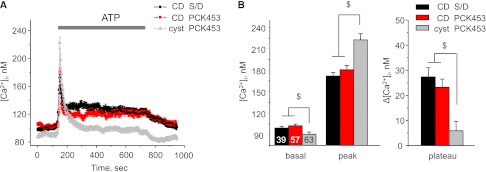
Lack of ATP-induced Ca2+ plateau in CD-derived cyst cells. (A) The average time course of [Ca2+]i changes in response to 10-minute application of ATP (10 μM, shown with a bar) for CD cells from Sprague-Dawley (S/D) rats (black), cells of nondilated CD from PCK453 rats (red), and CD-derived cyst cells from PCK453 rats (gray). (B) Summary graph of the basal [Ca2+]i values (basal), the magnitudes of ATP-induced Ca2+ peak (peak), and the absolute values of the sustained plateau phase (plateau) for cells described in Figure 6A. $Significant changes versus cyst PCK453.
Our results point to a severe TRPV4 dysfunction in CD-derived cysts of ARPKD. Physiologic stimuli, including flow-mediated shear stress and activation of purinergic cascade, fail to effectively stimulate TRPV4 to elicit proper mechanosensitive response. Moreover, impaired TRPV4 functional status contributes to the decreased resting [Ca2+]i levels (Figures 2 and 7). However, TRPV4 can still be activated to some extent by strong pharmacologic inputs, such as GSK1016790A (Figure 2D). We hypothesized that augmentation of the mechanosensitivity on the TRPV4 axis will interfere with ARPKD progression. Indeed, a recent report provided initial evidence that prolonged injection of GSK1016790A to PCK453 rats decreases renal cystic area.49 Therefore, we next tested if GSK1016790A treatment interferes with ARPKD progression by restoring mechanosensitive properties of the CD-derived cyst cells.
Systemic administration of GSK1016790A (3 μg⋅kg body weight per day) for 1 and 2 months (Figure 8, A and B) to 1-month-old PCK453 rats diminished renal cyst development and growth compared with the control vehicle-treated group. Specifically, we detected a significant reduction of relative cyst area (Figure 8C; also see low-powered section slices in Supplemental Figure 5) at both time points. We still observed the disease progression in GSK1016790A-treated animals between 1 and 2 months of treatment, although at a slower rate compared with that in vehicle-treated animals. The kidney/total body index was also reduced by GSK106790A treatment (Figure 8D). In contrast, GSK1016790A (1 month) had no significant effect on kidney/total body index in control S/D rats (0.89±0.08 versus 0.86±0.04 for treated and untreated animals, respectively).
Figure 8.

Systemic treatment with the TRPV4 activator GSK1016790A attenuates renal ARPKD manifestations. Representative kidney sections from PCK453 rats treated with vehicle and GSK1016790A for 1 month (A) and 2 months (B), respectively. (C) Summary graphs comparing relative visible cyst areas for kidney slices similar to that shown in A and B. (D) Summary graph of kidneys to total body weight ratios for PCK453 rats treated with vehicle and GSK1016790A for 1 and 2 months. *Significant decrease versus vehicle.
We next examined the ramifications of systemic GSK1016790A administration on [Ca2+]i signaling in cells from residual CD-derived cysts. We detected gradual restoration of the flow-mediated [Ca2+]i responses and return of resting [Ca2+]i levels to control values after 1 and 2 months of treatment, respectively (Figure 9A). We also found progressive augmentation of [Ca2+]i response to an acute application of GSK1016790A (Figure 9B). This suggests enhancement of functional TRPV4 status in GSK1016790A-treated animals, likely due to TRPV4 sensitization. Consistently, we detected appearance of the ATP-induced Ca2+ plateau (Figures 9C). Overall, we concluded that prolonged systemic administration of GSK1016790A recovers the bulk of mechanosensitive [Ca2+]i signaling in CD-derived cyst cells.
Figure 9.
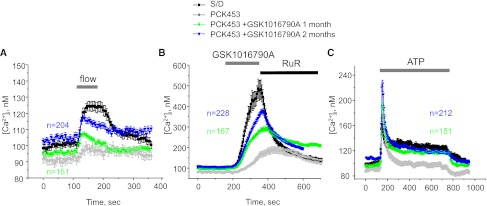
Mechanosensitive [Ca2+]i signaling is restored in CD-derived cyst cells of GSK1016790A-treated PCK453 rats. The average time course of [Ca2+]i changes in response to an abrupt 10× elevation in flow (A), application of GSK1016790A (30 nM) followed by 2 μM Ruthenium red (B), and application of 10 μM ATP (C) for individual cells within freshly isolated CD-derived cyst monolayers from PCK453 rats treated with GSK1016790A for 1 (green) and 2 months (blue), respectively. The time courses of control CD cells from Sprague-Dawley (S/D) rats (black) and CD-derived cyst monolayers from untreated PCK453 rats (gray) are reproduced from Figure 2A. Numbers of all individual recordings for each condition are shown in the respective color. Durations of treatments are shown on the top with bars.
We next subjected CD-derived cyst monolayers of GSK1016790A-treated PCK453 rats to immunofluorescence. Systemic GSK1016790A treatment for 1 month greatly reversed the apical TRPV4 translocation observed in cysts from untreated animals of the same age (Figure 10; see Figure 4B for comparison). Specifically, we detected a shift of the TRPV4-reporting fluorescent signal from apical membrane to cytosol. We also detected redistribution of AQP2 from the apical membrane, indicating a possible decrease in cellular cAMP levels. Treatment with GSK1016790A for 2 months yielded similar results (Supplemental Figure 6). However, GSK1016790A fails to restore TRPV4 glycosylation in the kidney of PCK453 rats (Supplemental Figure 7). Total TRPV4 levels were also not altered in GSK1016790A-treated PCK453 rats (87%±3% versus 100%±2% for treated and untreated rats, respectively). We concluded that TRPV4 distribution in cyst cells of GSK1016790A-treated animals mainly recapitulates TRPV4 localization in CD cells of control Sprague-Dawley rats (Figure 4A). This occurs independently of significant changes in renal TRPV4 levels and TRPV4 glycosylation status.
Figure 10.

Systemic GSK1016790A treatment normalizes subcellular distribution of TRPV4 and AQP2 in freshly isolated CD-derived cyst monolayers. Representative confocal plane micrographs (axes are shown) and corresponding cross-sections (arrows) of three-dimensional stacks of TRPV4 localization (pseudocolor green), AQP2 localization (pseudocolor red), and the combined image for a CD-derived cyst monolayer from PCK453 rat treated with GSK1016790A for 1 month. Nuclear staining with DAPI is shown by pseudocolor blue. a, apical side; b, basolateral side; DAPI, 4',6-diamidino-2-phenylindole.
Discussion
This study provides two major advances in our understanding of the relation between mechanosensitivity and cystogenesis in the CD. First, we demonstrate that the defect in the fibrocystin gene in PCK453 rats does not lead to immediate disruption of flow sensitivity and compromised TRPV4 activity in epithelial cells of nondilated CDs. Thus, the intact mechanosensitive [Ca2+]i signalization might have a permissive role for normal tubular function and its disruption specifically occurs before, or as an early event in, cyst development. Second, we provide evidence that restoration of mechanosensitivity and TRPV4 function in cyst cells drastically diminishes ARPKD progression. This strongly supports the concept that flow-dependent [Ca2+]i signaling plays an important modulatory role in renal cyst development. However, it does not seem that the disruption of mechanosensitivity per se initiates cystogenesis because TRPV4−/− mice50 and zebrafish29 do not have renal cysts.
We consistently observed diminished basal levels of [Ca2+]i in native CD-derived cyst cells of ARPKD rats (Figures 2 and 7). Similar decreases in resting [Ca2+]i values were reported for cultured human ADPKD and ARPKD cells.45 We further provide strong experimental support to the conception that TRPV4 activity is a critical determinant of basal [Ca2+]i. Inhibition of TRPV4 with HC067047 decreases basal [Ca2+]i recapitulating the values observed in cyst cells that have marked TRPV4 dysfunction (Figure 2, B–D). Restoration of functional TRPV4 status in GSK1016790A-treated animals brings [Ca2+]i back to the levels detected in normal CD cells. Decreased [Ca2+]i values during PKD may contribute to the abnormally elevated cAMP signaling.42,44,45 This is known to stimulate Cl− secretion inside the cyst lumen and to promote proliferation of cyst cells.51 By monitoring changes in AQP2 subcellular distribution patterns, we provide evidence that pharmacologic activation of TRPV4 blunts cAMP levels likely due to augmentation of basal [Ca2+]i (Figures 4 and 10). This explains, at least in part, the anticystogenic effect of GSK1016790A (Figure 8 and Supplemental Figure 5). The observation that systemic GSK1016790A treatment enhances TRPV4 activity requires a comment. Unlike many other Ca2+-permeable channels, TRPV4 can be activated by modest elevations in [Ca2+]i via mechanisms involving direct binding of calmodulin to the channel.52 Initial small activation of TRPV4 by GSK1016790A provides a source for such increases in [Ca2+]i leading to sustained potentiation of TRPV4 activity. On a longer timescale, elevations of [Ca2+]i will also promote phosphorylation of TRPV4 by Ca2+-dependent protein kinase C isoforms further activating the channel.53 In contrast, an excessive increase in [Ca2+]i will lead to inhibition/desensitization of TRPV4 via a negative feedback mechanism52 protecting cells from the cytotoxic effects of elevated [Ca2+]i.
We demonstrate that TRPV4 functionality determines the status of flow-mediated [Ca2+]i signaling in the CD. Diminished TRPV4 activity is associated with the lack of flow-sensitive [Ca2+]i elevations in freshly isolated CD-derived cyst monolayers, whereas TRPV4 sensitization after systemic GSK1016790A treatment rejuvenates [Ca2+]i responses to elevated flow in cyst cells. This enables us to correlate changes in mechanosensitivity at the cellular level with systemic progression of ARPKD. We found that TRPV4 dysfunction in cyst cells is not likely associated with decreased TRPV4 protein levels (Figure 3) but rather with impaired ability of the channel to be activated (Figure 2D). Our results point to dramatically reduced glycosylation of TRPV4 in PCK453 rats (Figure 3). Interestingly, a recent study demonstrated that mutations in TRPV4 associated with diminished glycosylation abolished responses to environmental stimuli (such as hypotonicity) and decreased responses to GSK1016790A.54 Such impaired post-translational TRPV4 processing has been implicated in the development of familial digital arthropathy-brachydactyly.54
Interestingly, the beneficial effects of TRPV4 activation during ARPKD might not be limited to the kidney. A recent study identifies an antiproliferative role for TRPV4 in cholangiocytes from ARPKD rats and TRPV4 activation has a tendency to decrease liver cysts.49 This study also provides initial observation that injection of GSK1016790A diminishes renal cyst area. In our study, we manage, for the first time, isolation of monolayers of CD-derived cysts to correlate TRPV4 function, mechanosensitivity of native CD cells, and renal cystogenesis at the timescale of ARPKD progression using per os administration of the TRPV4 activator, GSK1016790A. However, it should be noted that excessive activation of TRPV4 by an acute administration of high doses of GSK1016790A increases permeability of the pulmonary microvascular barrier leading to circulatory collapse.55 In contrast, gradual ingestion of small doses of the drug for up to 2 months, as used in this study, is well tolerated and does not produce any detrimental side effects.
This investigation also addresses the purinergic aspect of mechanosensitivity and its relation to TRPV4 function. ATP is constitutively released from CD cells and this process is strongly potentiated by mechanical stimuli.21,56 It becomes increasingly appreciated that paracrine purinergic signaling is an important facilitator of mechanosensitivity in the CD. Indeed, we demonstrated that genetic deletion of the major purinergic receptor, P2Y2, markedly blunts cellular responses to elevated flow and hypotonicity.18 Remodeling kidney tissue during development of PKD creates a virtually closed environment inside cysts that can trap ATP.14 Several reports suggest a marked augmentation of purinergic cascade and an increased ATP concentration inside the cyst lumen in PKD.47,48 This potentiation was shown to have detrimental renal effects by promoting Cl− secretion and cyst growth.47,51,57 Pharmacologic inhibition of purinergic signaling could be a putative pharmacologic target to manage PKD.58 Despite the fact that paracrine ATP release might be an important component of mechanosensitivity in normal CDs,18 this paradoxical upregulation of purinergic signaling in PKD pathology does not improve mechanosensitive properties of cyst cells. We found that the initial transient ATP-induced [Ca2+]i peak was elevated in CD-derived cyst cells. In contrast, the sustained plateau is nearly absent in cyst cells. This suggests that the chronic potentiation of purinergic cascade in ARPKD will have a minimal effect on [Ca2+]i levels. We identified a dominant role of TRPV4 in the generation of the ATP-induced [Ca2+]i plateau and found that genetic deletion of TRPV4 nearly abolishes the sustained elevation of [Ca2+]i in response to ATP.18 The absence of the plateau in cyst cells is consistent with TRPV4 dysfunction and impaired mechanical perception and vice versa, restoration of TRPV4 functionality in GSK1016790A-treated animals causes appearance of ATP-induced Ca2+ plateau (Figure 9C). Therefore, our results allow consolidating the upregulation of purinergic signaling inside cyst lumen and the loss of mechanosensitivity in ARPKD.
In summary, this study directly demonstrates the relation between TRPV4 activity, mechanosensitivity in CD cells, and renal cyst development in ARPKD. By identifying TRPV4 dysfunction as an underlying cause for disrupted flow perception by the cyst cells, we provide evidence that augmentation of TRPV4 status limits renal ARPKD manifestations. It is also tempting to speculate that pharmacologic targeting of signaling pathways that possess stimulatory actions on TRPV4 would yield similar results. Finally, although our investigation focuses exclusively on ARPKD, we anticipate that systemic enhancement of TRPV4 function will also be beneficial for treatment of ADPKD because both pathologies are associated with disrupted mechanosensitive [Ca2+]i signalization.
Concise Methods
Materials and Animals
All chemicals and materials were from Sigma (St. Louis, MO), VWR (Radnor, PA), and Tocris (Ellisville, MO) unless noted otherwise and were of reagent grade. Animal use and welfare adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals following a protocol reviewed and approved by the Institutional Laboratory Animal Care and Use Committee of the University of Texas Health Science Center at Houston. For experiments, 1- to 3-month-old control Sprague-Dawley (S/D) and ARPKD PCK453 rats (Charles River Laboratories, Wilmington, MA) were used. Animals were maintained on standard rodent regimen (Purina #5001) and had free access to tap water. For some experiments, 1-month-old PCK453 rats received either tap water or tap water containing GSK1016790A (3 μg⋅kg body weight per day) for either 1 or 2 months. This concentration is 100 times lower than the reported lethal dose for an acute intravenous injection of the substance.55 We did not observe any noticeable morbidity in the GSK1016790A-treated groups during the time course of the drug administration. After the end of the treatment, rats were sacrificed by CO2 administration followed by cervical dislocation and the kidneys were immediately removed and weighted.
Tissue Isolation
The procedure for isolation of the CDs from Sprague-Dawley rats and nondilated CDs from PCK453 rats suitable for Ca2+ imaging and immunofluorescence closely follows our previously reported rat and mouse protocols.18,59–61 Kidneys were cut into thin slices (<1 mm), with slices placed into ice-cold physiologic saline solution buffered with HEPES (pH 7.4). The CD was visually identified by its morphologic features (pale color, coarse surface, and, in some cases, bifurcations) and was mechanically isolated from kidney slices by microdissection using watchmaker forceps under a stereomicroscope. Isolated CDs were attached to 5×5 mm coverglass coated with poly-l-lysine. A coverglass containing CD was placed in a perfusion chamber mounted on an inverted Nikon Eclipse Ti microscope and perfused with room temperature HEPES-buffered (pH 7.4) saline solution. CDs were split-opened with two sharpened micropipettes, controlled with different micromanipulators, to gain access to the apical membrane. The nephrons were used within 1–2 hours of isolation.
Using a similar approach, CD-derived cyst monolayers were gently teased from open cyst cavities in kidney slices from PCK453 rats using watchmaker forceps under a stereomicroscope. ARPKD cyst monolayers were further mechanically separated from surrounding tissue/nephrons and attached with the basal side to 5×5 mm coverglass coated with poly-l-lysine.
[Ca2+]i Measurements
Unless otherwise noted, 2-month-old Sprague-Dawley and PCK453 rats were used for experiments. Intracellular calcium levels were measured in individual cells of the split-opened CDs and CD-derived cyst monolayers using Fura-2 fluorescence radiometric imaging as described previously.18,34,38 Split-opened CDs and cysts fragments were loaded with Fura-2 by incubation with 2 μM Fura-2/AM in a bath solution for 45 minutes at room temperature. Subsequently, the tissue samples were washed and incubated for an additional 10–15 minutes before experimentation. The CDs and cyst monolayers were then placed in an open-top imaging study chamber (Warner RC-10) with a bottom coverslip viewing window and the chamber attached to the microscope stage of an InCa Imaging Workstation (Intracellular Imaging Inc, Cincinnati, OH). Cells were imaged with a 20× Nikon Super Fluor objective and regions of interest drawn for individual cells. The Fura-2 fluorescence intensity ratio was determined by excitation (an average for approximately 300 ms) at 340 nm and 380 nm and calculating the ratio of the emission intensities at 511 nm in the usual manner every 5 seconds. We observed no significant Fura-2 bleaching and minimal Fura-2 leakage at both wavelengths during experiments. The changes in the ratio are converted to intracellular Ca2+ concentrations using the calibration methods as previously described.18,59,62 At least three individual CDs or cysts from three rats were used for each experimental set.
Western Blot Analyses
Immediately after dissection, kidneys were placed on ice, decapsulated, and homogenized in 3 volumes of ice-cold hypotonic lysis buffer containing 50 mM Tris, 1% Triton X-100, 5 mM EDTA (pH 7.4) supplemented with 1 mM PMSF and 2 mg/ml protease inhibitor cocktail (Complete Mini; Roche Diagnostics, Indianapolis, IN). Protein concentration was determined with a Bradford assay using IgG as a standard. The samples were diluted with hypotonic lysis buffer, denatured, and reduced in Laemmli buffer supplemented with 10 mM of dithiothreitol at +75°C for 10 minutes to obtain the final protein concentration of 4 mg/ml. The samples (10 μg/lane) were separated on 9% polyacrylamide gels at 150 V for 1.75 hours and transferred to nitrocellulose membrane for 1.5 hours at 100 V. Subsequently, the nitrocellulose membrane was incubated with anti-TRPV4 (1:500; Alomone Labs, Jerusalem, Israel) and anti-actin (1:1000; Abcam, Cambridge, UK) primary antibodies for 2 hours at room temperature. We previously tested specificity of the TRPV4 antibodies in expression systems,38,63 cultured M1 cells,34 and TRPV4−/− mice.30 Upon washout (three times for 10 minutes in TBS-Tween), the membrane was incubated with peroxidase-conjugated goat anti-rabbit secondary antibodies (1:30,000; Bio-Rad, Hercules, CA) for 1 hour at room temperature. Blots were quantified using ImageJ 1.47 software (National Institutes of Health, Bethesda, MD). The intensities for TRPV4 protein bands were normalized to the intensities of the corresponding actin bands, used as a loading control. All experiments were repeated three times.
Immunofluorescence and Renal Histology
Kidneys from PCK453 rats were briefly fixed in situ via intracardiac perfusion and subjected to standard immunohistochemical procedures as done before.30 Briefly, animals were anesthetized by isoflurane inhalation and kidneys prepared by intracardiac perfusion by a fixation solution (20 ml, 4% paraformaldehyde and 0.1 M cacodylate buffer in ice-cold PBS, pH 7.4). Kidneys were removed and placed in 4% paraformaldehyde overnight, and then mounted in tissue freezing medium (Tissue Tek; Sakura Finetek Inc., Torrance, CA) and frozen at −30°C. Sagittal sections (10 μm thick) were obtained, using an OTF 5000 cryostat (Bright Instrument, Huntingdon, UK). Sections were allowed to warm to room temperature and washed in PBS. Sections were then blocked with 1% donkey serum, and incubated overnight at 4°C with primary antibody: anti-TRPV4 (1:500; Alomone Labs) and anti-AQP2 tagged with ATTO 550 (1:200; Alomone). Sections were subsequently washed and incubated at room temperature for 3 hours with secondary antibody Cy2 anti-rabbit (1:500; Jackson ImmunoResearch, West Grove, PA). The tissue was then mounted in ProLong Gold antifade reagent with 4',6-diamidino-2-phenylindole (Invitrogen, Carlsbad, CA).
For histologic assessment of renal ARPKD manifestation, kidney sections (30 μM) from untreated and GSK106790A-treated PCK453 rats were obtained as described above. Sections were stained with hematoxylin and eosin with standard procedures and used to quantify cystic area. Image analysis was performed using NIS elements 4.00 software (Nikon, Nelville, NY). At least four different sections from three different animals were used for the assessment.
Freshly isolated CD-derived cyst monolayers and split-opened CDs were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 20 minutes at room temperature (RT). After fixation the samples were permeabilized by addition of 0.1% Triton in PBS for 5 minutes and washed in PBS three times for 5 minutes. Nonspecific staining was blocked with 10% normal goat serum (NGS; Jackson ImmunoResearch) in PBS for 30 minutes at RT. After washing with PBS (three times for 5 minutes) the samples were incubated for 3 hours at RT in dark with the mix of anti-TRPV4 (1:500; Alomone Labs), anti-AQP2 tagged with ATTO 550 (1:100; Alomone Labs) in 1% serum + 0.1% Triton in PBS. Subsequently, samples were washed three times with PBS and incubated for 1.5 hours at RT in dark with goat anti-rabbit IgG labeled with Alexa Fluor 488 (1:400 dilution; Invitrogen) in 1% NGS + 0.1% Triton in PBS. After washing with PBS (three times for 5 min) the samples were stained with 4',6-diamidino-2-phenylindole (300 nM concentration; Calbiochem, San Diego, CA) to visualize nuclei. Subsequently the samples were dehydrated, and mounted with permanent mounting media (Thermo Scientific, Pittsburg, PA). Labeled tissue samples were examined with an inverted Nikon Eclipse Ti fluorescent microscope using a 40× Plan-Fluor oil-immersion (1.3 NA) objective. Samples were excited with 405, 488, and 561 nm laser diodes and emission captured with a 16-bit Cool SNAP HQ2 camera (Photometrics, Tucson, AZ) interfaced to a PC running NIS 4.00 elements software.
Solutions
Typical bath solution was as follows: 150 mM NaCl, 5 mM KCl, 1 mM CaCl2, 2 mM MgCl2, 5 mM glucose, and 10 mM HEPES (pH 7.4). All reagents were applied by perfusing the experimental chamber at 1.5 ml/min. To test the effect of elevated flow on [Ca2+]i, the rate of perfusion was instantly increased from 1.5 ml/min (approximately 15 mm H2O) to 15 ml/min (approximately 80 mm H2O). Using a parallel plate chamber, we recently estimated that this maneuver produces shear stress of approximately 3 dyn/cm2.30 This value fits well within the physiologic range of shear stress present in the rat and mouse CD as assessed previously.34,64
Statistical Analyses
All summarized data are reported as mean ± SEM. Data were compared using the t test or an one-way ANOVA as appropriate. P≤0.05 was considered significant.
Disclosures
None.
Acknowledgments
This research was supported by grants from the American Heart Association (SDG2230391 to O.P.) and the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (DK095029 to O.P.), as well as a Carl W. Gottschalk research scholar grant from the American Society of Nephrology (to O.P.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2012050442/-/DCSupplemental.
References
- 1.Harris PC, Torres VE: Polycystic kidney disease. Annu Rev Med 60: 321–337, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siroky BJ, Guay-Woodford LM: Renal cystic disease: The role of the primary cilium/centrosome complex in pathogenesis. Adv Chronic Kidney Dis 13: 131–137, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Torres VE, Harris PC: Polycystic kidney disease in 2011: Connecting the dots toward a polycystic kidney disease therapy. Nat Rev Nephrol 8: 66–68, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torres VE, Harris PC: Mechanisms of disease: Autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2: 40–55, quiz 55, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Wilson PD: Polycystic kidney disease. N Engl J Med 350: 151–164, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Sweeney WE, Jr, Avner ED: Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res 326: 671–685, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Torres VE, Harris PC, Pirson Y: Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Bergmann C, Senderek J, Küpper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schöneborn S, Kirfel J, Furu L, Onuchic LF, Rossetti S, Harris PC, Somlo S, Guay-Woodford L, Germino GG, Moser M, Büttner R, Zerres K: PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat 23: 453–463, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Wallace DP: Cyclic AMP-mediated cyst expansion. Biochim Biophys Acta 1812: 1291–1300, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim I, Fu Y, Hui K, Moeckel G, Mai W, Li C, Liang D, Zhao P, Ma J, Chen XZ, George AL, Jr, Coffey RJ, Feng ZP, Wu G: Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am Soc Nephrol 19: 455–468, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim I, Li C, Liang D, Chen XZ, Coffy RJ, Ma J, Zhao P, Wu G: Polycystin-2 expression is regulated by a PC2-binding domain in the intracellular portion of fibrocystin. J Biol Chem 283: 31559–31566, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J: Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol 27: 3241–3252, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geyti CS, Odgaard E, Overgaard MT, Jensen ME, Leipziger J, Praetorius HA: Slow spontaneous [Ca2+] i oscillations reflect nucleotide release from renal epithelia. Pflugers Arch 455: 1105–1117, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Hovater MB, Olteanu D, Welty EA, Schwiebert EM: Purinergic signaling in the lumen of a normal nephron and in remodeled PKD encapsulated cysts. Purinergic Signal 4: 109–124, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leipziger J: Control of epithelial transport via luminal P2 receptors. Am J Physiol Renal Physiol 284: F419–F432, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM: Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol 285: F998–F1012, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Liu W, Morimoto T, Woda C, Kleyman TR, Satlin LM: Ca2+ dependence of flow-stimulated K secretion in the mammalian cortical collecting duct. Am J Physiol Renal Physiol 293: F227–F235, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Mamenko M, Zaika O, Jin M, O’Neil RG, Pochynyuk O: Purinergic activation of Ca2+-permeable TRPV4 channels is essential for mechano-sensitivity in the aldosterone-sensitive distal nephron. PLoS ONE 6: e22824, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J: Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33: 129–137, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Praetorius HA, Leipziger J: Released nucleotides amplify the cilium-dependent, flow-induced [Ca2+]i response in MDCK cells. Acta Physiol (Oxf) 197: 241–251, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Praetorius HA, Leipziger J: Intrarenal purinergic signaling in the control of renal tubular transport. Annu Rev Physiol 72: 377–393, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Satlin LM, Carattino MD, Liu W, Kleyman TR: Regulation of cation transport in the distal nephron by mechanical forces. Am J Physiol Renal Physiol 291: F923–F931, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Woda CB, Leite M, Jr, Rohatgi R, Satlin LM: Effects of luminal flow and nucleotides on [Ca(2+)](i) in rabbit cortical collecting duct. Am J Physiol Renal Physiol 283: F437–F446, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Xu C, Shmukler BE, Nishimura K, Kaczmarek E, Rossetti S, Harris PC, Wandinger-Ness A, Bacallao RL, Alper SL: Attenuated, flow-induced ATP release contributes to absence of flow-sensitive, purinergic Cai2+ signaling in human ADPKD cyst epithelial cells. Am J Physiol Renal Physiol 296: F1464–F1476, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hovater MB, Olteanu D, Hanson EL, Cheng NL, Siroky B, Fintha A, Komlosi P, Liu W, Satlin LM, Bell PD, Yoder BK, Schwiebert EM: Loss of apical monocilia on collecting duct principal cells impairs ATP secretion across the apical cell surface and ATP-dependent and flow-induced calcium signals. Purinergic Signal 4: 155–170, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siroky BJ, Ferguson WB, Fuson AL, Xie Y, Fintha A, Komlosi P, Yoder BK, Schwiebert EM, Guay-Woodford LM, Bell PD: Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am J Physiol Renal Physiol 290: F1320–F1328, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Liu W, Murcia NS, Duan Y, Weinbaum S, Yoder BK, Schwiebert E, Satlin LM: Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. Am J Physiol Renal Physiol 289: F978–F988, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Du J, Wong WY, Sun L, Huang Y, Yao X: Protein kinase G inhibits flow-induced Ca2+ entry into collecting duct cells. J Am Soc Nephrol 23: 1172–1180, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Köttgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G: TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol 182: 437–447, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berrout J, Jin M, Mamenko M, Zaika O, Pochynyuk O, O’Neil RG: Function of transient receptor potential cation channel subfamily V member 4 (TRPV4) as a mechanical transducer in flow-sensitive segments of renal collecting duct system. J Biol Chem 287: 8782–8791, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song MY, Yuan JX: Introduction to TRP channels: Structure, function, and regulation. Adv Exp Med Biol 661: 99–108, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Goel M, Sinkins WG, Zuo CD, Hopfer U, Schilling WP: Vasopressin-induced membrane trafficking of TRPC3 and AQP2 channels in cells of the rat renal collecting duct. Am J Physiol Renal Physiol 293: F1476–F1488, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Goel M, Sinkins WG, Zuo CD, Estacion M, Schilling WP: Identification and localization of TRPC channels in the rat kidney. Am J Physiol Renal Physiol 290: F1241–F1252, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Wu L, Gao X, Brown RC, Heller S, O’Neil RG: Dual role of the TRPV4 channel as a sensor of flow and osmolality in renal epithelial cells. Am J Physiol Renal Physiol 293: F1699–F1713, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD: OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2: 695–702, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Liedtke W, Choe Y, Martí-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S: Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103: 525–535, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, Liedtke W, Hoyer J, Köhler R: Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS ONE 2: e827, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao X, Wu L, O’Neil RG: Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem 278: 27129–27137, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Taniguchi J, Tsuruoka S, Mizuno A, Sato J, Fujimura A, Suzuki M: TRPV4 as a flow sensor in flow-dependent K+ secretion from the cortical collecting duct. Am J Physiol Renal Physiol 292: F667–F673, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG: Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408: 990–994, 2000 [DOI] [PubMed] [Google Scholar]
- 41.Patel V, Chowdhury R, Igarashi P: Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens 18: 99–106, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gattone VH, 2nd, Wang X, Harris PC, Torres VE: Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9: 1323–1326, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Yamaguchi T, Nagao S, Kasahara M, Takahashi H, Grantham JJ: Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis 30: 703–709, 1997 [DOI] [PubMed] [Google Scholar]
- 44.Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH, 2nd: Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med 10: 363–364, 2004 [DOI] [PubMed] [Google Scholar]
- 45.Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP: Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol 17: 178–187, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM, Jr, Grantham JJ: Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 66: 964–973, 2004 [DOI] [PubMed] [Google Scholar]
- 47.Schwiebert EM, Wallace DP, Braunstein GM, King SR, Peti-Peterdi J, Hanaoka K, Guggino WB, Guay-Woodford LM, Bell PD, Sullivan LP, Grantham JJ, Taylor AL: Autocrine extracellular purinergic signaling in epithelial cells derived from polycystic kidneys. Am J Physiol Renal Physiol 282: F763–F775, 2002 [DOI] [PubMed] [Google Scholar]
- 48.Wilson PD, Hovater JS, Casey CC, Fortenberry JA, Schwiebert EM: ATP release mechanisms in primary cultures of epithelia derived from the cysts of polycystic kidneys. J Am Soc Nephrol 10: 218–229, 1999 [DOI] [PubMed] [Google Scholar]
- 49.Gradilone SA, Masyuk TV, Huang BQ, Banales JM, Lehmann GL, Radtke BN, Stroope A, Masyuk AI, Splinter PL, LaRusso NF: Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology 139: 304–314, e2, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liedtke W, Friedman JM: Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci U S A 100: 13698–13703, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sullivan LP, Wallace DP, Grantham JJ: Epithelial transport in polycystic kidney disease. Physiol Rev 78: 1165–1191, 1998 [DOI] [PubMed] [Google Scholar]
- 52.Strotmann R, Schultz G, Plant TD: Ca2+-dependent potentiation of the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin binding site. J Biol Chem 278: 26541–26549, 2003 [DOI] [PubMed] [Google Scholar]
- 53.Fan HC, Zhang X, McNaughton PA: Activation of the TRPV4 ion channel is enhanced by phosphorylation. J Biol Chem 284: 27884–27891, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamandé SR, Yuan Y, Gresshoff IL, Rowley L, Belluoccio D, Kaluarachchi K, Little CB, Botzenhart E, Zerres K, Amor DJ, Cole WG, Savarirayan R, McIntyre P, Bateman JF: Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat Genet 43: 1142–1146, 2011 [DOI] [PubMed] [Google Scholar]
- 55.Willette RN, Bao W, Nerurkar S, Yue TL, Doe CP, Stankus G, Turner GH, Ju H, Thomas H, Fishman CE, Sulpizio A, Behm DJ, Hoffman S, Lin Z, Lozinskaya I, Casillas LN, Lin M, Trout RE, Votta BJ, Thorneloe K, Lashinger ES, Figueroa DJ, Marquis R, Xu X: Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J Pharmacol Exp Ther 326: 443–452, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Rieg T, Vallon V: ATP and adenosine in the local regulation of water transport and homeostasis by the kidney. Am J Physiol Regul Integr Comp Physiol 296: R419–R427, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mangoo-Karim R, Ye M, Wallace DP, Grantham JJ, Sullivan LP: Anion secretion drives fluid secretion by monolayers of cultured human polycystic cells. Am J Physiol 269: F381–F388, 1995 [DOI] [PubMed] [Google Scholar]
- 58.Chang MY, Lu JK, Tian YC, Chen YC, Hung CC, Huang YH, Chen YH, Wu MS, Yang CW, Cheng YC: Inhibition of the P2X7 receptor reduces cystogenesis in PKD. J Am Soc Nephrol 22: 1696–1706, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O: Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem 287: 660–671, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, Stockand JD: Acute regulation of the epithelial Na+ channel by phosphatidylinositide 3-OH kinase signaling in native collecting duct principal cells. J Am Soc Nephrol 18: 1652–1661, 2007 [DOI] [PubMed] [Google Scholar]
- 61.Zaika O, Mamenko M, O’Neil RG, Pochynyuk O: Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. Am J Physiol Renal Physiol 300: F1105–F1115, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin M, Wu Z, Chen L, Jaimes J, Collins D, Walters ET, O’Neil RG: Determinants of TRPV4 activity following selective activation by small molecule agonist GSK1016790A. PLoS ONE 6: e16713, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin M, Berrout J, Chen L, O’Neil RG: Hypotonicity-induced TRPV4 function in renal collecting duct cells: Modulation by progressive cross-talk with Ca2+-activated K+ channels. Cell Calcium 51: 131–139, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cai Z, Xin J, Pollock DM, Pollock JS: Shear stress-mediated NO production in inner medullary collecting duct cells. Am J Physiol Renal Physiol 279: F270–F274, 2000 [DOI] [PubMed] [Google Scholar]