

Abstract
High levels of homocysteine (Hcy) known as hyperhomocysteine-mia (HHcy), contribute to autophagy and ischemia/reperfusion injury (I/R). Previous studies have shown that I/R injury and HHcy cause increased cerebrovascular permeability; however, the associated mechanism remains obscure. Interestingly, during HHcy, cytochome-c becomes homocysteinylated (Hcy-cyto-c). Cytochrome-c (cyto-c) transports electrons and facilitates bioenergetics in the system. However, its role in autophagy during ischemia/reperfusion injury is unclear. Tetrahydrocurcumin (THC) is a major herbal antioxidant and anti-inflammatory agent. Therefore, the objective of this study was to determine whether THC ameliorates autophagy during ischemia/reperfusion injury by reducing homocysteinylation of cyto-c in hyperhomocysteinemia pathological condition. To test this hypothesis we employed 8–10 weeks old male cystathionine-beta-synthase heterozygote knockout (CBS+/−) mice (genetically hyperhomocystemic mice). Experimental group was: CBS+/−, CBS+/−+THC (25mg/kg in 0.1%DMSO dose); CBS (+/−)/I/R and CBS (+/−)/I/R+THC(25mg/kg in 0.1%DMSO dose). Ischemia was performed for 30 min and reperfusion for 72 hours. THC was injected intra-peritoneally (I.P.) once daily for a period of 3 days after 30 of ischemia. The infarct area was measured using 2,3,5-triphenyltetrazolium chloride staining. Permeability was determined by brain edema and Evans Blue extravasation. The brain tissues were analyzed for oxidative stress, matrix metalloproteinase-9 (MMP-9), damage-regulated autophagy modulator (DRAM), and microtubule-associated protein 1 light chain 3 (LC3) by Western blot. The mRNA levels of S-Adenosyl-L-homocysteine hydrolases (SAHH) and Methylenetetrahydrofolate reductase (MTHFR) genes were measured by Quantitative real-time polymerase chain reaction. Co-immunoprecipitation was used to determine the homocysteinylation of cyto-c. We found that brain edema and Evans Blue leakage were reduced in I/R+THC treated groups as compared to sham operated groups along with reduced brain infarct size. THC also decreased oxidativedamage and ameliorated the homocysteinylation of cyto-c in-part by MMP-9 activation which leads to autophagy in I/R groups as compared to sham operated groups. This study suggests a potential therapeutic role of dietary THC in cerebral ischemia.
Keywords: Stroke, Blood Brain Barrier, Redox Stress, Methionine, Curcumin
INTRODUCTION
Cerebral stroke is a major cause of morbidity and mortality worldwide. Many studies indicate a plethora of risk factors for cerebral stroke (i.e., hypertension, obesity, smoking, and diabetes mellitus) (Manolescu et al., 2010). Nevertheless, cerebrovascular events can occur sometimes in individuals without any of the previously mentioned risk factors. Homocysteine is a thiol-containing amino acid formed during the methionine metabolism via methylation to homocysteine which requires S-Adenosyl-L-homocysteine hydrolases (SAHH), remethylation to methionine, which requires methylenetetrahydrofolate reductase (MTHFR); and transsulfuration to cysteine which requires cystathionine beta synthase (CBS) enzymes (Obeid and Herrmann, 2006). Severe hyperhomocysteinemia (HHcy), which results in abnormally high plasma homocysteine concentration, is usually caused by cystathionine beta synthase (CBS) deficiency (Selhub, 1999; Watanabe et al., 1995). HHcy is a common risk factor for stroke, vascular and possibly neurodegenerative diseases (Beard, Jr. and Bearden, 2011; Kraus et al., 1999).
HHcy causes increased oxidative stress by differential expressions of oxidant and anti-oxidant enzymes (Tyagi et al., 2005b). Oxidative stress has been suggested to be involved in brain edema and post-ischemic neuronal damage (Abe et al., 1988). Ischemia rapidly consumes endogenous antioxidants and produces excessive amounts of toxic free radicals(Chen et al., 2011). The accumulation of these toxic free radicals plays an essential role in blood brain barrier (BBB) disruption through matrix metalloproteinases (MMPs) activation (Gasche et al., 1999; Romanic et al., 1998). MMPs are membrane-bound, zinc-binding proteolytic enzymes are essential for the breakdown of extracellular matrix (ECM) components of the basement membrane around cerebral blood vessels and neurons. MMPs are synthesized as pre-proenzymes, secreted from cells as proenzymes, and activated by other proteases and free radicals in the extracellular compartment(Lee et al., 2005). Among these MMPs, MMP-2 and MMP-9 are key enzymes (Romanic et al., 1998). Several reports have suggested that MMP-9 plays a significant role in brain injury after cerebral ischemia (Fujimura et al., 1999; Lee et al., 2004; Rosenberg et al., 1998). Pharmacological inhibition of MMP-9 as well as targeted deletion of the MMP-9 gene in mice resulted in substantial reductions of brain damage after ischemia (Asahi et al., 2000; Wang et al., 2000).
Jakubowski demonstrated that Hcy-induced vascular damage could be related to modification of proteins by a metabolite of Hcy, Hcy-thiolactone (N-homocysteinylation) (Jakubowski, 2007; Jakubowski, 2008). Indeed N-homocysteinylation is elevated in CBS deficient patients and is more toxic than Hcy itself (Perla-Kajan et al., 2008). N-homocysteinylation impairs protein function and leads to cell and tissue damage. In vitro studies have demonstrated homocysteinylation of cyto-c. Cyto-c is a mitochondrial peripheral membrane protein and is essential for mitochondrial function. Cyto-c is very susceptible to the modification by N-homocysteinylation (Perla-Kajan et al., 2007; Zhao et al., 2010). The role of N-Hcy-Cyto-c in autophagy during genetic HHcy is not clear.
Autophagy is a regulated process of degradation and recycling of cellular constituents (Rami et al., 2008). Autophagy may help to promote cell survival (Rami et al., 2008) or may also promote cell death (Reggiori and Klionsky, 2002). There were evidences that autophagy increases in neurons in the periphery of the ischemia area after 6–24h post injury(Rami et al., 2008). In contrast post-ischemic injections of the autophagy inhibitor 3-methyladenine block the ischemia-induced increase in LC3-II and DRAM (autophagy marker). These results point out the need to develop autophagy inhibitors that could be more suitable than 3-methyladenine (3-MA, an autophagy inhibitor) for therapeutic purpose(Rami et al., 2008).
Tetrahydrocurcumin (THC) is one of the major metabolites of curcumin with potent antioxidant and anti-inflammatory activity (Pan et al., 1999). Curcumin (diferuloyl methane) is a polyphenol compound, extracted from the rhizomes of the Curcuma longa plant (turmeric). Structurally, THC and curcumin are similar expect THC lacks the double bonds of curcumin (Okada et al., 2001). However, recent attention has been focused on THC because it appears to exert a greater antioxidant activity in both in-vitro and in-vivo systems (Okada et al., 2001; Pari and Murugan, 2004). Unlike curcumin, THC is stable in all physiological buffers. In recent years, curcumin has been shown to be neuro-protective and also lower blood glucose levels in type 2 diabetes mice (Nishiyama et al., 2005). However, the effect of THC on various aspects of HHcy has not been investigated. Thus we explore in the present study the effect of THC on homocysteinylated cyto-c mediated autophagy in the well characterized genetic model of severe HHcy in mice after cerebral ischemia.
EXPERIMENTAL PROCEDURES
Mice
All experimental procedures were approved and carried out in accordance with the Institutional Animal Care and Use Committee of the University of Louisville. 8–10 weeks old CBS heterozygous knockout mice (CBS+/−) were obtained from Jackson Laboratory (Bar Harbor, ME) and maintained in the animal facility center at University of Louisville. The mice were fed with regular mice chow PMI®LabDiet®St Louis, MO (Cat # 5015) and water ad libitum. Mice genotypes were determined by a polymerase chain reaction of DNA obtained from tail biopsies with a specific set of primers (Kumar et al., 2008).
Focal Cerebral Ischemia
Animals weighing 22–25 g were anesthetized with sodium pentobarbital (70 mg/kg bd wt). Body temperature was controlled using a rectal temperature probe and maintained at 37°C±0.5°C by a thermostat-controlled heated blanket. Ischemia was induced by occlusion of the middle cerebral artery (MCAO), using a modified intraluminal technique. MCAO was performed with a silicone resin-coated 6-0 nylon monofilament (Ethicon, Titusville, N.J) which was introduced into a small incision of the left common carotid artery and advanced distal to the carotid bifurcation for temporary occlusion of the middle cerebral artery (30 min). Regional cerebral blood flow (CBF) was continuously monitored using a laser doppler probe (Moor Instruments, moor FLPI) to verify ischemia and reperfusion. The occlusion was established when the CBF dropped below 20% of the pre-MCAO flow-value. After 30 minutes of middle cerebral artery occlusion, blood flow was restored by withdrawal of the monofilament. Sham-operated mice were subjected to the same anesthesia and surgical procedure, except MCAO (Longa et al., 1989).
Administration of THC
THC used in the study was purchased from Sabinsa Corporation, USA. (1) CBS+/− heterozygous knockout mice with vehicle treatment; (2) CBS+/− heterozygous knockout mice with THC treatment; (3) CBS+/− heterozygous knockout mice with ischemia/reperfusion injury (I/R); (4) CBS+/− heterozygous knockout mice with ischemia/reperfusion injury with THC treatment. THC was dissolved in 0.1% dimethyl sulfoxide (DMSO; Sigma, USA). 30 mins after MCAO, THC (25mg/kg/day) or vehicle was given for 3 days by intra-peritoneal injection.
Assessment of Neurological functional
After recovery from anesthesia and again after 72hrs, neurological function was assessed according to the method of Longa etal. (Longa et al., 1989). Neurological findings were scored on a 5-point scale. No neurological deficit = 0, failure to extend right paw fully = 1, circling to right = 2, falling to right = 3, did not walk spontaneously and has depressed levels of consciousness = 4.
Determination of infarct volume
Animals were sacrificed 72hrs after MCAO. Brains were quickly removed and frozen at − 20 °C for 10 min. Coronal sections were cut into 2 mm slice using a mouse brain slice matrix (Harvard Apparatus, Holliston, MA, USA). The slices were stained for 30 min at 37°C with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, Taufkirchen, Germany) in PBS to visualize the infarctions (Bederson et al., 1986; Kleinschnitz et al., 2010).
Measurement of Brain water content
The water content of the brain tissue was measured by the wet and dry weight method as described previously (Fukui et al., 2003). Briefly, Brain tissue samples from CBS+/− + vehicle, CBS+/−+THC, CBS+/− + I/R and CBS+/− I/R+ THC mice were immediately weighed to obtain wet weight (WW). The tissue was then dried in an oven at 95°C for 72 hrs and weighed again to obtain the dry weight (DW). The formula (WW−DW)/WW×100% was used to calculate the water content and expressed as a percentage of wet weight.
Evaluation of BBB Integrity
We investigated the integrity of the BBB in mice by measuringthe extravasation of Evans blue (Belayev et al., 1996; Uyama et al., 1988). Evans blue dye (2%, 4 mL/kg body weight) was slowly administered via carotid artery and allowed to circulate for 3 hrs in mice with or without THC treatment. At the end of the experiment, the mice were perfused with saline to wash away any remaining dye in the blood vessels. The dissected brain wereweighed and extracted in 1.0ml of 50% trichloroacetic acid solution. After homogenization and centrifugation, the extract was dilutedwith ethanol (1:3), and its fluorescence was determined at 620 nm (excitation) and 680nm (emission) with a spectrophotometer (Spectra Max 3000, CA, USA). The amount of Evans blue was expressed as was expressed as nm/ischemic hemisphere by using a standardized curve.
In Situ Zymography
In situ zymography gelatinolytic activity was assessed using a commercially available kit (Molecular Probes) as described previously (Gasche et al., 2001) with some modification. After 30 min of ischemia or 30 min of ischemia followed by 72hrs of reperfusion, the animals were scarified, and the brains were frozen in 2-methylbutane with liquid nitrogen. Frozen brain were sectioned with a cryostat to a thickness of 30μm and incubated for 3 hours at 37°C with 400μg/mL DQ gelatin conjugate (Molecular probe), a fluorogenic substrate. After 3hr washed with PBS and fixed in 4% paraformaldehyde in PBS. DQ gelatin cleaved by MMPs resulted in a green fluorescent product (excitation, 495nm; emission, 515nm).
Western Blot Analysis
Change in protein content during ischemia was assessed by Western blot analysis (Tyagi et al., 2010). Briefly, frozen brain tissue washed twice with ice-cold PBS and lysed with ice-cold RIPA buffer (containing 5mM of ethylenediaminetetraacetic acid), which was supplemented with phenylmethylsulfonyl fluoride (1 mM) and protease inhibitor cocktail (1ml/ml of lysis buffer). Protein content of the lysate was determined using the Bicinchronic Acid protein assay kit (BCA, Pierce, Rockford, IL). Equal amounts of protein (60ug) were resolved on 10–15% SDS-PAGE and transferred onto a polyvinylidene difluoride membrane (PVDF, Bio-Rad) as described (Qipshidze et al., 2010). The membranes were blocked with 5% non-fat dry milk in TBS-T (50 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20, pH 7.4), incubated with respective primary antibodies for 3 hrs at room temperature. After probing with appropriate secondary antibodies for 1.5 hrs at room temperature the blots were analyzed with Gel-Pro Analyzer software (Media Cybernetics, Silver Spring, MD) as described earlier (Qipshidze et al., 2010). Membranes were stripped and re-probed for GAPDH as a loading control. The protein expression intensity was assessed by integrated optical density (IOD) of the area of the band in the lane profile. To account for possible differences in the protein load, measurements presented are the IOD of each band under study (protein of interest) divided by the IOD of the respective GAPDH band.
Confocal Microscopy
Hcy level in brain tissue during ischemia was assessed by immunohistochemistry analysis as described earlier (Qipshidze et al., 2010). Samples of brain tissue from all experimental groups were immediately placed onto freezing media and stored at −70°C until they were used. Briefly, sections were post-fixed in 4% of paraformaldehyde, and labeled with Hcy antibody (Abcam Antibodies, Cambridge, MA). After an overnight incubation, sections were washed with PBS and incubated with appropriate secondary antibody conjugated with Texas Red. After washing, sections were mounted with FluoroGel mounting medium (GeneTex, Inc) and visualized with confocal microscopy (Olympus, FluoView 1000, objective 60x). DAPI was used for nuclei staining. Total fluorescence (red) intensity in 5 random fields (for each experiment) was measured with image analysis software (Image-Pro Plus, Media Cybernetics) and expressed in fluorescence intensity units (FIU). The fluorescence intensity values were averaged for each experimental group.
Reverse transcription-polymerase chain reaction
The RT-PCR was performed for mRNA expression of SAHH, MTHFR and GAPDH in all experimental group using ImProm-II™ Reverse Transcription system kit (Promega Corporation, Madison, WI, USA, cat # A3800) as described elsewhere (Mishra et al., 2010). For gene amplification the RT-PCR program was 95 °C–7.00 min [95 °C–0.50 min, 55 °C–1.00 min, 72 °C–1.00 min] × 34, 72 °C–5.00 min, 4 °C-∞. The primers for RT-PCR are: MTHFR- Forward 5′-CTACCAGAGCCCAAGACAGC-3′; reverse 5′-ATTGCAGTTGCTCCTTGTCC SAHH-Forward 5′-CGATTCTGTCACCAAGAGCA-3′; reverse 5′-TGAAGGAGTTGCTCATCACG-3′; GAPDH Forward5′-TGTTGCTGTAGCCGTATTCA-3′; reverse 5′-CAACAGCAACTCCCACTCTT-3′. The RT-PCR product was electrophoresed on 1% agarose gel in TAE with 0.008% ethidium bromide. All of the data were analyzed using GAPDH as loading control.
Quantitative real-time PCR
Quantitative real-time PCR (QRT-PCR) for SAHH and MTHFR genes was performed using an Mx3000p QPCR method as described previously (Tyagi N. et. al.2010). The first-strand cDNA reaction (0.5 μl) was subjected to real-time PCR amplification using gene-specific primers. The sequences of the primers used are as follows: SAHH: 5′-ACTGAGAAGCAGGCCCAGTA -3′ (forward) and 5′-CACTGTCCAGGCTACTGCAA-3′ (reverse) and MTHFR: 5′-CCATCCTCAGACCCTGTTGT -3′ (forward) and 5′-CGTCCACGATGTGGTAGTTG -3′ (reverse); GAPDH 5′-CAACAGCAACTCCCACTCTT-3′ (forward) and 5′-TGTTGCTGTAGCCGTATTCA -3′ (reverse). Real-time PCR assays were erformed in 25-μl reactions, consisting of 2× (12.5 μl) Brilliant SYBR Green QPCR Master Mix (Stratagene), 400 nmol/L primers (0.5 μl each from the stock), 11 μl of water, and 0.5 μl of template. The thermal conditions consisted of an initial denaturation at 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 15 s, annealing, and extension at 56°C for 1 min, and, for a final step, a melting curve of 95°C for 15 s, 56°C for 15 s, and 95°C for 15 s. All reactions were performed in triplicate to reduce variation. Data normalization was accomplished using GAPDH as an endogenous control, and the normalized values were subjected to a 2−ΔΔCt formula to calculate the fold change between the control and experimental groups.
Co-immunoprecipitation
Briefly, tissue extract from all groups (150mg protein) were pre-incubated for 1 h with 40 μl of Protein A–Sepharose beads at 4°C. Samples were centrifuged for 2 min at 1500 g, and the supernatants were further incubated with the rabbit anti-cyto-c antibody (Abcam Antibodies, Cambridge, MA) (Damdimopoulos et al., 2002). Protein A–Sepharose (40 μl) was added to each sample and the mixture was incubated for 3 h, at 4 °C. Protein A–Sepharose was pelleted by centrifugation at 1500g for 2 min and washed three times in PBS. The samples were boiled in SDS/PAGE loading buffer, in the presence of 5% (v/v) 2-mercaptoethanol, for 4 min and centrifuged at 1500g for 2 min. The resulting supernatants were subjected to SDS/PAGE (12% gel), and Western blotting was performed with specific antibodies against cyto-c and homocysteine (rabbit polyclonal). Membranes were re-probed with antibody against the GAPDH.
Statistical Analysis
Results are expressed as mean ± SD. Statistical analysis used Student t test or ANOVA to compare quantitative data with normal distribution and equal variance. A p value <0.05 was consider statistically significant unless otherwise specified.
RESULTS
Effect of THC on cerebral infarction and neuroscore
To determine the neuroprotective effect of THC against ischemia/reperfusion insult, we measured the Infarct volume and neuroscroe with or without THC administration. The infarct volume and neuroscore were significantly increased in CBS+/− ischemic group as compared to sham-operated group (CBS+/−) (Fig.1A, B). While the infarct volume and neuroscore were significantly decreased in THC treated mice as compared to CBS+/− ischemic mice (Fig.1A, B). As shown in Fig. 1A, a remarkably decreased pale-colored region was observed in the THC-treated mice brain as compared with CBS+/− ischemia/reperfusion. These observations indicate that THC can prevent the ischemia/reperfusion induced brain injury.
Figure 1. Neuroprotective effect of THC against ischemia/reperfusion insult.
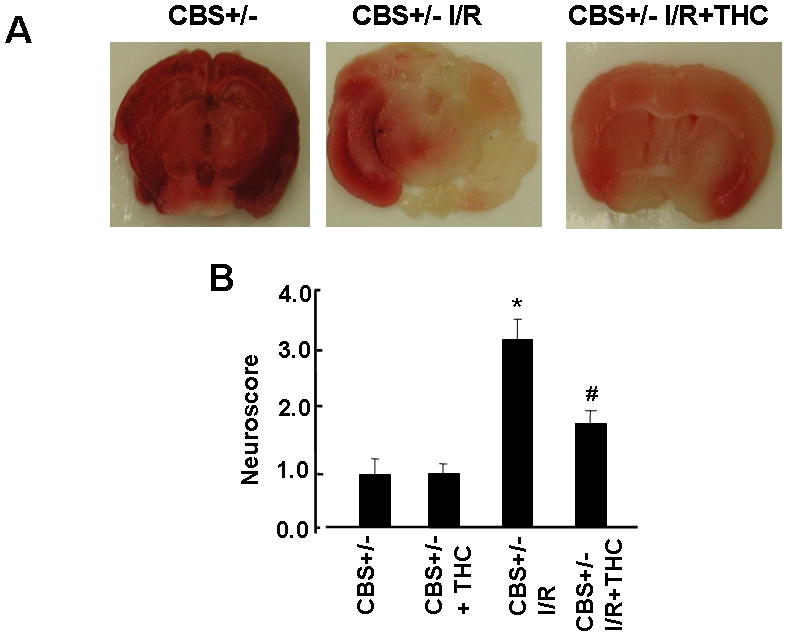
A: Representative coronal sections stained with 2% TTC after 45 min of middle cerebral artery occlusion and 72 hrs of reperfusion showing infarction. Dark colored region indicates no-ischemic portion of brain and pale-colored region indicates ischemic portion of the brain. B: Neuroscore of vehicle and THC-treated mice measured 3day after middle cerebral artery occlusion. *P<0.05 values significantly different from CBS+/− mice, #P<0.05 values significantly different from CBS+/− I/R mice; n=4 animals/group.
Effect of THC on blood brain barrier integrity after focal cerebral ischemia
Increased vascular permeability and disruption of the blood brain barrier (BBB) could be initiating factors for the development of cerebral injury and critical factors for the determination of post-ischemic severity. To elicit the effect of THC administration on BBB permeability, we quantified the extravasation of Evans blue in the brain as an indicator of BBB breakdown. In the sham operated group (CBS+/−), the content of Evans blue was unchanged (Fig. 2A). The levels of Evans blue were significantly increased in CBS+/− ischemic group as compared to sham-operated group (CBS+/−) (Figs. 2A). While the extravasation of Evans blue were significantly decreased in THC treated mice as compared to CBS+/− ischemic mice (Fig.2A). Increased vascular permeability results in edema formation, which is a major complication in strokes. Brain water content, as the index of cerebral edema, was higher in the CBS+/− ischemic group as compared to sham-operated group (CBS+/−). Administration of THC produced a significant decrease in post-ischemic cerebral edema (Fig. 2B) as compared to CBS+/− ischemic group. These results indicate that THC could improve post-ischemic brain injury by decreasing the vascular permeability and edema.
Figure 2. Effect of THC on blood-brain barrier integrity.
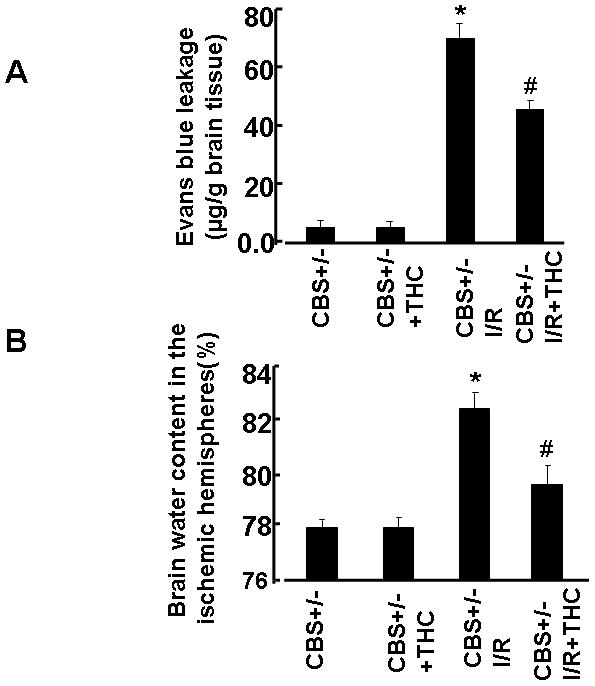
A: Quantitative analysis of the extravasation of Evens blue dye showing integrity of the blood–brain barrier after 45min of MACO and 72hrs of reperfusion. B: Brain edema formation after 45 min of MACO and 72 hrs of reperfusion. *P<0.05 values significantly different from CBS+/− mice, #P<0.05 values significantly different from CBS+/− I/R mice; n=4 animals/group.
Effect of THC on Homocysteine Metabolism
Because stress decreases MTHFR and increases SAHH echelons thus causes an increase in total Hcy levels in tissue. We measured mRNA expression of Hcy metabolic enzymes MTHFR and SAHH. Induction of ischemia significantly decreases the expression of MTHFR whereas SAHH expression was increased in CBS+/− ischemic group as compared to sham-operated group (CBS+/−) (Fig.3A, B,C,D). Supplementation with THC restored expression of SAHH and MTHFR in ischemic brain (Fig. 3A, B,C,D). Impairment in Hcy metabolism causes an increased in total Hcy levels. To assess the effect of THC on total Hcy levels during cerebral ischemia, we evaluated total Hcy levels in brain tissue by immunohistochemistry in all the groups. Total Hcy levels were robustly increased in ischemia groups as compared to sham operator groups (Fig.3E, F). The THC treatment significantly reduced total Hcy level in CBS+/− I/R+THC group as compared to CBS+/− I/R groups. These results suggest the inhibitory action of THC on Hcy levels (Fig.3).
Figure 3. Effect of THC on Hcy metabolism and levels.
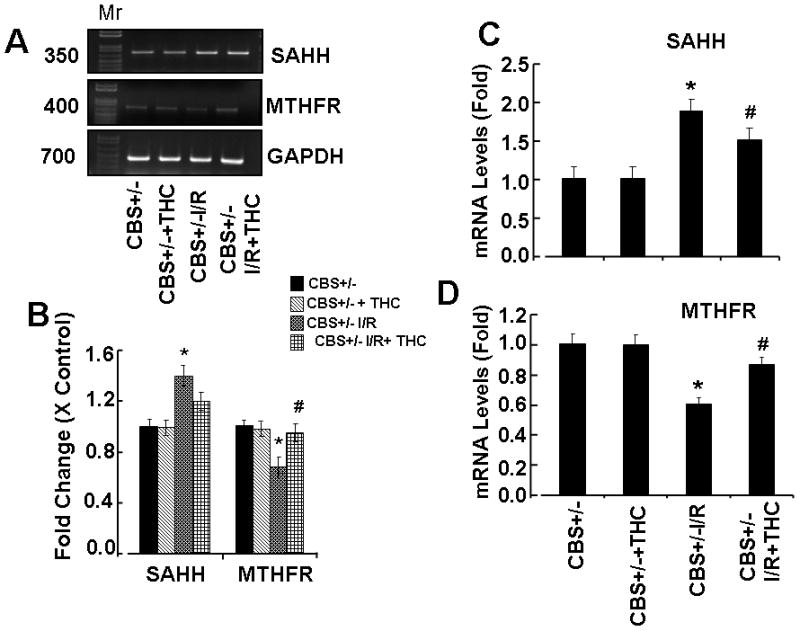
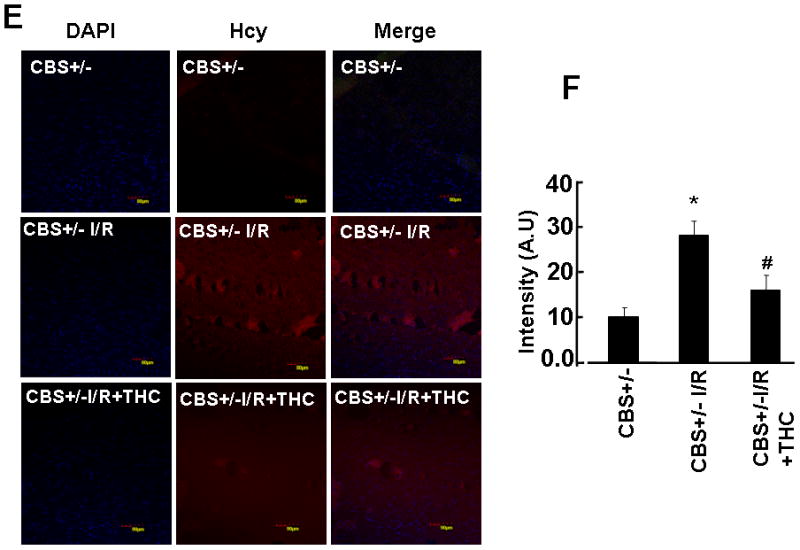
A: Represented semi quantitative mRNA expression for SAHH, MTHFR and with GAPDH control. B: Densitometric analysis of SAHH and MTHFR mRNA expressions as represented in the bar diagram. C & D: Total mRNA was isolated from the brain of all experimental groups and the mRNA levels of MTHFR and SAHH was measured by QRT-PCR. *P<0.01 values significantly different from CBS+/− mice, #P<0.01 values significantly different from CBS+/−I/R mice; n=4 animals/group. E: Immunostaining results demonstrate Hcy expression in the brain sections from CBS+/−, CBS+/− + THC, CBS+/− I/R and CBS+/− I/R+THC group. Cellular nuclei in all experiments were stained with 4′,6-diamidino-2-phenylindole (DAPI). F: Bar diagram represents quantification of immunostaining using image pro software. *P<0.05 values significantly different from CBS+/− mice, *P<0.05 values significantly different from CBS+/− mice, #P<0.05 values significantly different from CBS+/− I/R mice; n=3 animals/group.
Effect of THC on Oxidative Stress
To test whether elevated levels of homocysteine caused oxidative stress during ischemia/reperfusion injury. We measured protein expression of p47phox (oxidant enzyme) and catalase (antioxidant enzyme). Protein level of p47phox was significantly up-regulated in CBS+/− ischemic group as compared to the sham operator groups (CBS+/−) whereas protein level of catalase was significantly down-regulated in CBS+/− ischemic group as compared to sham operator groups (Figs.4A, B). Treatment with THC mitigates these effects. These results suggested that during brain injury, increased total Hcy level caused elevation in oxidative stress and THC treatment attenuated these effects (Fig.4).
Figure 4. Effect of THC on oxidative stress.
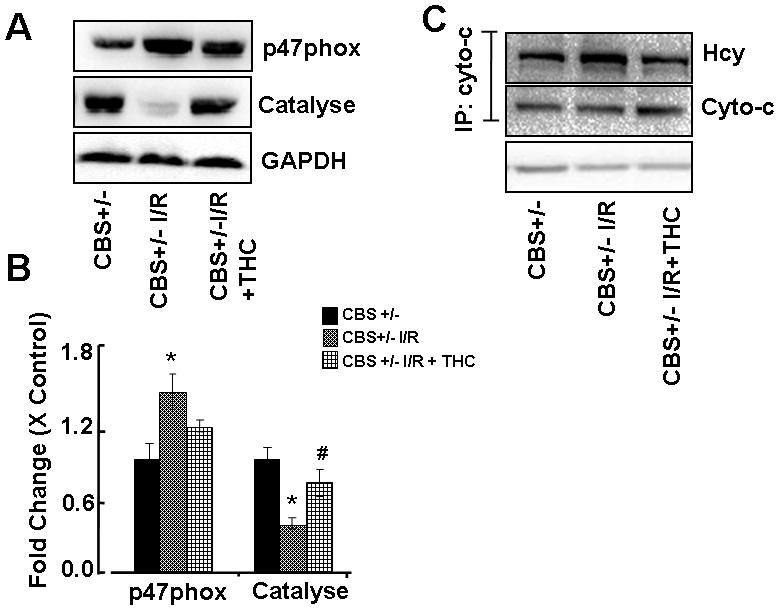
A: Representative immunoblots for p47phox, catalyse and GAPDH control. B: Densitometric analysis of p47phox and catalyse protein expressions using Image Lab software as shown in the bar diagram. C: Brain tissue lysate was immunoprecipitated with anti-cyto-C antibody. Immunoprecipitated samples were subjected to Western blot with anti-Hcy antibody and with anti-cyto-C antibody. GAPDH was used as a loading control. *P<0.05 values significantly different from CBS+/− mice, #P<0.05 values significantly different from CBS+/− I/R mice; n=4 animals/group.
Effect of THC on Homocystenylation of Cytochrome-c
To determine whether an increase in Hcy levels cause protein N-homocysteinylation of cyto-C during ischemia/reperfusion injury and induce autophagy. In the ischemic brain, the levels of cyto-c homocystenylation were increases as compared to sham-operated group. THC treatment inhibits the homocystenylation of cyto-c (Fig.4C). N-linked Hcy thiol group was blocked by the treatment with THC (Fig.4).
Effect of THC on MMPs
MMP-9 is a known marker for cerebral injury. To assess total MMPs activity, in-situ DQ-gelatin zymography was performed in fresh brain slices from all groups. There was a marked increase in gelatinolytic activity in the ischemic group as compared to sham operator groups (CBS+/−) (Figs.5A, B, C, and D). Gelatinolytic activity was reduced in the THC-treated groups (Figs.5A, B, C,D). To verify the gelatinolytic activity result, we performed Western blot in all groups (Figs.5E, F) Interestingly, MMP-9 protein expression was markedly increased in CBS+/− ischemia groups as compared to sham operator groups (CBS+/−) (Figs.5E,F). These results suggested that THC administration significantly inhibited the induction of MMP-9 (Fig.5).
Figure 5. Effect of THC on MMP-9.
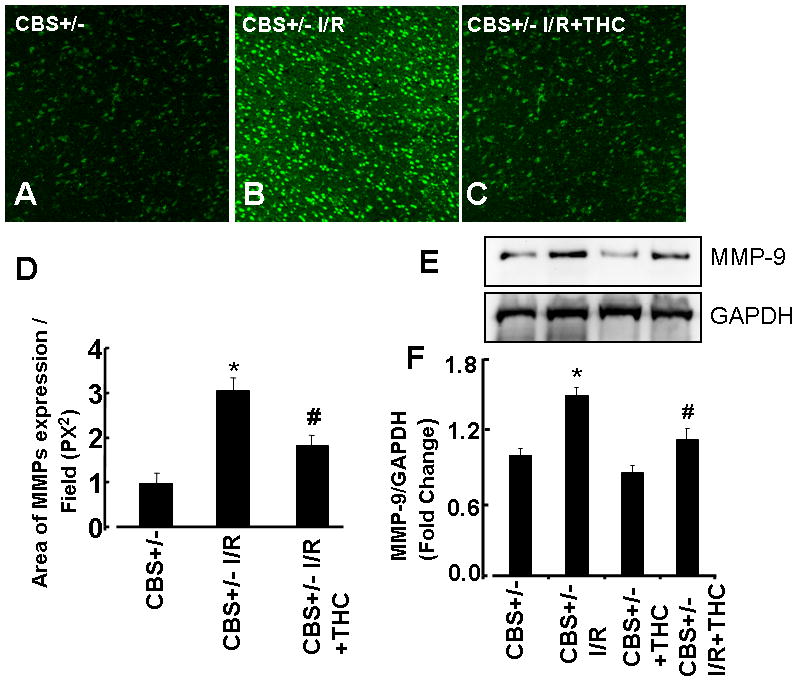
A, B, and C: Representative photomicrographs of gelatinase activity. D: Bar graph of changes in integrated optical density (IOD) in activity of MMP-9. The micrographs were taken under the identical set of conditions for all groups. E: Representative immunoblots for MMP-9 protein and GAPDH control. F: Densitometric analysis of MMP-9 protein expressions using Image Lab software as shown in the bar diagram. *P<0.05 values significantly different from CBS+/− mice, #P<0.05 values significantly different from CBS+/− I/R mice; n=3 animals/group.
Anti-autophagy effect of THC
Ischemia/reperfusion injury induces autophagy. Our results showed LC3-II levels and DRAM protein expression was increased in CBS+/− ischemic group as compared to sham operator groups (Figs.6). Administration of THC inhibited the conversion of LC3-I to LC3II (Figs.6A, C) and also decreased the protein expression of DRAM in CBS+/− ischemic group as compared to sham operator groups (Figs.6B,D). These results suggested that protective effects of THC are associated with the antiautophagic effect after cerebral ischemia/reperfusion injury, in part, by homocystenylation of cyto-c and activation MMP-9.
Figure 6. Effect of THC on autophagy.
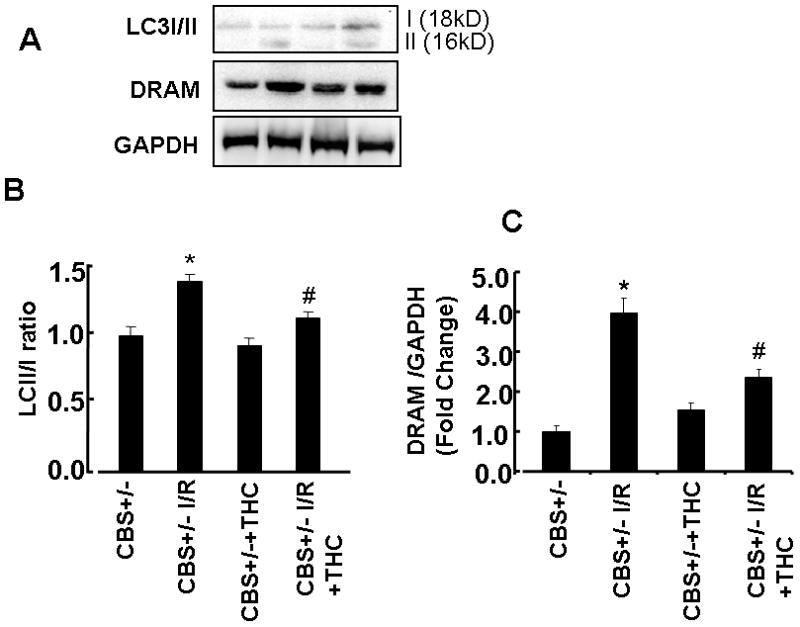
A: Representative immunoblots for cleaved LC3 (LC3-II), DRAM and control GAPDH protein control. B: Densitometric analysis of MMP-9 protein expressions using Image Lab software as shown in the bar diagram. *P<0.05 values significantly different from CBS+/− mice, #P<0.05 values significantly different from CBS+/− I/R mice; n=4 animals/group.
DISCUSSION
The aim of this study was to investigate the neuroprotective effects of tetrahydrocurcumin, a polyphenolic non-enzymatic antioxidant agent, against Hcy induced neurotoxicity. The goal was to establish an animal model of genetically hyperhomocystemia. Previously Jiang etal. Showed the neuroprotective effect of curcumin during cerebral ischemia/reperfusion injury in rat (Jiang et al., 2007; Thiyagarajan and Sharma, 2004).
Results of the present study shows that THC decreased infarct volume and improved neurological outcomes after ischemic/reperfusion injury (I/R) (Fig.1) in genetically hyperhomocystemia mice. Tetrahydrocurcumin diminishes homocystenylation of cyto-c by decreasing oxidative stress, MMP-9 and provides neuronal protection through autophagy mechanisms. Disruption of the blood brain barrier occurs under various pathological conditions such as I/R injury. Which causing to an increased vascular permeability with subsequent development of brain edema (Utepbergenov et al., 1998). Protection of the blood–brain barrier has become an important experimental therapeutically target during I/R injury (Veltkamp et al., 2005). In the present study, our results indicate that THC inhibited the neurotoxicity of Hcy by decreasing water content of the brain and prevented the absorbance of Evans blue dye after I/R injury, suggesting that THC protects blood–brain barrier integrity by reducing endothelial cells damage (Fig.2) (Jiang et al., 2007).
In the body Hcy is formed from methionine and can be metabolized to cystathionine or methylated to methionine by three enzymes MTHFR, SAHH and CBS. CBS enzyme is the first enzyme of the tran-sulfuration pathway (Kleinschnitz et al., 2010) and directly involved in the removal of Hcy from the methionine cycle (Finkelstein, 1998). Deficiency of CBS enzyme caused homocystinuria (Kleinschnitz et al., 2010). In order to understand the role of THC treatment in expression of Hcy metabolic pathway genes in brain tissue, we measured the mRNA expression of MTHFR and SAHH. QRT-PCR results revealed down-regulation of MTHFR and up-regulation of SAHH genes in CBS+/− +I/R group which was mitigated with THC supplementation (Fig 3A,B,C, D). These results suggest that THC retain transulfuration and remethylation pathways, thus reduce the total Hcy level (Fig.3E,F).
It is well known that Hcy causes cell detachment and cell death by creating oxidative stress (Tyagi et al., 2005b; Tyagi et al., 2006). Thus, the generation of excessive free radicals (ROS) during I/R injury in genetic HHcy condition plays a major role in brain injury associated with stroke. Our result showed that tetrahydrocurcumin treatment significantly decreased oxidative stress by down-regulating oxidant enzyme (phox47) and up-regulating anti-oxidant enzyme (catalyse) expression after I/R injury. Our results are consistent with earlier observations regarding the antioxidant role of THC (Fig.4).
It is known that Hcy induces cerebral arteriolar stiffness and finally brain dysfunction (Nappo et al., 1999). The mechanism involves 1) Hcy toxicity is the modification of proteins by a Hcy metabolite i:e Hcy- thiolactone (HTL); 2)protein modification by HTL is N-homocysteinylation. The N-homocysteinylation has detrimental effects on protein structure and function (Jakubowski, 2006) causing vascular diseases. In this study, we examined the effect of HHcy on homocysteinylation of cyto-c during I/R injury (Fig.5). Our findings demonstrate that THC restrain homocysteinylation of cyto-c by decreasing oxidative stress, which in turn initiate MMP-9 activation.
Increasing evidence has indicated that MMP-2 and MMP-9 level increases in brain injury (Mun-Bryce and Rosenberg, 1998; Strickland et al., 1996; Wang and Tsirka, 2005; Zhao et al., 2007). In HHcy conditions, early appearance of MMP-2, MMP-9 has been reported in cardiovascular diseases and neurovascular diseases (Lominadze et al., 2006; Sen et al., 2007). MMPs, especially MMP-9, have a critical function in proteolytic degradation of the BBB in the pathological process. The lack of MMP-9 gene showed less edema than their WT littermates in a model of ICH (Tejima et al., 2007; Xue et al., 2006), as well as in cerebral ischemia (Asahi et al., 2001). In this study, we showed that increased gelatinolytic activity as well as protein expression in CBS+/− ischemic mice was significantly reduced by THC treatment. The results suggest that MMPs are also implicated in I/R injury induced brain dysfunction, at least in part, through MMP-9 activation. These data collectively support the neuro-protective effect of THC (Fig.6). Previous studies have demonstrated that THC down regulates the MMP-2 and MMP-9 expression (Yodkeeree et al., 2008; Yodkeeree et al., 2009) during cancer cell invasion. Cellular organelles are the prime target for Hcy toxicity. However, the effect of HHcy on autophagy particularly in ischemic condition and its consequence on brain function is not ever been questioned. However, the role of autophagy under HHcy conditions was controversial. It has been reported that Hcy induced apoptosis (Tyagi et al., 2005a; Tyagi et al., 2006) or autophagy/mitophagy (Dennis and Mercer, 2009). Uchiyama’s group reported that in a mouse ischemia-hypoxia model, many damaged neurons showed some features of autophagic/lysosomal cell death (Adhami et al., 2006; Adhami et al., 2007; Ventruti and Cuervo, 2007). In contrast, one study showed that autophagy is associated with neuroprotection (Ventruti and Cuervo, 2007). Thus, the role of autophagy in cell death and survival in cerebral ischemia remains to be defined. In this study, we have demonstrated that THC ameliorates autophagy in genetically HHcy mice after cerebral ischemia/reperfusion injury. Our results indicated that autophagy occurred with increase in the protein level of DRAM and degradation of LC3-I to LC3-II (Fig.7). Autophagy may represent a novel mechanism by which permanent ischemic stroke induces neuronal death, and inhibition of the activation and maturation of autophagy may help reduce ischemic injury. In conclusion, if hyperhomocysteinemia is one of the pathological reasons for neurodegenerative disorders (stroke), THC may be an effective prophylactic agent in the prevention of oxidative stress by Hcy. Furthermore, data of this study suggest that antioxidant property of THC may be responsible for protection against Hcy oxidative stress, possibly by increasing the endogenous defenses against oxidative stress and hence improves brain functions. Further studies are required to reveal the exact mechanism of Hcy in cell death process (apoptosis or autophagy) and the neuroprotective properties of THC must be studied in more detail.
Rational.
Cystathionine-beta synthase deficient homozygous mice suffer from severe growth retardation and a majority of them are dead by 5 weeks of age. Histological examination showed that the hepatocytes of homozygotes were enlarged, multinucleated, and filled with microvesicular lipid droplets. Plasma homocysteine levels of the homozygotes were approximately 40 times normal. Heterozygous mutants have an approximately 50% reduction in cystathionine beta-synthase mRNA and enzyme activity and have twice normal plasma homocysteine levels. Thus, the heterozygous mutants are promising for studying the in vivorole of elevated levels of homocysteine.
Acknowledgments
This work was supported by NIH grants: HL-71010, NS-51568 to SCT and HL-107640 to NT.
Reference List
- 1.Abe K, Yuki S, Kogure K. Strong attenuation of ischemic and postischemic brain edema in rats by a novel free radical scavenger. Stroke. 1988;19:480–485. doi: 10.1161/01.str.19.4.480. [DOI] [PubMed] [Google Scholar]
- 2.Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adhami F, Schloemer A, Kuan CY. The roles of autophagy in cerebral ischemia. Autophagy. 2007;3:42–44. doi: 10.4161/auto.3412. [DOI] [PubMed] [Google Scholar]
- 4.Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beard RS, Jr, Bearden SE. Vascular complications of cystathionine beta-synthase deficiency: future directions for homocysteine-to-hydrogen sulfide research. Am J Physiol Heart Circ Physiol. 2011;300:H13–H26. doi: 10.1152/ajpheart.00598.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17:1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- 8.Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739:88–96. doi: 10.1016/s0006-8993(96)00815-3. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, Chan PH. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal. 2011;14:1505–1517. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damdimopoulos AE, Miranda-Vizuete A, Pelto-Huikko M, Gustafsson JA, Spyrou G. Human mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J Biol Chem. 2002;277:33249–33257. doi: 10.1074/jbc.M203036200. [DOI] [PubMed] [Google Scholar]
- 11.Dennis PB, Mercer CA. The GST-BHMT assay and related assays for autophagy. Methods Enzymol. 2009;452:97–118. doi: 10.1016/S0076-6879(08)03607-0. [DOI] [PubMed] [Google Scholar]
- 12.Finkelstein JD. The metabolism of homocysteine: pathways and regulation. Eur J Pediatr. 1998;157(Suppl 2):S40–S44. doi: 10.1007/pl00014300. [DOI] [PubMed] [Google Scholar]
- 13.Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- 14.Fukui S, Fazzina G, Amorini AM, Dunbar JG, Marmarou A. Differential effects of atrial natriuretic peptide on the brain water and sodium after experimental cortical contusion in the rat. J Cereb Blood Flow Metab. 2003;23:1212–1218. doi: 10.1097/01.WCB.0000088762.02615.30. [DOI] [PubMed] [Google Scholar]
- 15.Galluzzi L, Morselli E, Vicencio JM, Kepp O, Joza N, Tajeddine N, Kroemer G. Life, death and burial: multifaceted impact of autophagy. Biochem Soc Trans. 2008;36:786–790. doi: 10.1042/BST0360786. [DOI] [PubMed] [Google Scholar]
- 16.Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, Chan PH. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999;19:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Jakubowski H. Pathophysiological consequences of homocysteine excess. J Nutr. 2006;136:1741S–1749S. doi: 10.1093/jn/136.6.1741S. [DOI] [PubMed] [Google Scholar]
- 19.Jakubowski H. The molecular basis of homocysteine thiolactone-mediated vascular disease. Clin Chem Lab Med. 2007;45:1704–1716. doi: 10.1515/CCLM.2007.338. [DOI] [PubMed] [Google Scholar]
- 20.Jakubowski H. The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease. J Physiol Pharmacol. 2008;59(Suppl 9):155–167. [PubMed] [Google Scholar]
- 21.Jiang J, Wang W, Sun YJ, Hu M, Li F, Zhu DY. Neuroprotective effect of curcumin on focal cerebral ischemic rats by preventing blood-brain barrier damage. Eur J Pharmacol. 2007;561:54–62. doi: 10.1016/j.ejphar.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 22.Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, Austinat M, Nieswandt B, Wiendl H, Stoll G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115:3835–3842. doi: 10.1182/blood-2009-10-249078. [DOI] [PubMed] [Google Scholar]
- 23.Kraus JP, Janosik M, Kozich V, Mandell R, Shih V, Sperandeo MP, Sebastio G, de FR, Andria G, Kluijtmans LA, Blom H, Boers GH, Gordon RB, Kamoun P, Tsai MY, Kruger WD, Koch HG, Ohura T, Gaustadnes M. Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat. 1999;13:362–375. doi: 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 24.Kumar M, Tyagi N, Moshal KS, Sen U, Kundu S, Mishra PK, Givvimani S, Tyagi SC. Homocysteine decreases blood flow to the brain due to vascular resistance in carotid artery. Neurochem Int. 2008;53:214–219. doi: 10.1016/j.neuint.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JE, Yoon YJ, Moseley ME, Yenari MA. Reduction in levels of matrix metalloproteinases and increased expression of tissue inhibitor of metalloproteinase-2 in response to mild hypothermia therapy in experimental stroke. J Neurosurg. 2005;103:289–297. doi: 10.3171/jns.2005.103.2.0289. [DOI] [PubMed] [Google Scholar]
- 26.Lee SR, Tsuji K, Lee SR, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi SC. Homocysteine causes cerebrovascular leakage in mice. Am J Physiol Heart Circ Physiol. 2006;290:H1206–H1213. doi: 10.1152/ajpheart.00376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 29.Manolescu BN, Oprea E, Farcasanu IC, Berteanu M, Cercasov C. Homocysteine and vitamin therapy in stroke prevention and treatment: a review. Acta Biochim Pol. 2010;57:467–477. [PubMed] [Google Scholar]
- 30.Mishra PK, Givvimani S, Metreveli N, Tyagi SC. Attenuation of beta2-adrenergic receptors and homocysteine metabolic enzymes cause diabetic cardiomyopathy. Biochem Biophys Res Commun. 2010;401:175–181. doi: 10.1016/j.bbrc.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mun-Bryce S, Rosenberg GA. Matrix metalloproteinases in cerebrovascular disease. J Cereb Blood Flow Metab. 1998;18:1163–1172. doi: 10.1097/00004647-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Nappo F, De RN, Marfella R, De LD, Ingrosso D, Perna AF, Farzati B, Giugliano D. Impairment of endothelial functions by acute hyperhomocysteinemia and reversal by antioxidant vitamins. JAMA. 1999;281:2113–2118. doi: 10.1001/jama.281.22.2113. [DOI] [PubMed] [Google Scholar]
- 33.Nishiyama T, Mae T, Kishida H, Tsukagawa M, Mimaki Y, Kuroda M, Sashida Y, Takahashi K, Kawada T, Nakagawa K, Kitahara M. Curcuminoids and sesquiterpenoids in turmeric (Curcuma longa L.) suppress an increase in blood glucose level in type 2 diabetic KK-Ay mice. J Agric Food Chem. 2005;53:959–963. doi: 10.1021/jf0483873. [DOI] [PubMed] [Google Scholar]
- 34.Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006;580:2994–3005. doi: 10.1016/j.febslet.2006.04.088. [DOI] [PubMed] [Google Scholar]
- 35.Okada K, Wangpoengtrakul C, Tanaka T, Toyokuni S, Uchida K, Osawa T. Curcumin and especially tetrahydrocurcumin ameliorate oxidative stress-induced renal injury in mice. J Nutr. 2001;131:2090–2095. doi: 10.1093/jn/131.8.2090. [DOI] [PubMed] [Google Scholar]
- 36.Pan MH, Huang TM, Lin JK. Biotransformation of curcumin through reduction and glucuronidation in mice. Drug Metab Dispos. 1999;27:486–494. [PubMed] [Google Scholar]
- 37.Pari L, Murugan P. Protective role of tetrahydrocurcumin against erythromycin estolate-induced hepatotoxicity. Pharmacol Res. 2004;49:481–486. doi: 10.1016/j.phrs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 38.Perla-Kajan J, Marczak L, Kajan L, Skowronek P, Twardowski T, Jakubowski H. Modification by homocysteine thiolactone affects redox status of cytochrome C. Biochemistry. 2007;46:6225–6231. doi: 10.1021/bi602463m. [DOI] [PubMed] [Google Scholar]
- 39.Perla-Kajan J, Stanger O, Luczak M, Ziolkowska A, Malendowicz LK, Twardowski T, Lhotak S, Austin RC, Jakubowski H. Immunohistochemical detection of N-homocysteinylated proteins in humans and mice. Biomed Pharmacother. 2008;62:473–479. doi: 10.1016/j.biopha.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Qipshidze N, Tyagi N, Sen U, Givvimani S, Metreveli N, Lominadze D, Tyagi SC. Folic acid mitigated cardiac dysfunction by normalizing the levels of tissue inhibitor of metalloproteinase and homocysteine-metabolizing enzymes postmyocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2010;299:H1484–H1493. doi: 10.1152/ajpheart.00577.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132–141. doi: 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell. 2002;1:11–21. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 44.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 45.Selhub J. Homocysteine metabolism. Annu Rev Nutr. 1999;19:217–246. doi: 10.1146/annurev.nutr.19.1.217. [DOI] [PubMed] [Google Scholar]
- 46.Sen U, Herrmann M, Herrmann W, Tyagi SC. Synergism between AT1 receptor and hyperhomocysteinemia during vascular remodeling. Clin Chem Lab Med. 2007;45:1771–1776. doi: 10.1515/CCLM.2007.354. [DOI] [PubMed] [Google Scholar]
- 47.Strickland S, Gualandris A, Rogove AD, Tsirka SE. Extracellular proteases in neuronal function and degeneration. Cold Spring Harb Symp Quant Biol. 1996;61:739–745. [PubMed] [Google Scholar]
- 48.Tejima E, Zhao BQ, Tsuji K, Rosell A, van LK, Gonzalez RG, Montaner J, Wang X, Lo EH. Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J Cereb Blood Flow Metab. 2007;27:460–468. doi: 10.1038/sj.jcbfm.9600354. [DOI] [PubMed] [Google Scholar]
- 49.Thiyagarajan M, Sharma SS. Neuroprotective effect of curcumin in middle cerebral artery occlusion induced focal cerebral ischemia in rats. Life Sci. 2004;74:969–985. doi: 10.1016/j.lfs.2003.06.042. [DOI] [PubMed] [Google Scholar]
- 50.Tyagi N, Givvimani S, Qipshidze N, Kundu S, Kapoor S, Vacek JC, Tyagi SC. Hydrogen sulfide mitigates matrix metalloproteinase-9 activity and neurovascular permeability in hyperhomocysteinemic mice. Neurochem Int. 2010;56:301–307. doi: 10.1016/j.neuint.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tyagi N, Moshal KS, Ovechkin AV, Rodriguez W, Steed M, Henderson B, Roberts AM, Joshua IG, Tyagi SC. Mitochondrial mechanism of oxidative stress and systemic hypertension in hyperhomocysteinemia. J Cell Biochem. 2005a;96:665–671. doi: 10.1002/jcb.20578. [DOI] [PubMed] [Google Scholar]
- 52.Tyagi N, Ovechkin AV, Lominadze D, Moshal KS, Tyagi SC. Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. J Cell Biochem. 2006;98:1150–1162. doi: 10.1002/jcb.20837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol. 2005b;289:H2649–H2656. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- 54.Utepbergenov DI, Mertsch K, Sporbert A, Tenz K, Paul M, Haseloff RF, Blasig IE. Nitric oxide protects blood-brain barrier in vitro from hypoxia/reoxygenation-mediated injury. FEBS Lett. 1998;424:197–201. doi: 10.1016/s0014-5793(98)00173-2. [DOI] [PubMed] [Google Scholar]
- 55.Uyama O, Okamura N, Yanase M, Narita M, Kawabata K, Sugita M. Quantitative evaluation of vascular permeability in the gerbil brain after transient ischemia using Evans blue fluorescence. J Cereb Blood Flow Metab. 1988;8:282–284. doi: 10.1038/jcbfm.1988.59. [DOI] [PubMed] [Google Scholar]
- 56.Veltkamp R, Siebing DA, Sun L, Heiland S, Bieber K, Marti HH, Nagel S, Schwab S, Schwaninger M. Hyperbaric oxygen reduces blood-brain barrier damage and edema after transient focal cerebral ischemia. Stroke. 2005;36:1679–1683. doi: 10.1161/01.STR.0000173408.94728.79. [DOI] [PubMed] [Google Scholar]
- 57.Ventruti A, Cuervo AM. Autophagy and neurodegeneration. Curr Neurol Neurosci Rep. 2007;7:443–451. doi: 10.1007/s11910-007-0068-5. [DOI] [PubMed] [Google Scholar]
- 58.Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005;128:1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci. 2000;20:7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xue M, Hollenberg MD, Yong VW. Combination of thrombin and matrix metalloproteinase-9 exacerbates neurotoxicity in cell culture and intracerebral hemorrhage in mice. J Neurosci. 2006;26:10281–10291. doi: 10.1523/JNEUROSCI.2806-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yodkeeree S, Chaiwangyen W, Garbisa S, Limtrakul P. Curcumin, demethoxycurcumin and bisdemethoxycurcumin differentially inhibit cancer cell invasion through the down-regulation of MMPs and uPA. J Nutr Biochem. 2009;20:87–95. doi: 10.1016/j.jnutbio.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 63.Yodkeeree S, Garbisa S, Limtrakul P. Tetrahydrocurcumin inhibits HT1080 cell migration and invasion via downregulation of MMPs and uPA. Acta Pharmacol Sin. 2008;29:853–860. doi: 10.1111/j.1745-7254.2008.00792.x. [DOI] [PubMed] [Google Scholar]
- 64.Zhao BQ, Tejima E, Lo EH. Neurovascular proteases in brain injury, hemorrhage and remodeling after stroke. Stroke. 2007;38:748–752. doi: 10.1161/01.STR.0000253500.32979.d1. [DOI] [PubMed] [Google Scholar]
- 65.Zhao J, Zhu W, Liu T, Yang J, Li G. Electrochemical probing into cytochrome c modification with homocysteine-thiolactone. Anal Bioanal Chem. 2010;397:695–701. doi: 10.1007/s00216-010-3553-7. [DOI] [PubMed] [Google Scholar]