

Abstract
Current models of necrotizing enterocolitis (NEC) propose intraluminal microbes destroy intestinal mucosa and activate an inflammatory cascade that ends in necrosis. We suggest an alternate hypothesis wherein NEC is caused by injury to Paneth cells (PCs) in the intestinal crypts. PCs are specialized epithelia that protect intestinal stem cells from pathogens, stimulate stem cell differentiation, shape the intestinal microbiota, and assist in repairing the gut. Our novel model of NEC uses neonatal mice and ablates Paneth cells followed by enteral infection. We contrast this model with other animal examples of NEC and the clinical disease. Selective destruction of PCs using dithizone likely releases TNF-alpha and other inflammatory mediators. We propose this event produces inflammation in the submucosa, generates platelet activating factor, and induces a coagulopathy. The role of PCs in NEC is consistent with the onset of disease in preterm infants after a period of PC-related maturation, the central role of PCs in crypt-related homeostasis, the anatomic location of pneumatosis intestinalis close to the crypts, and the proximity of Paneth cells to occluded blood vessels that cause coagulation necrosis of the intestinal villi. We offer this hypothesis to promote new thought about how NEC occurs and its potential prevention.
Keywords: dithizone, alpha-defensins, enteric infection, ileal necrosis, intestinal crypts, Paneth cell ablation, Paneth cell-related gut injury and repair, tumor necrosis factor-alpha
Introduction
Over the past four decades, necrotizing enterocolitis (NEC) has been the most highly investigated, widely debated, and clinically devastating gastrointestinal disease of preterm infants [1, 2]. Epidemiologic associations have identified: a) preterm birth, b) enteral feedings, c) dysfunctional intestinal motility, d) reduced intestinal blood flow, e) microbe-induced, pro-inflammatory epithelial signaling, and f) enteroinvasive microbes as factors in the pathogenesis of NEC [3–7]. Among these connections, significant inflammation in response to the intestinal microbiome is a vital contributor to the onset of NEC. Genetic predispositions to NEC have been described infrequently and require more research [8, 9].
NEC causes: a) increased hospital-related costs and heightened expenditures after discharge [10, 11], b) adverse neurodevelopmental outcomes [12, 13], c) long-term neurologic and intestinal rehabilitative needs [11–14], and d) a mortality that can range from 15 to 30% [2, 6]. Vital statistics in the United States [15, 16] show that infant mortality caused by NEC increased between 2000 and 2010 [table 1]. On the other hand, infant mortality associated with early-onset neonatal sepsis and respiratory distress syndrome significantly declined over the same period. Because of high morbidity, mortality and health care costs, prevention of NEC is considered urgent [4]. Only nutrition with human milk reduces the prevalence of NEC [4], while oral probiotic prophylaxis requires more research and evaluation before its routine clinical use [2].
Table 1.
| Neonatal Disease | Necrotizing Enterocolitis | Early-onset Neonatal Sepsis | Respiratory Distress Syndrome |
|---|---|---|---|
| Year – 2000 | 348 deaths and 8.7 per 100,000 births | 734 deaths and 18.3 per 100,000 births | 958 deaths and 23.8 per 100,000 births |
| Year – 2010 | 470 deaths and 11.7 per 100,000 births | 596 deaths and 14.2 per 100,000 births | 496 deaths and 12.4 per 100,000 births |
A decisive understanding of the pathogenesis of NEC is essential to developing effective prevention and treatment approaches. Many reviews appreciate the multifactorial nature of the disorder, yet recent overviews of NEC clearly state its pathophysiology is incompletely understood [2, 5]. Recent historic [17] and scientific [18] perspectives express frustration with our comprehension of this common intestinal disease of neonates.
The Traditional Hypothesis Related to the Pathogenesis of NEC
The principal hypothesis linked to the pathogenesis of NEC can be described as a “top down scenario” [2, 3, 5, 19]. Published flow diagrams or illustrations show intra-luminal bacteria disrupting and invading epithelia at the tips of intestinal villi (figure 1). After endotoxin binds to Toll-like receptor 4 (TLR4) on enterocytes and/or following activation of other pathogen-associated molecular pattern (PAMP) receptors on intestinal epithelia, the gut barrier appears to fail and bacterial translocation takes place. An intense inflammatory response ensues in the lamina propria and is mediated by tumor necrosis factor-alpha (TNF-α) and interleukin-1beta (IL-1β) [20]. These events also stimulate the release of vasoactive substances that can initiate either potent vasoconstriction or excessive vasodilatation in the intestine. Vasoactive substances associated with NEC include platelet activating factor (PAF), endothelin-1 (ET-1), and the nitric oxide (NO)/peroxynitrite (OONO−) couple [3, 7]. Inflammation in the intestinal wall also triggers activation of the complement and coagulation systems [3]. Stimulated leukocytes and platelets adhere to endothelium causing a relative anatomical obstruction to blood flow in the microvasculature of the small bowel. Additional damage to endothelia caused by adherence of neutrophils and platelets additionally impairs generation of NO and thereby hinders vasorelaxation in the presence of vasospasm. During the pathogenesis of NEC, a life-threatening element is vascular thrombosis in the intestinal submucosa and this event produces so called coagulation necrosis of the intestinal villi [21–23]. This ‘top down’ scheme for the pathogenesis of NEC involves a logical cascade of events; however, the validity of this pattern that ends in NEC can be questioned. In the next section, we will emphasize why pneumatosis intestinalis and thrombosis of submucosal vessels may arise from pathophysiology in the crypts of Lieberkühn compared to far away inflammation in the tips of villi as a response to events in the intestinal lumen.
Fig 1. Traditional “Top Down” Hypothesis of Necrotizing Enterocolitis.

Intraluminal bacterial dysbiosis causes activation of epithelial cell pathogen-associated molecular pattern receptors, especially Toll-like Receptor 4, and may induce either apoptosis (identified as small nucleated cells) or necrosis (identified as ghost cells). Bacterial invasion of epithelia can also initiate anoikis (apoptosis and cell detachment from the basement membrane – cells with bacteria and micronuclei). Failure of the epithelial barrier allows bacterial translocation and results in a vigorous inflammatory response and causes coagulation necrosis, a constant submucosal finding in necrotizing enterocolitis (NEC). The invading bacteria also gain access to the submucosa, where they ferment sugar and make gas-filled bullae called pneumatosis intestinalis. Please note there is no mention of Paneth cells in this conceptual pathophysiology for NEC.
A “Bottom Up” Hypothesis to Explain the Onset of NEC
The following rationale contests the traditional beliefs about the origins of NEC. First, the distance from the apex of the villus wherein bacterial invasion is proposed to occur to the arteriolar plexus in the submucosa where thrombosis begins is substantial [22]. The close proximity of the crypts of Lieberkühn to the lamina propria and submucosal arterioles suggests that bacterial translocation through the crypts is a more plausible reason for an inflammatory response in the submucosa compared to pathogens entering the body at the tips of villi. This pathway would rapidly activate the coagulation system in major blood vessels penetrating the bowel wall from the mesentery. Based on the anatomic pathology, it is reasonable to postulate that bacterial toxins act within the crypts and bacterial invasion occurs after epithelial damage takes place. Such a ‘bottom up’ pathogenesis for NEC may better explain rapid occlusion of perforating arteries in the submucosa which in turn causes coagulation necrosis of the intestinal villi [21–25]. Intense inflammation in the lamina propria below the crypts can also initiate a coagulopathy in submucosal vessels that proliferates along extended segments of small bowel and renders the tissues gangrenous. This phenomenon may well be the mechanism whereby necrotizing enterocolitis totalis is found during either surgery or a post-mortem examination.
Second, pneumatosis intestinalis, the pathognomonic sign for NEC, is not identified in intestinal villi of infants or animal models with NEC. Rather pneumatosis intestinalis occurs in the submucosa of the bowel where bullae are identified by gross anatomic, low power microscopic or abdominal ultrasound examinations [21–25]. Crypt penetration provides microbes with easy access to submucosal tissues where intramural gas production by bacteria [26, 27] causes pneumatosis. From this location, rather than the villi, pneumatosis can track along splanchnic vessels to the liver and is manifest by portal venous air.
The ‘bottom up’ hypothesis states NEC begins in the crypts of Lieberkühn. Our hypothesis originated from investigations of Paneth cells (PCs) in the immature intestine of rodents. When neonatal rats or mice had PCs destroyed by dithizone [28] followed by gut infection with enteric pathogens [29, 30], the ileum became grossly necrotic in a significant number of newborn animals. The anatomic appearance of the ileal necrosis was consistent with NEC observed in human preterm infants. This finding fostered our rethinking of events related to the pathogenesis of NEC. The rest of this review a) discusses Paneth cells in health and disease, b) describes the role of PCs in intestinal development, and c) details the mechanisms whereby we propose these specialized epithelia mediate the onset of NEC.
Paneth Cells in Human Health and Disease
Paneth cells were described in the crypts of Lieberkühn in 1867 [31], yet it took another 125 years before interest in these cells intensified. In 1992, PCs of mice [32] and man [33] were shown to contain antimicrobial peptides (AMPs). In response to lipoteichoic acid, lipopolysaccharide, Gram-negative, Gram-positive bacteria, and other agonists, murine PCs secrete phospholipase A2 and cryptdins (known as alpha-defensins in man) [34–36]. Transgenic mice expressing the human defensin 5 gene (HD-5) are protected from death during enteral infection with Salmonella typhimurium [37]. By expressing PAMP receptors on their luminal surface to recognize microbes, and by releasing AMPs after these receptors are engaged, Paneth cells are a major facilitator of homeostasis in the gut-associated microbiome [38, 39]. For these reasons, PCs have been properly called ‘guardians’ of intestinal stem cells in the niche called the crypts of Lieberkühn where these cells live and work in cooperation [40].
It is appreciated that PCs have functions beyond innate immunity in the intestine. Figure 2 A displays the intimate relationship between arterioles entering and venules leaving individual villi and the nearby Paneth cells [41]. The anatomic location of PCs is within one to several cell diameters of these vessels and allows these specialized epithelia to regulate postnatal vascular growth by secreting angiogenins through an enteric bacteria-sensing mechanism [42]. Interestingly, angiogenins also have antimicrobial actions [43]. Figure 2B depicts how PC-related control of the intestinal microbiota is fundamental to orderly renewal of epithelia on the surface of villi [44]. PCs are a key constituent of the mammalian intestinal stem-cell niche, and they augment stem-cell function in response to calorie restriction [45]. PCs express epidermal growth factor, transforming growth factor-α, Wnt3, and the Notch ligand Dll4, all essential signals for stem-cell maintenance in culture. Genetic removal of PCs in vivo results in the concomitant loss of Lgr5 stem cells [46]. These observations emphasize the importance of PCs in crypt-related homeostasis and epithelial renewal in the villi. When PCs undergo destruction after dithizone exposure, they may release TNF-α [47]. This constituent pro-inflammatory cytokine of PCs is responsible for activation of nuclear factor-kappaB in other intestinal epithelia, an event associated with a subsequent proliferative effect in these cells [47].
Figure 2. Paneth Cells (PCs) – Normal Anatomy and Function.

In addition to their antimicrobial functions, Paneth cells play a vital role regulating the intestinal architecture. A) PCs are located near the vasculature entering and leaving the villi and they regulate blood vessel growth through the secretion of angiogenins and B) PCs are found intertwined with intestinal stem cells in the crypts. PCs are critical to maintaining stem cell homeostasis through Wnt signaling pathways. Cooperation between PCs and stem cells allows orderly regeneration of intestinal surface epithelia on a continuous basis.
Crohn’s disease or regional enteritis is associated with failure of the antimicrobial functions of PCs in children and adults. Mechanisms responsible for defective antimicrobial function in PCs are complex and include: a) mutations in the expression of α-defensins or their intracellular inactivation, b) mutations in nucleotide-binding oligomerization domain 2 (NOD2) and loss of function, c) a disturbance of the Wnt pathway transcription factor TCF7L2 (also known as TCF4), d) defects in the autophagy factor ATG16L1 that destroys invading intracellular bacteria, e) alterations in the endosomal stress protein XBP1, f) loss of Toll-like receptor 9 (TLR9)-related function, and g) abnormalities in the calcium-mediated potassium channel KCNN4 [48]. While these disturbances in PC-associated functions in Crohn’s disease have not been identified to date in patients with NEC, Crohn’s disease and necrotizing enterocolitis do share some pathogenic features. Chronic inflammation in Crohn’s disease and acute inflammation in NEC are significantly related to tumor necrosis factor-alpha (TNF-α) secretion by PCs [49–52]. Secretion of TNF-α may be heightened when PCs are stressed by bacterial overload, virulence, their toxins and/or other microbial components in the intestinal crypts [53]. The next section will discuss this pathophysiology in relationship to NEC.
Paneth Cells and Necrotizing Enterocolitis: A Final Pathway to Disease Onset
The bottom up hypothesis related to the pathogenesis of NEC emerged when we were studying the role of PCs in bacterial translocation from the neonatal intestine. The zinc chelator dithizone selectively alters or kills Paneth cells in the small bowel [28, 29, 47] and was used to diminish the host defense functions of these specialized epithelia in neonatal rats. Six hours after administering dithizone, the newborn animals underwent a significant enteral infection with Escherichia coli [29]. Jejunal and ileal fluid and the intestinal wall of jejunum had significantly increased numbers of E. coli, while the wall of ileum approached a statistically noteworthy result (P = 0.06). In delineating why the ileum did not have higher numbers of E. coli, we noted that 8 of 32 ileal samples (25%) had transmural necrosis. Two hypotheses flowed from this pathologic finding and they are the topics of this review. First, in killing PCs with dithizone, we propose substantial amounts of TNF-α may have been released [47] and could explain death of the ileum. Alternatively, in the presence of a significant infection with Escherichia coli in the intestinal lumen, necrosis of the ileum may be an attempt by the host to limit translocation and systemic bacteremia. The latter concept is consistent with reports that bacteremia is lower than expected during necrotizing enterocolitis [54].
Confirmation that Paneth cells may be a central figure in the pathogenesis of NEC came from a novel model of necrotizing enterocolitis in neonatal mice [30]. This model used 14 to 16-day-old mice that were treated with dithizone [75 mg/kg] and 6 hours later infected enterally by instilling 1 x 1011 colony-forming units per kg body weight of Klebsiella pneumoniae into the stomach. Ten hours after infection, the neonatal mice treated with dithizone or control conditions had gross and microscopic examination of the bowel. Only those mice given dithizone followed by enteral infection had significant evidence of transmural necrosis in the ileum (figure 4). Dithizone treatment was shown to deplete the Paneth cell population by fifty percent. Dithizone or enteral infection alone was unable to significantly initiate the onset of NEC in immature mice. Rather, dithizone treatment followed by enteral infection with a pathogen was required to alter the presence and physiology of Paneth cells in the small bowel [29].
Fig 4. Selective ablation of Paneth cells followed by enteric infection with Klebsiella induces a NEC-like injury in 14- to 16-day-old mice.
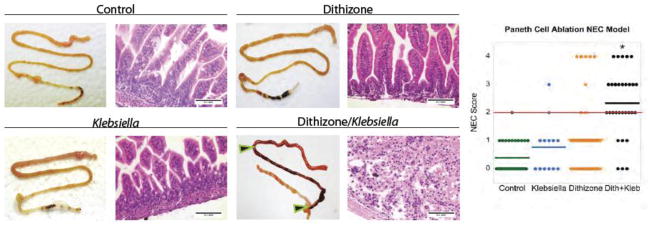
P14 – P16 CD-1 mice were divided into four groups (control n = 31, dithizone only n = 42, Klebsiella only n = 13 and dithizone/Klebsiella {designated Dith/Kleb}, n = 30). Mice were given either an intraperitoneal of dithizone (75 mg/kg) or an equivalent volume of Li2CO3 diluent. At 6 hours after the injection, mice were given either a gavage feeding of 1 x 1011 CFU Klebsiella pneumoniae per kg body weight suspended in nutrient broth or nutrient broth only. Mice were monitored for 10 hours following gavage and then underwent euthanasia for harvesting of the small intestine. A blinded investigator applied a Lickert system that evaluated villus integrity on a scale of 0 to 4. Mice treated with Dith/Kleb were significantly different from the other groups and their average score was >2 (see red line on scoring graphic) and was considered significant for NEC-like damage (* P<0.001 vs. other groups). The loss of intestinal villi on histologic analyses of the Dith/Kleb-treated pups is remarkable compared to the control groups. Horizontal lines for each group indicate the mean histologic score. In the Dith/Kleb group, the terminal ileum showed transmural necrosis and is displayed between the black and red arrows; intestinal damage is also seen proximally. Internal size scale: bars are 0.1 mm. Permission granted for use of this illustration from Diseases Models and Mechanisms.
Extremely-low-birth-weight human infants generally have the longest postnatal interval before the onset of NEC [55, 56]. This dependable observation suggests that maturation of innate and/or adaptive immunity must occur before classic NEC becomes possible. Examining the development of PCs in the intestinal crypts offers insight into this delay in appearance of NEC after birth. In humans, PCs appear early in the second trimester but have markedly diminished expression of HD-5 and HD-6 until the middle of the third trimester [57]. Term infants have PCs with an increased HD-5 and HD-6 content compared to premature infants, although the concentrations do not approach levels seen in human adults [57]. In mice and rats, PCs are sparse at birth and their AMP content is low, but after birth there are logarithmic increases in expression of cryptdins, lysozyme, and secretory phospholipase A2 as pups wean from suckling [58]. In the dithizone-Klebsiella model, younger mice do not yet have functional PCs, and they do not develop NEC [30]. We propose that in immature prematurely-born human infants, a period of time is required for PCs to mature. Paneth cells may be enabled to initiate NEC after they become full-fledged pro-inflammatory and host defense cells during postnatal life [51, 57].
PCs have a 3-fold amplification of defensin expression in surgical specimens from neonates with NEC compared to controls [59]. Other studies found the lysozyme content of PCs is virtually absent in surgical tissues recovered from cases with NEC [60, 61]. Two hypotheses are proposed to explain these findings [62]. One theory says the initial paucity of AMPs in PCs creates an abnormal microflora in the intestine of preterm infants. This pathologic microbiota invades intestinal epithelium. The second model theorizes bacterial dysbiosis in the gut enhances the expression of defensins in PCs around the time that NEC has its onset. If defensins are sufficient in PCs, this finding suggests other events occur in the final pathway. These two disparate hypotheses related to the content of AMPs in neonatal PCs and the onset of NEC are better understood if α-defensins are simply a surrogate for other PC-associated molecules that are central to the pathogenesis of NEC. We suggest those molecules in PCs are pro-inflammatory cytokines with the major effector being TNF-α [51, 52]. Release of TNF-α from PCs may occur when these epithelia are either disrupted by bacterial toxins or experience microbial invasion and death [63]. Degradation of PCs is also consistent with the reduced numbers of PCs found in surgical samples from NEC cases [30, 61]. The recent description that HD6 secreted from PCs forms nanonets with an enteric pathogen and hinders invasion suggests trapping microbes and their toxins in the crypts may be an important defense mechanism (64). The effectiveness or side effects related to this newly reported host defense in the small intestine deserves immediate study in neonates. Its relationship to the pathogenesis of NEC must be defined.
We propose that PCs are the key cellular mediator related to the onset of NEC because a) PCs can sense microbial invasion via several surface and intracellular PAMP receptors and b) PCs can initiate a local and a systemic inflammatory response because they contain appropriate cytokines and are next to arterioles and venules that perfuse and drain the intestinal villi (figures 2 and 3). This scenario is in harmony with the suggestion that PCs are akin to stationary leukocytes [49]. Paneth cells are known to store and secrete TNF-α and interleukin-17 (IL-17) from their basolateral surface when PAMP receptors are stimulated or intracellular bacterial invasion occurs [49, 65]. Combined release of TNF-α and IL-17 from PCs can initiate shock [65]. Once intense inflammation begins in blood vessels entering and leaving the intestinal villi, the adherence of leukocytes and platelets to endothelium would significantly elevate platelet activating factor (PAF), endothelin-1 (ET-1), and other vasoactive agents in the microvasculature of bowel [3]. Damage to endothelium of the villus reduces the production of nitric oxide and accentuates a low flow state in the intestinal microvasculature. In concert with the inflammatory milieu and vasoactive events, the intravascular complement and coagulation systems are activated in the intestinal blood vessels. PCs at the base of crypts are adjacent to the arteries and veins in the lamina propria (41), and this supports the bottom up hypothesis where thrombosis of these vessels may cause coagulation necrosis of the intestinal villi.
Fig 3. Paneth Cells -- The “Bottom Up” Hypothesis of Necrotizing Enterocolitis.

In this figure, the sequence of events associated with the ‘Bottom Up’ hypothesis is identified by bold numbers. ➊ Microbial toxins and invasion cause disruption of Paneth cells (PCs). On the luminal surface of PCs, lipopolysaccharide (LPS) receptors CD-14 and Toll-like receptor-4 (TLR-4) are identified. Additionally, the TLR-5 receptor that binds flagellin and TLR-9 receptor that binds CpG are shown. ➋ Disruption of PCs releases their granules into the crypt space, while cytokines likely undergo basolateral discharge into the lamina propria and its adjacent microvasculature. ➌ Released cytokines (tumor necrosis factor-alpha, TNF-α; interleukin-17, IL-17; interleukin-1 beta, IL-1β) and other mediators (phospholipase A2, PLA2) activate inflammation and injure endothelium. ➍ Altered endothelia enhance neutrophil and platelet attachment to their surface. ➎ Intravascular inflammation initiates fibrin aggregation to endothelium, adherent platelets and adherent neutrophils. ➏ A disseminated intravascular coagulopathy ensues with damaging effects to erythrocytes. ➐ Endothelial damage is magnified and markedly decreases production of nitric oxide (NO) which additionally hinders perfusion of the intestinal villi. ➑ This phenomenon also generates platelet activating factor (PAF) and other mediators that cause downstream vasoconstriction in the intestinal villi. ➒ The low flow state and intravascular coagulopathy extends to the submucosal vessels and thrombotic occlusion takes place. This vascular obstruction causes coagulation necrosis of the intestinal mucosa, a common histologic finding in necrotizing enterocolitis (NEC). ➓ Finally, microbes that have invaded the disrupted crypts, move to the submucosa, ferment sugars and produce gas, and thus are the origins of pneumatosis intestinalis, the pathognomonic sign of NEC.
In the neonatal rat [29] and mouse [30] models that use dithizone treatment followed by enteral infection to create a necrotizing enterocolitis-like illness, the distal ileum is the principal anatomic location with complete intestinal necrosis. The predilection to the ileum is an interesting one and two hypotheses predominate. The first is that functional differences between the jejunum, ileum, and colon predispose the ileum to NEC (i.e., the main difference is in the host), while the second is that regional differences in the microbiota are of primary importance [66]. In support of the first hypothesis, Paneth cells may be more numerous in the ileum compared to jejunum of neonates, but the definitive comparative studies at each location have not been performed. Quantitative stereologic methods to define the numbers of PCs in neonatal jejunum and ileum exist, but have not been reported to date. Moreover, data from mice suggest there is increased expression of many Paneth cell products in the ileum compared to the jejunum [67]. To investigate the second hypothesis, careful metagenomic studies of intestinal loop models will be necessary to determine regional differences in the microbiota. Changes in the fecal microbiota appear to precede the onset of NEC [68], although how well the fecal microbiota reflects inflammatory changes in the small bowel has yet to be proven. It is also not clear that changes in the intestinal microbiome have a cause and effect relationship. Another research question to be explored is whether incompetency of the ileo-cecal valve with reflux of colonic bacteria into the ileum plays a role in the pathogenesis of neonatal NEC. Colonic imaging studies in premature infants suggest that the ileocecal valve is reasonably competent despite immaturity; however, the impact of inflammation on ileocecal valve competence in premature infants is unknown.
Clinical Evidence Supporting a Central Role for Paneth Cells in NEC
There is other evidence that PCs are involved in the pathogenesis of NEC. Antenatal corticosteroids reduce the incidence of respiratory distress syndrome in preterm infants, but they also lower the frequency of necrotizing enterocolitis [69]. This effect corresponds with accelerated maturation of Paneth cells after immature opossums are given hydrocortisone [70].
Epidermal growth factor (EGF) is present in human milk and reduces the incidence of NEC in the neonatal rat model of disease [4]. Autophagy is activated in the intestinal epithelium of NEC patients and in the ileum of rats with NEC. Enteral administration of EGF blocks intestinal autophagy both in vivo and in vitro [71]. Thus, EGF-mediated protection from NEC-related injury is associated with regulation of intestinal autophagy. Interestingly, EGF is localized to Paneth cells in neonatal rats [72]. These concurrences become more compelling because autophagy is associated with the pathogenesis of Crohn’s disease [73]. The concept of disordered autophagy in Paneth cells and the pathogenesis of NEC require future basic and clinical studies.
Hospitalized preterm infants that are fed probiotic bacteria have a reduced prevalence of NEC [74, 75]. When diabetes-prone rats are fed Lactobacillus johnsonii, an increased number of Paneth cells are seen in the intestine [76]. Based on reduced numbers of PCs in the bowel of infants with NEC [30, 61], one must consider that probiotics may afford protection against the disease by enhancing the Paneth cell population. However, an effective and safe licensed supplement of probiotic bacteria that can be used enterally in preterm infants to achieve this goal is currently not available.
Lastly, intelectin is a specific lectin expressed on the surface of Paneth and goblet cells in the small intestine. Intelectin is proposed to serve a protective role in the innate immune response [77]. Bovine lactoferrin binds to human intelectin [78]. These two observations provide a possible mechanism whereby lactoferrin in breast milk binds to PCs and enhances their integrity and/or functions. Since lactoferrin binds endotoxin, its binding to the intelectin receptor may prime host defenses in epithelia and dendritic cells for Th1 responses rather than create a pro-inflammatory state [79]. This conjecture comes to life because enterally-administered bovine lactoferrin reduces late-onset sepsis, and there is a trend towards decreasing the occurrence of NEC in very preterm infants [80].
The aforementioned scientific observations are persuasive reasons why PCs may have a major role in preventing NEC through a variety of already recognized interventions. These findings will hopefully focus research on PCs and the crypts of Lieberkühn as an anatomic location wherein the final pathway to NEC originates.
Conclusions
Paneth cells utilize the secretion of defensins and other microbicidal peptides to a) kill invasive pathogens, b) shape the intestinal microbiota, and c) protect intestinal stem cells from harm. At the base of the crypts, PCs are strategically placed to secrete angiogenins during vascular development of the villi and to regenerate blood vessels after injuries to the gut. At the base of the crypts, PCs can also send danger signals to the villi and the submucosa when host defenses are failing. The mediators include cytokines and vasoactive mediators. The inflammation causes vascular obstructions that devitalize the intestine. This vascular occlusion, however, may be effective in limiting massive microbial entry from intestinal lumen into the bloodstream. Cooperation between stem cells and PCs in a niche localized to the crypts is essential for normal renewal of epithelia needed to cover the intestinal surface. Moreover, after the intestine is seriously damaged by infection or other insults, PCs play an equally key role in initiating and maintaining healing.
Given the anatomic location of Paneth cells, one can reasonably envision how destruction of PCs leads to bacterial invasion, intense inflammation, pneumatosis intestinalis, and vascular occlusion in the submucosa. These events are the final pathway to necrotizing enterocolitis. Based on these arguments, we suggest that the intestinal crypt rather than the apex of the villus holds the key to understanding NEC. We hope that future studies aimed at elucidating the role of the Paneth cell in NEC will lead to new and important advances in the field, and in turn, help protect prematurely-born infants from this devastating disease.
The traditional hypothesis or the “top down” model of NEC has been proposed for many years (Fig. 1). This review hypothesizes an alternative pathophysiology for NEC that involves Paneth cells and the crypts of Lieberkuhn in a “bottom up” scenario (Fig. 3). Investigators should consider both theories regarding the pathogenesis of NEC as researchers and caregivers move forward to understand and prevent this disease and improve the health outcomes of preterm infants.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) K08 DK083677 to S.J.M, NIH RO1 HD059127 to M.A.U., and NIH R44 HD057744 and a Gerber Foundation grant PN002-1214-2708 to M.P.S.
Footnotes
Disclosure Statement
Drs. McElroy, Underwood and Sherman have no financial conflicts of interest to report.
References
- 1.Kliegman RM, Fanaroff AA. Necrotizing enterocolitis. N Engl J Med. 1984;310:1093–1103. doi: 10.1056/NEJM198404263101707. [DOI] [PubMed] [Google Scholar]
- 2.Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med. 2011;364:255–264. doi: 10.1056/NEJMra1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsueh W, De Plaen IG, Caplan MS, Qu XW, Tan XD, Gonzalez-Crussi F. Neonatal necrotizing enterocolitis: clinical aspects, experimental models and pathogenesis. World J Pediatr. 2007;3:17–29. [Google Scholar]
- 4.Grave GD, Nelson SA, Walker WA, Moss RL, Dvorak B, Hamilton FA, Higgins R, Raju TN. New therapies and preventive approaches for necrotizing enterocolitis: report of a research planning workshop. Pediatr Res. 2007;62:510–514. doi: 10.1203/PDR.0b013e318142580a. [DOI] [PubMed] [Google Scholar]
- 5.Afrazi A, Sodhi CP, Richardson W, Neal M, Good M, Siggers R, Hackam DJ. New insights into the pathogenesis and treatment of necrotizing enterocolitis: Toll-like receptors and beyond. Pediatr Res. 2011;69:183–188. doi: 10.1203/PDR.0b013e3182093280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman L, Moss RL. Necrotizing enterocolitis: an update. Semin Fetal Neonatal Med. 2011;16:145–150. doi: 10.1016/j.siny.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Carlisle EM, Poroyko V, Caplan MS, Alverdy JA, Liu D. Gram negative bacteria are associated with the early stages of necrotizing enterocolitis. PLoS One. 2011;6:e18084. doi: 10.1371/journal.pone.0018084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sampath V, Le M, Lane L, Patel AL, Cohen JD, Simpson PM, Garland JS, Hines RN. The NFKB1 (g-24519delATTG) variant is associated with necrotizing enterocolitis (NEC) in premature infants. J Surg Res. 2011;169:e51–e57. doi: 10.1016/j.jss.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 9.Morrow AL, Meinzen-Derr J, Huang P, Schibler KR, Cahill T, Keddache M, Kallapur SG, Newburg DS, Tabangin M, Warner BB, Jiang X. Fucosyltransferase 2 non-secretor and low secretor status predicts severe outcomes in premature infants. J Pediatr. 2011;158:745–751. doi: 10.1016/j.jpeds.2010.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bisquera JA, Cooper TR, Berseth CL. Impact of necrotizing enterocolitis on length of stay and hospital charges in very low birth weight infants. Pediatrics. 2002;109:423–428. doi: 10.1542/peds.109.3.423. [DOI] [PubMed] [Google Scholar]
- 11.Spencer AU, Kovacevich D, McKinney-Barnett M, Hair D, Canham J, Maksym C, Teitelbaum DH. Pediatric short-bowel syndrome: the cost of comprehensive care. Am J Clin Nutr. 2008;88:1552–1559. doi: 10.3945/ajcn.2008.26007. [DOI] [PubMed] [Google Scholar]
- 12.Hintz SR, Kendrick DE, Stoll BJ, Vohr BR, Fanaroff AA, Donovan EF, Poole WK, Blakely ML, Wright L, Higgins R, NICHD Neonatal Research Network Neurodevelopmental and growth outcomes of extremely low birth weight infants after necrotizing enterocolitis. Pediatrics. 2005;115:696–703. doi: 10.1542/peds.2004-0569. [DOI] [PubMed] [Google Scholar]
- 13.Martin CR, Dammann O, Allred EN, Patel S, O'Shea TM, Kuban KC, Leviton A. Neurodevelopment of extremely preterm infants who had necrotizing enterocolitis with or without late bacteremia. J Pediatr. 2010;157:751–756.e1. doi: 10.1016/j.jpeds.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torres C, Sudan D, Vanderhoof J, Grant W, Botha J, Raynor S, Langnas A. Role of an intestinal rehabilitation program in the treatment of advanced intestinal failure. J Pediatr Gastroenterol Nutr. 2007;45:204–212. doi: 10.1097/MPG.0b013e31805905f9. [DOI] [PubMed] [Google Scholar]
- 15.Kochanek KD, Smith BL. Deaths: Preliminary Data for 2002. National Vital Statistics Reports. 2002;52:1–47. [PubMed] [Google Scholar]
- 16.Murphy SL, Xu J, Kochanek KD. Deaths: Preliminary Data for 2010. National Vital Statistics Reports. 2012;60:1–68. [PubMed] [Google Scholar]
- 17.Obladen M. Necrotizing enterocolitis -- 150 years of fruitless search for the cause. Neonatology. 2009;96:203–210. doi: 10.1159/000215590. [DOI] [PubMed] [Google Scholar]
- 18.Schnabl KL, Van Aerde JE, Thomson AB, Clandinin MT. Necrotizing enterocolitis: a multifactorial disease with no cure. World J Gastroenterol. 2008;14:2142–2161. doi: 10.3748/wjg.14.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emami CN, Petrosyan M, Giuliani S, Williams M, Hunter C, Prasadarao NV, Ford HR. Role of the host defense system and intestinal microbial flora in the pathogenesis of necrotizing enterocolitis. Surg Infect (Larchmt) 2009;10:407–417. doi: 10.1089/sur.2009.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viscardi RM, Lyon NH, Sun CC, Hebel JR, Hasday JD. Inflammatory cytokine mRNAs in surgical specimens of necrotizing enterocolitis and normal newborn intestine. Pediatr Pathol Lab Med. 1997;17:547–559. [PubMed] [Google Scholar]
- 21.Sántulli TV, Schullinger JN, Heird WC, Gongaware RD, Wigger J, Barlow B, Blanc WA, Berdon WE. Acute necrotizing enterocolitis in infancy: a review of 64 cases. Pediatrics. 1975;55:376–387. [PubMed] [Google Scholar]
- 22.Nowicki PT. Ischemia and necrotizing enterocolitis: where, when, and how. Semin Pediatr Surg. 2005;14:152–158. doi: 10.1053/j.sempedsurg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Nankervis CA, Giannone PJ, Reber KM. The neonatal intestinal vasculature: contributing factors to necrotizing enterocolitis. Semin Perinatol. 2008;32:83–91. doi: 10.1053/j.semperi.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Ballance WA, Dahms BB, Shenker N, Kliegman RM. Pathology of neonatal necrotizing enterocolitis: a ten-year experience. J Pediatr. 1990;117(1 Pt 2):S6–S13. doi: 10.1016/s0022-3476(05)81124-2. [DOI] [PubMed] [Google Scholar]
- 25.Epelman M, Daneman A, Navarro OM, Morag I, Moore AM, Kim JH, Faingold R, Taylor G, Gerstle JT. Necrotizing enterocolitis: review of state-of-the-art imaging findings with pathologic correlation. Radiographics. 2007;27:285–305. doi: 10.1148/rg.272055098. [DOI] [PubMed] [Google Scholar]
- 26.Hill HR, Hunt CE, Matsen JM. Nosocomial colonization with Klebsiella, type 26, in a neonatal intensive-care unit associated with an outbreak of sepsis, meningitis, and necrotizing enterocolitis. J Pediatr. 1974;85:415–419. doi: 10.1016/s0022-3476(74)80133-2. [DOI] [PubMed] [Google Scholar]
- 27.Smith B, Bodé S, Petersen BL, Jensen TK, Pipper C, Kloppenborg J, Boyé M, Krogfelt KA, Mølbak L. Community analysis of bacteria colonizing intestinal tissue of neonates with necrotizing enterocolitis. BMC Microbiol. 2011;11:73. doi: 10.1186/1471-2180-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sawada M, Takahashi K, Sawada S, Midorikawa O. Selective killing of Paneth cells by intravenous administration of dithizone in rats. Int J Exp Pathol. 1991;72:407–421. [PMC free article] [PubMed] [Google Scholar]
- 29.Sherman MP, Bennett SH, Hwang FF, Sherman J, Bevins CL. Paneth cells and antibacterial host defense in neonatal small intestine. Infect Immun. 2005;73:6143–6146. doi: 10.1128/IAI.73.9.6143-6146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C, Sherman MP, Prince LS, Bader D, Weitkamp JH, Slaughter JC, McElroy SJ. Paneth cell ablation in the presence of Klebsiella pneumoniae induces necrotizing enterocolitis (NEC)-like injury in the small intestine of immature mice. Dis Models Mechanisms. 2012;5:1–11. doi: 10.1242/dmm.009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ouellette AJ, Niklas V. Paneth cells in intestinal health and disease in the newborn. NeoReviews. 2010;11:e551–e557. [Google Scholar]
- 32.Selsted ME, Miller SI, Henschen AH, Ouellette AJ. Enteric defensins: antibiotic peptide components of intestinal host defense. J Cell Biol. 1992;118:929–936. doi: 10.1083/jcb.118.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones DE, Bevins CL. Paneth cells of the human small intestine express an antimicrobial peptide gene. J Biol Chem. 1992;267:23216–23225. [PubMed] [Google Scholar]
- 34.Qu XD, Lloyd KC, Walsh JH, Lehrer RI. Secretion of type II phospholipase A2 and cryptdin by rat small intestinal Paneth cells. Infect Immun. 1996;64:5161–5165. doi: 10.1128/iai.64.12.5161-5165.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 36.Lehrer RI, Lu W. α-Defensins in human innate immunity. Immunol Rev. 2012;245:84–112. doi: 10.1111/j.1600-065X.2011.01082.x. [DOI] [PubMed] [Google Scholar]
- 37.Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. 2003;422(6931):522–526. doi: 10.1038/nature01520. [DOI] [PubMed] [Google Scholar]
- 38.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci USA. 2008;105:20858–863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjöberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganz T. Paneth cells--guardians of the gut cell hatchery. Nat Immunol. 2000;1:99–100. doi: 10.1038/77884. [DOI] [PubMed] [Google Scholar]
- 41.Behnke O, Moe H. An electron microscope study of mature and differentiating Paneth cells in the rat, especially of their endoplasmic reticulum and lysozymes. J Cell Biol. 1964;22:633–652. doi: 10.1083/jcb.22.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci USA. 2002;99:15451–5455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 44.Vereecke L, Beyaert R, van Loo G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol Med. 2011;17:584–593. doi: 10.1016/j.molmed.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Yilmaz OH, Katajisto P, Lamming DW, Gültekin Y, Bauer-Rowe KE, Sengupta S, Birsoy K, Dursun A, Yilmaz VO, Selig M, Nielsen GP, Mino-Kenudson M, Zukerberg LR, Bhan AK, Deshpande V, Sabatini DM. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012;486(7404):490–495. doi: 10.1038/nature11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seno H, Sawada M, Fukuzawa H, Morita-Fujisawa Y, Takaishi S, Hiai H, Chiba T. Involvement of tumor necrosis factor alpha in intestinal epithelial cell proliferation following Paneth cell destruction. Scand J Gastroenterol. 2002;37:154–60. doi: 10.1080/003655202753416803. [DOI] [PubMed] [Google Scholar]
- 48.Wehkamp J, Stange EF. Paneth's disease. J Crohns Colitis. 2010;4:523–531. doi: 10.1016/j.crohns.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 49.Keshav S. Paneth cells: leukocyte-like mediators of innate immunity in the intestine. J Leukoc Biol. 2006;80:500–508. doi: 10.1189/jlb.1005556. [DOI] [PubMed] [Google Scholar]
- 50.Ordás I, Feagan BG, Sandborn WJ. Early use of immunosuppressives or TNF antagonists for the treatment of Crohn's disease: time for a change. Gut. 2011;60:1754–1763. doi: 10.1136/gutjnl-2011-300934. [DOI] [PubMed] [Google Scholar]
- 51.Tan X, Hsueh W, Gonzalez-Crussi F. Cellular localization of tumor necrosis factor (TNF)-alpha transcripts in normal bowel and in necrotizing enterocolitis. TNF gene expression by Paneth cells, intestinal eosinophils, and macrophages. Am J Pathol. 1993;142:1858–1865. [PMC free article] [PubMed] [Google Scholar]
- 52.McElroy SJ, Prince LS, Weitkamp JH, Reese J, Slaughter JC, Polk DB. Tumor necrosis factor receptor 1-dependent depletion of mucus in immature small intestine: a potential role in neonatal necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G656–G666. doi: 10.1152/ajpgi.00550.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rumio C, Sommariva M, Sfondrini L, Palazzo M, Morelli D, Viganò L, De Cecco L, Tagliabue E, Balsari A. Induction of Paneth cell degranulation by orally administered Toll-like receptor ligands. J Cell Physiol. 2012;227:1107–1113. doi: 10.1002/jcp.22830. [DOI] [PubMed] [Google Scholar]
- 54.Geffers C, Baerwolff S, Schwab F, Gastmeier P. Incidence of healthcare-associated infections in high-risk neonates: results from the German surveillance system for very-low-birthweight infants. J Hosp Infect. 2008;68:214–221. doi: 10.1016/j.jhin.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 55.González-Rivera R, Culverhouse RC, Hamvas A, Tarr PI, Warner BB. The age of necrotizing enterocolitis onset: an application of Sartwell's incubation period model. J Perinatol. 2011;31:519–523. doi: 10.1038/jp.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yee WH, Soraisham AS, Shah VS, Aziz K, Yoon W, Lee SK Canadian Neonatal Network. Incidence and timing of presentation of necrotizing enterocolitis in preterm infants. Pediatrics. 2012;129:e298–304. doi: 10.1542/peds.2011-2022. [DOI] [PubMed] [Google Scholar]
- 57.Mallow EB, Harris A, Salzman N, Russell JP, DeBerardinis RJ, Ruchelli E, Bevins CL. Human enteric defensins. Gene structure and developmental expression. J Biol Chem. 1996;271:4038–4045. doi: 10.1074/jbc.271.8.4038. [DOI] [PubMed] [Google Scholar]
- 58.Underwood MA, Kananurak A, Coursodon CF, Adkins-Reick CK, Chu H, Bennett SH, Wehkamp J, Castillo PA, Leonard BC, Tancredi DJ, Sherman MP, Dvorak B, Bevins CL. Bifidobacterium bifidum in a rat model of necrotizing enterocolitis: antimicrobial peptide and protein responses. Pediatr Res. 2012;71:546–551. doi: 10.1038/pr.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salzman NH, Polin RA, Harris MC, Ruchelli E, Hebra A, Zirin-Butler S, Jawad A, Martin Porter E, Bevins CL. Enteric defensin expression in necrotizing enterocolitis. Pediatr Res. 1998;44:20–26. doi: 10.1203/00006450-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 60.Coutinho HB, da Mota HC, Coutinho VB, Robalinho TI, Furtado AF, Walker E, King G, Mahida YR, Sewell HF, Wakelin D. Absence of lysozyme (muramidase) in the intestinal Paneth cells of newborn infants with necrotising enterocolitis. J Clin Pathol. 1998;51:512–514. doi: 10.1136/jcp.51.7.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schaart MW, de Bruijn AC, Bouwman DM, de Krijger RR, van Goudoever JB, Tibboel D, Renes IB. Epithelial functions of the residual bowel after surgery for necrotising enterocolitis in human infants. J Pediatr Gastroenterol Nutr. 2009;49:31–41. doi: 10.1097/MPG.0b013e318186d341. [DOI] [PubMed] [Google Scholar]
- 62.Underwood MA, Bevins CL. Defensin-barbed innate immunity: clinical associations in the pediatric population. Pediatrics. 2010;125:1237–1247. doi: 10.1542/peds.2009-3289. [DOI] [PubMed] [Google Scholar]
- 63.Günther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2012 Jun 27; doi: 10.1136/gutjnl-2011-301364. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 64.Chu H, Pazgier M, Jung G, Nuccio SP, Castillo PA, de Jong MF, Winter MG, Winter SE, Wehkamp J, Shen B, Salzman NH, Underwood MA, Tsolis RM, Young GM, Lu W, Lehrer RI, Bäumler AJ, Bevins CL. Human α-Defensin 6 Promotes Mucosal Innate Immunity Through Self-Assembled Peptide Nanonets. Science. 2012;337(6093):477–481. doi: 10.1126/science.1218831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takahashi N, Vanlaere I, de Rycke R, Cauwels A, Joosten LA, Lubberts E, van den Berg WB, Libert C. IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med. 2008;205:1755–1761. doi: 10.1084/jem.20080588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang T, Desimone RA, Jiao X, Rohlf FJ, Zhu W, Gong QQ, Hunt SR, Dassopoulos T, Newberry RD, Sodergren E, Weinstock G, Robertson CE, Frank DN, Li E. Host genes related to paneth cells and xenobiotic metabolism are associated with shifts in human ileum-associated microbial composition. PLoS One. 2012;7:e30044. doi: 10.1371/journal.pone.0030044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karlsson J, Pütsep K, Chu H, Kays RJ, Bevins CL, Andersson M. Regional variations in Paneth cell antimicrobial peptide expression along the mouse intestinal tract. BMC Immunol. 2008;9:37. doi: 10.1186/1471-2172-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mai V, Young CM, Ukhanova M, Wang X, Sun Y, Casella G, Theriaque D, Li N, Sharma R, Hudak M, Neu J. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One. 2011;6:e20647. doi: 10.1371/journal.pone.0020647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bauer CR, Morrison JC, Poole WK, Korones SB, Boehm JJ, Rigatto H, Zachman RD. A decreased incidence of necrotizing enterocolitis after prenatal glucocorticoid therapy. Pediatrics. 1984;73:682–688. [PubMed] [Google Scholar]
- 70.Coutinho VB, Coutinho HB, Coutinho EM. Effects of hydrocortisone acetate treatment on the small intestine of the lactent marsupial Didelphis albiventris. Anat Anz. 1991;172:213–221. [PubMed] [Google Scholar]
- 71.Maynard AA, Dvorak K, Khailova L, Dobrenen H, Arganbright KM, Halpern MD, Kurundkar AR, Maheshwari A, Dvorak B. Epidermal growth factor reduces autophagy in intestinal epithelium and in the rat model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G614–G622. doi: 10.1152/ajpgi.00076.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raaberg L, Nexø E, Damsgaard Mikkelsen J, Seier Poulsen S. Immunohistochemical localisation and developmental aspects of epidermal growth factor in the rat. Histochemistry. 1988;89:351–356. doi: 10.1007/BF00500636. [DOI] [PubMed] [Google Scholar]
- 73.Thachil E, Hugot JP, Arbeille B, Paris R, Grodet A, Peuchmaur M, Codogno P, Barreau F, Ogier-Denis E, Berrebi D, Viala J. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn's disease. Gastroenterology. 2012;142:1097–1099.e4. doi: 10.1053/j.gastro.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 74.Deshpande G, Rao S, Patole S, Bulsara M. Updated meta-analysis of probiotics for preventing necrotizing enterocolitis in preterm neonates. Pediatrics. 2010;125:921–930. doi: 10.1542/peds.2009-1301. [DOI] [PubMed] [Google Scholar]
- 75.Wang Q, Dong J, Zhu Y. Probiotic supplement reduces risk of necrotizing enterocolitis and mortality in preterm very low-birth-weight infants: an updated meta-analysis of 20 randomized, controlled trials. J Pediatr Surg. 2012;47:241–248. doi: 10.1016/j.jpedsurg.2011.09.064. [DOI] [PubMed] [Google Scholar]
- 76.Kingma SD, Li N, Sun F, Valladares RB, Neu J, Lorca GL. Lactobacillus johnsonii N6. 2 stimulates the innate immune response through Toll-like receptor 9 in Caco-2 cells and increases intestinal crypt Paneth cell number in biobreeding diabetes-prone rats. J Nutr. 2011;141:1023–1028. doi: 10.3945/jn.110.135517. [DOI] [PubMed] [Google Scholar]
- 77.Wrackmeyer U, Hansen GH, Seya T, Danielsen EM. Intelectin: a novel lipid raft-associated protein in the enterocyte brush border. Biochemistry. 2006;45:9188–9197. doi: 10.1021/bi060570x. [DOI] [PubMed] [Google Scholar]
- 78.Shin K, Wakabayashi H, Yamauchi K, Yaeshima T, Iwatsuki K. Recombinant human intelectin binds bovine lactoferrin and its peptides. Biol Pharm Bull. 2008;31:1605–1608. doi: 10.1248/bpb.31.1605. [DOI] [PubMed] [Google Scholar]
- 79.Sherman MP, Adamkin DH, Radmacher PG, Sherman J, Niklas V. Protective Proteins in Mammalian Milks: Lactoferrin Steps Forward. NeoReviews. 2012;13:e293–e300. [Google Scholar]
- 80.Manzoni P, Rinaldi M, Cattani S, Pugni L, Romeo MG, Messner H, Stolfi I, Decembrino L, Laforgia N, Vagnarelli F, Memo L, Bordignon L, Saia OS, Maule M, Gallo E, Mostert M, Magnani C, Quercia M, Bollani L, Pedicino R, Renzullo L, Betta P, Mosca F, Ferrari F, Magaldi R, Stronati M, Farina D Italian Task Force for the Study and Prevention of Neonatal Fungal Infections, Italian Society of Neonatology. Bovine lactoferrin supplementation for prevention of late-onset sepsis in very low-birth-weight neonates: a randomized trial. JAMA. 2009;302:1421–1428. doi: 10.1001/jama.2009.1403. [DOI] [PubMed] [Google Scholar]