

Abstract
Accumulation of misfolded proinsulin in the β-cell leads to dysfunction induced by endoplasmic reticulum (ER) stress, with diabetes as a consequence. Autophagy helps cellular adaptation to stress via clearance of misfolded proteins and damaged organelles. We studied the effects of proinsulin misfolding on autophagy and the impact of stimulating autophagy on diabetes progression in Akita mice, which carry a mutation in proinsulin, leading to its severe misfolding. Treatment of female diabetic Akita mice with rapamycin improved diabetes, increased pancreatic insulin content, and prevented β-cell apoptosis. In vitro, autophagic flux was increased in Akita β-cells. Treatment with rapamycin further stimulated autophagy, evidenced by increased autophagosome formation and enhancement of autophagosome–lysosome fusion. This was associated with attenuation of cellular stress and apoptosis. The mammalian target of rapamycin (mTOR) kinase inhibitor Torin1 mimicked the rapamycin effects on autophagy and stress, indicating that the beneficial effects of rapamycin are indeed mediated via inhibition of mTOR. Finally, inhibition of autophagy exacerbated stress and abolished the anti-ER stress effects of rapamycin. In conclusion, rapamycin reduces ER stress induced by accumulation of misfolded proinsulin, thereby improving diabetes and preventing β-cell apoptosis. The beneficial effects of rapamycin in this context strictly depend on autophagy; therefore, stimulating autophagy may become a therapeutic approach for diabetes.
In eukaryotic cells, secreted proteins undergo cotranslational folding in the endoplasmic reticulum (ER) lumen. The β-cell ER faces a high protein-folding burden due to the high proinsulin biosynthesis rate: proinsulin mRNA may reach 20% of total mRNA (1) and proinsulin production 50% of total protein synthesis in stimulated β-cells (2). Furthermore, correct folding of proinsulin is difficult due to its complex tertiary structure, containing three disulfide bonds that depend on the redox state of the ER, which can be altered by the inflammation and oxidative stress of nutrient overload and obesity (3,4). Indeed, several reports showed that ER stress is linked to β-cell dysfunction in type 2 diabetes (4–7).
The causality between proinsulin misfolding and β-cell failure is epitomized in the mutant INS gene–induced diabetes of youth syndrome, in which mutations in proinsulin trigger irreparable misfolding (8,9). As an example, the C(A7)Y proinsulin mutation results in severe congenital diabetes in man and in the Akita mouse. The pathophysiology of β-cell failure in Akita is complex and involves trapping of nonmutant proinsulin in the ER, leading to impaired β-cell function, stress, and apoptosis (10–12). Notably, a subset of Akita β-cells can compensate for proinsulin misfolding, thereby avoiding diabetes (13). Therefore, unraveling the adaptive mechanisms that operate in stressed β-cells may have important implications for diabetes treatment.
Accumulation of misfolded proteins in the ER stimulates the unfolded protein response (UPR), an adaptive homeostatic signaling pathway aimed to reduce stress. The UPR increases the expression of ER chaperones and oxireductases, inhibits mRNA translation, and stimulates ER-associated degradation, thus reducing ER protein load and enhancing folding capacity and clearance of misfolded proteins. However, if ER stress is not subdued, its continuous activation results in apoptosis. ER stress induces autophagy to eliminate damaged organelles and protein aggregates, thus improving cell function and survival (14). This comprises the transport of cytosolic portions and entire organelles to lysosomes via double-membrane vesicles called autophagosomes. Lysosomal degradation recycles amino and fatty acids for energy production in starvation, but also serves an important homeostatic function in response to stress in nutrient abundance (15). Transgenic mice with impaired β-cell autophagy exhibited decreased insulin secretion, glucose intolerance, and islet degeneration, indicating that basal autophagy is required for β-cell well being (16,17). The nutrient-sensing kinase mammalian target of rapamycin complex 1 (mTORC1) is an important regulator of autophagy (18–20). Under nutrient availability, mTORC1 phosphorylates Atg13, which prevents binding to Atg1 (ULK1 in mammals) and hence reduced formation of the Atg1–Atg13–Atg17 complex (21). Conversely, mTORC1 inhibition during starvation or by rapamycin administration stimulates initiation of autophagosome budding.
In this study, we studied in Akita β-cells the effects of proinsulin misfolding on autophagy and whether stimulating autophagy using mTORC1 inhibitors attenuates stress and prevents diabetes progression in Akita mice in vivo.
RESEARCH DESIGN AND METHODS
Animals.
C57BL/6J wild-type (WT) and diabetic heterozygous Akita (Ins2WT/C96Y) mice were from The Jackson Laboratory (Bar Harbor, ME). Male Ins2WT/C96Y were bred with female WT mice at The Hebrew University of Jerusalem. Presence of Akita mutation was verified by the absence of an Fnu4HI restriction site in the 280-bp PCR product of the Ins2 gene. Four- to 8-week-old female Ins2WT/C96Y and their littermate WT (Ins2WT/WT) mice were used. Diabetic Akita mice were treated by daily intraperitoneal (IP) injection of 0.2 mg/kg rapamycin (Sigma-Aldrich, Rehovot, Israel) or saline as control (controls: diabetic Akita mice and WT mice) for 15 days. Body weight, food intake, and tail blood glucose were monitored every other day (Accuchek; Roche Diagnostics GmbH, Mannheim, Germany). Insulin sensitivity was assessed by insulin tolerance test. Fasted animals were given 0.75 U/kg regular insulin IP followed by consecutive blood glucose measurements. Animals underwent also an IP glucose tolerance test (2 g/kg) after a 16-h fast. At termination of the studies, animals were killed by cervical dislocation. The head of the pancreas was frozen in liquid nitrogen and analyzed for insulin content by radioimmunoassay (RIA). The pancreas tail was immersion-fixed in 4% formalin for morphometric analysis. Animal use was approved by the institutional animal care and use committee of The Hebrew University-Hadassah Medical Organization.
Islet isolation, β-cell line culture, and experimental protocol.
Akita and WT β-cells were generated as described (22). Cells were cultured in Dulbecco’s modified Eagle’s medium containing 150 μmol/L β-mercaptoethanol/mL at 25 mmol/L glucose. Islets were isolated from Akita and WT mice by collagenase injection to the bile duct (Collagenase P; Roche Diagnostics) and cultured in RPMI 1640 with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L l-glutamine (Biological Industries, Beit Haemek, Israel) at different glucose concentrations. To study autophagy, Akita and WT islets and β-cell lines were treated with and without rapamycin (50 nmol/L) or Torin1 (250 nmol) (Tocris Bioscience, Bristol, U.K.). Bafilomycin A1 (100 nmol/L) and chloroquine (50 μmol/L) (Sigma-Aldrich) were used to inhibit lysosomal degradation. The chemical chaperones 4-phenyl butyrate (10 mmol/L) and tauroursodeoxycholic acid (0.5 mg/mL) were used to study the role of ER stress in autophagy (Sigma-Aldrich).
Western blots.
The following antibodies were used: p-S6 ribosomal protein (p-S6; Ser235/236), insulin receptor substrate 2 (IRS2), p-Akt/protein kinase B (p-PKB; Ser473), rictor, p-eukaryotic initiation factor 2α (p-eIF2α), p-protein kinase RNA-like endoplasmic reticulum kinase (p-PERK), p-stress-activated protein kinase/Jun NH2-terminal kinase (p-SAPK/JNK; Thr183/Tyr185), SAPK/JNK, c-JUN, IRE1α, total/cleaved caspase 3, LC3-I/II (Cell Signaling Technology, Beverly, MA); glyceraldehyde-3-phosphate dehydrogenase (GAPDH), GADD153/C/EBP homologous protein (CHOP), and P62/SQSTM1 (Santa Cruz Biotechnology, Santa Cruz, CA). Peroxidase-conjugated AffiniPure goat anti-rabbit and anti-mouse IgG from Jackson ImmunoResearch Laboratories (West Grove, PA) were secondary antibodies.
Quantitative real-time RT-PCR.
RNA was extracted using TRI Reagent (Biolab, Jerusalem, Israel); samples of 1 μg total RNA were reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI). Quantitative real-time RT-PCR for spliced Xbp1 was performed on a Prism 7000 Sequence Detection System using the Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). All samples were analyzed in triplicate and corrected for glyceraldehyde-3-phosphate dehydrogenase. The following oligonucleotides were used for the PCR of spliced Xbp1: forward, 5′-GAGTCCGCAGCAGGTG-3′ and reverse, 5′-GAAGAGGCAACAGCG-TCAGA-3′.
Immunofluorescence staining and analysis.
Paraffin sections were rehydrated and antigens retrieved. The following antibodies were used: guinea pig anti-insulin 1:200 (DakoCytomation, Glostrup, Denmark), rabbit anti-glucagon 1:200 (DakoCytomation), rabbit anti-Ki67 1:200 (Thermo Scientific, Kalamazoo, MI), and guinea pig anti-Pdx1 1:2500 (DakoCytomation). Propidium iodide, 2 μg/mL (Invitrogen, Carlsbad, CA), was used for DNA counterstain. TUNEL staining was done with the Roche Cell Death Detection Kit (Roche Diagnostics). Secondary antibodies were all from Jackson ImmunoResearch Laboratories. To determine β-cell mass, consecutive paraffin sections 75 μm apart spanning the entire pancreas (nine sections per pancreas) were stained for insulin and hematoxylin. Digital images were obtained at an original magnification of ×40 with a Nikon C1 confocal microscope, stitched using NIS-Elements software (Nikon, Melville, NY), and the percent area covered by insulin was determined. β-Cell mass was calculated as the product of pancreas weight and percentage insulin area.
Live cell imaging.
Autophagosome and autolysosome formation was imaged in live WT and Akita β-cell lines using a Zeiss LSM 710 confocal microscope equipped with an incubator providing 37°C and 5% CO2 (Carl Zeiss, Oberkochen, Germany). Cells were seeded on eight-chamber borosilicate coverglass plates (Nunc, Rochester, NY) and transfected the subsequent day with the following constructs using Lipofectamine 2000 and Opti-MEM (Invitrogen): lysosomal-associated membrane protein 1 (LAMP-1)–enhanced green fluorescent protein (eGFP) (23), GFP-LC3 (24), mCherry-P62 (24), WT–insulin-GFP, Akita insulin-GFP (25), and RFP-ER (26). The day after, cells were treated as indicated and imaged with a 63× objective. GFP was excited using a 488-nm argon laser and mCherry with a 561-nm helium/neon laser. Image processing and lysosome size measurement were conducted with Metamorph software (Molecular Devices, Sunnyvale, CA).
Oxygen consumption.
Oxygen consumption was measured in β-cells in V7 culture plates in an XF24 respirometer (Seahorse Bioscience, Billerica, MA). One hour prior to measurements, culture medium was replaced with assay medium containing 3 mmol/L glucose, 0.8 mmol/L Mg2+, 1.8 mmol/L Ca2+, 143 mmol/L NaCl, 5.4 mmol/L KCl, 0.91 mmol/L NaH2PO4, and 15 mg/mL phenol red (Seahorse Bioscience). Measurements were recorded before and consecutively after stimulation with 20 mmol/L glucose.
Metabolic labeling.
WT and Akita β-cell lines were incubated for 18 h in Dulbecco’s modified Eagle’s medium at 25 mmol/L glucose with and without 50 nmol/L rapamycin and then pulse-labeled for 45 min with [35S]-Met/Cys (7.5 mCi/500 μL) (Amersham Biosciences) in Krebs-Ringer bicarbonate HEPES (KRBH)-BSA buffer containing same agents. Labeling was terminated by ice-cold wash with glucose-free KRBH-BSA buffer. The cells were scraped, centrifuged, and pellets subjected to immunoprecipitation (27). Briefly, the cells were resuspended in 450 mL 0.2 mol/L glycine buffer containing 0.1% RIA-grade BSA and 0.5% Nonidet P-40 (pH 8.8) (glycine buffer/Nonidet P-40 buffer) and subjected to four freeze-thaw cycles in liquid nitrogen. Each sample was pretreated with protein A-Sepharose (Sigma-Aldrich) before immunoprecipitation with anti-insulin serum (Sigma-Aldrich) to correct for nonspecific binding. Insulin-like peptides were resolved on reducing Tris–SDS-PAGE gel.
Apoptosis ELISA.
Cells were lysed and oligonucleosomes in the cytosol, indicative of apoptosis-induced DNA degradation, quantified using the Cell Death ELISAPLUS assay (Roche Diagnostics).
Insulin assay.
Mouse insulin immunoreactivity was determined using rat insulin RIA kits from Linco Research Inc. (St. Charles, MO). The routine intra-assay coefficient of variation was 4–6%, and interassay coefficient of variation was 6–10%.
Data presentation and statistical analysis.
Data shown are means ± SE. Statistical significance of differences between groups was determined by one-way ANOVA followed by Newman-Keuls test using the InStat statistical program from GraphPad Software Inc. (San Diego, CA). A paired-sample t test was used when the difference between a reference (taken as 100%) and a test was analyzed. A P value of <0.05 was considered significant.
RESULTS
Rapamycin improves diabetes in Akita mice.
WT and female diabetic Akita mice were given daily IP 0.2 g/kg rapamycin or vehicle for 15 days (Fig. 1). In WT animals, random blood glucose was unchanged in rapamycin-treated littermates. When challenged with glucose (IP glucose tolerance test), rapamycin induced mild glucose intolerance. In contrast, robust effects of rapamycin were seen in Akita mice. At 2 weeks after randomization, random blood glucose in controls was 519 ± 34 mg/dL, with grossly impaired glucose tolerance. Rapamycin markedly decreased blood glucose in Akita mice to 182 ± 13 mg/dL (Fig. 1A), similar to that of control groups, with marked improvement of glucose tolerance (Fig. 1B); also, fasting blood glucose was normalized in rapamycin-treated Akita mice (94 ± 15 vs. 252 ± 50 mg/dL).
FIG. 1.
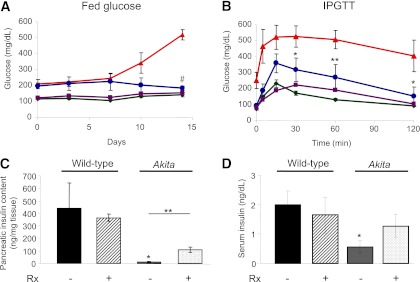
Rapamycin improves diabetes in Akita mice. Fed blood glucose (A), glucose tolerance (B), pancreatic insulin content (C), and serum insulin (D) of wild-type and Akita mice treated by daily IP injection of 0.2 g/kg rapamycin or the vehicle for 15 days. Animals underwent IP glucose tolerance test (IPGTT) by IP injection of 2 g/kg glucose at day 14. At termination, pancreatic insulin content and serum insulin were analyzed by RIA. Untreated wild-type mice, green; rapamycin-treated wild-type mice, purple; untreated Akita mice, red; and rapamycin-treated Akita mice, blue. Results are means ± SE of three mice in the wild-type groups and five in the Akita groups. *P < 0.05, **P < 0.01, #P < 0.001 for the difference between rapamycin-treated Akita mice and untreated controls (A and B) and between untreated wild-type and Akita mice (C and D).
Pancreatic insulin content was 100-fold reduced in Akita mice compared with WT, explaining the ∼95% decreased insulin secretion. Rapamycin partially prevented the decrease in insulin content and serum insulin (Fig. 1C and D), independently of food intake and changes in body weight (Supplementary Fig. 1). Insulin-induced glucose clearance (insulin tolerance test) was mildly decreased in rapamycin-treated WT mice, suggesting impaired insulin sensitivity (Supplementary Fig. 1). Fasting blood glucose in rapamycin-treated Akita diabetic mice being much lower than in untreated mice, the impact of rapamycin on insulin sensitivity could not be reliably assessed.
In Akita mice, islets were insulin-depleted, with disrupted architecture; rapamycin partially reversed these changes (Fig. 2A). β-Cell apoptosis (TUNEL) and proliferation (Ki67) were increased in Akita islets, which was prevented by rapamycin (Fig. 2B and C). Intriguingly, β-cell mass was not reduced in Akita mice despite diabetes, neither was it affected by rapamycin (Fig. 2D), suggesting that the improvement of diabetes by rapamycin is due to amelioration of β-cell function.
FIG. 2.

Rapamycin restores islet morphology, replenishes islet insulin content, and reduces β-cell turnover in diabetic Akita mice. Islet morphology (A), β-cell proliferation (A and B), apoptosis (A and C), and mass (D) in wild-type and Akita mice treated with and without rapamycin for 15 days. A: Pancreatic sections were immunostained for insulin, glucagon (top), and PDX1 (middle). Proliferation was assessed by staining for Ki67 (middle) and apoptosis by TUNEL assay (bottom). Scale bar, 20 μm. Total of 3,400–3,800 β-cells were counted per treatment (B–D). **P < 0.01, #P < 0.001 for the difference between the indicated groups. Rx, treatment with rapamycin.
In vitro effects of rapamycin on β-cell ER stress.
To examine the effects of rapamycin on β-cell functions, nonmutant WT proinsulin and the Akita mutant, conjugated to GFP, were transfected to WT β-cells. Consistent with previous studies (26), WT (pro)insulin was located both in the ER and secretory granules, whereas the Akita mutant was ER-trapped and did not reach the granules (Supplementary Fig. 2). To study the impact of proinsulin misfolding on UPR activation, we used stable β-cell lines derived from Akita and control mice. UPR markers CHOP, p-PERK, p-JNK, and p-c-JUN were augmented in Akita β-cells (Fig. 3); associated with induction of caspase 3 cleavage and apoptosis. Surprisingly, IRE1α and Xbp1s were lower in Akita than WT cells (Fig. 3A and C). IRS2 expression and mTORC1 signaling (S6 phosphorylation) were also lower in Akita cells. As expected, rapamycin effectively inhibited mTORC1 (decreased S6 phosphorylation). Prolonged treatment with rapamycin was recently shown to inhibit mTORC2, resulting in β-cell toxicity (28). In this study, however, rapamycin treatment of Akita and WT cells for 16 and 72 h did not affect rictor expression and Akt/PKB phosphorylation, suggesting unchanged mTORC2 signaling (Fig. 3 and Supplementary Fig. 3). Although mTORC1 exerts negative feedback on IRS2 (29), its inhibition did not rescue IRS2 expression. Rapamycin attenuated cellular stress and apoptosis in Akita β-cells and islets, evidenced by decreased CHOP expression and JNK and c-JUN phosphorylation, as well as caspase 3 cleavage (Fig. 3 and Supplementary Figs. 3 and 4).
FIG. 3.

Rapamycin reduces cellular stress and prevents apoptosis in Akita β-cells. Akita and wild-type β-cells were treated with and without 50 nmol/L rapamycin (R) for 16 h, followed by analysis for different markers of insulin signaling and stress by Western blot and qPCR (spliced Xbp1 [Xbp1s]). Insulin signaling (IRS2, p-Akt/PKB, and p-S6) and UPR (A–C) and JNK and c-JUN phosphorylation (D and E). F: Apoptosis was assessed by Western blot for cleaved caspase 3 and quantification of cytosolic oligonucleosomes by ELISA (bottom). Representative blots and quantifications are shown. Results are means ± SE of three to five separate experiments. *P < 0.05, **P < 0.01, #P < 0.001 for the difference between the indicated groups. a.u., arbitrary units; Cl., cleaved; eIF2α, eukaryotic initiation factor 2α; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
mTORC1 stimulates protein synthesis by phosphorylating the translation inhibitor 4EBP1 (30); therefore, rapamycin is expected to inhibit protein synthesis. Metabolic labeling indicated increased proinsulin synthesis by rapamycin in both WT and Akita β-cells (Fig. 4A). This unexpected result suggests that β-cells escape rapamycin’s inhibitory effect on protein synthesis; thus, rapamycin-induced attenuation of stress is not via decreasing the ER proinsulin cargo load.
FIG. 4.

Treatment with rapamycin increases proinsulin biosynthesis and glucose-stimulated oxygen consumption in Akita β-cells. Akita and wild-type β-cells were treated with and without 50 nmol/L rapamycin (Rapa.) for 16 h and then pulse-labeled for 45 min with [35S]-Met/Cys in KRBH-BSA buffer containing similar treatment. The labeling was terminated by ice-cold washout in glucose-free KRBH-BSA buffer. A: Proinsulin biosynthesis was determined by immunoprecipitation with anti-insulin serum (similar quantity of protein extracts were used for immunoprecipitation in the different experiments). Results are expressed as fold of proinsulin biosynthesis in untreated wild-type cells and are means ± SE of four individual experiments. B: Oxygen consumption was measured in wild-type and Akita cells in an XF24 respirometer. After basal measurements were recorded, cells were stimulated with 20 mmol/L glucose. Basal oxygen consumption is shown in the top panel and the increase in oxygen consumption in response to glucose in the bottom panel. *P < 0.05, **P < 0.01 for the difference between the indicated groups. PI, proinsulin.
ER stress may increase the ER–mitochondrial coupling, promoting mitochondrial respiration (31). Indeed, basal oxygen consumption at 3 mmol/L glucose was higher in Akita than WT cells; however, glucose-induced respiration at high glucose (20 mmol/L) was decreased (Fig. 4B). Rapamycin reversed these changes, indicating normalized mitochondrial function.
Collectively, rapamycin reduced stress in Akita cells, reversed the alterations in mitochondrial function in ER stress, and increased proinsulin biosynthesis.
Effects on autophagy.
Autophagy was quantified by cotransfection with the autophagosome markers LC3 conjugated to GFP (LC3-GFP) and P62/SQSTM1 conjugated to mCherry (P62-mCherry), followed by overnight treatment with rapamycin. P62/SQSM1 is an adaptor protein that interacts with lipidated LC3, LC3-II (32). Autophagosomes appear as puncta by confocal microscopy; the number of autophagosomes (LC3-GFP+ and/or P62-mCherry+ puncta) was twofold higher in Akita than in WT cells (Fig. 5). Increase in autophagosomes could result from block of autophagic degradation or enhanced autophagic flux. To distinguish between these, we treated β-cells with the lysosome inhibitor bafilomycin A1. LC3-II was markedly increased in WT and Akita β-cells and islets, indicating that the autophagic machinery is not impaired. Moreover, LC3-II accumulation in presence of bafilomycin A1 was greater in Akita islets and β-cells than in WT controls (Fig. 6), suggesting that proinsulin misfolding stimulates autophagy. The autophagosome content (LC3-GFP+ and/or P62-mCherry+ puncta) of Akita and control cells was increased fourfold by rapamycin (Fig. 5A and B). Rapamycin increased the percentage of P62-mCherry+/LC3-GFP− puncta (Fig. 5C); since GFP is degraded in the lysosome acidic environment, this suggests that rapamycin increased autophagosome–lysosome fusion. This was confirmed by cotransfection of cells with the autophagosome marker P62-mCherry and the lysosome marker LAMP-1–GFP. Rapamycin increased the number of autolysosomes (P62-mCherry+/LAMP-1–GFP+ puncta) by fivefold (Fig. 7A and B). Notably, rapamycin stimulation of autophagosome–lysosome fusion (P62-mCherry/LAMP-1–GFP colocalization) was twofold higher in Akita than in WT cells (Fig. 7B). In addition, stimulation of autophagy by rapamycin resulted in generation of giant autolysosomes (LAMP-1–GFP+) engulfing P62 (mCherry+), more so in Akita than in WT cells (Fig. 7C). Rapamycin decreased P62/SQSTM1 expression (Western blot) in mouse β-cells and islets (Fig. 6). Since P62/SQSTM1 is degraded via autophagy, this indicates stimulation of autophagic flux. LC3-II was markedly increased by inhibition of lysosomal degradation (bafilomycin A1), but not with rapamycin. This probably results from rapamycin stimulation of autophagosome turnover, preventing the accumulation of LC3-II–positive autophagosomes.
FIG. 5.

Rapamycin stimulates autophagy in wild-type and Akita β-cells. WT and Akita β-cells were cotransfected with LC3-GFP and P62-mCherry and then treated with and without rapamycin (Rapa.) for 16 h. Starved cells treated with the lysosome inhibitor bafilomycin A (Bafilo. A1) for 4 h were used as positive controls. Autophagosomes appear as LC3-GFP+ and/or P62-Cherry+ puncta (A). Quantification of number of autophagosomes per cell is shown in B (n = 10–13 cells per each treatment). Wild-type cells, white bars; Akita cells, black bars. C: Distribution of autophagosome types according to the expression of LC3-ΙΙ and P62/SQSTM1. Autophagosomes expressing LC3-ΙΙ or P62/SQSTM1 alone are shown in green and red, respectively; autophagosomes coexpressing LC3-ΙΙ and P62/SQSTM1 are shown in yellow (n = 10–13 cells/treatment). *P < 0.05, **P < 0.01, #P < 0.001 for the differences between the indicated groups (B) or between untreated (UT) Akita and wild-type cells or between rapamycin-treated cells and their matched controls (C). Scale bars, 5 μm.
FIG. 6.

Western blot analysis of autophagy in Akita and wild-type β-cells (A) and islets (B). Wild-type and Akita β-cells and islets were treated without (UT) and with rapamycin (R) for 16 h or with bafilomycin A1 (B) for 4 h. Wild-type and Akita β-cells were incubated at 25 mmol/L glucose and islets at 22.2 mmol/L glucose. Representative blots of LC3-Ι/ΙΙ and P62/SQSTM1 expression and quantification of the experiments in wild-type and Akita β-cell lines are shown. Wild-type cells, white bars; Akita cells, black bars. Results are expressed as means ± SE of five separate experiments. *P < 0.05, **P < 0.01, #P < 0.001 for the difference between the indicated groups or between rapamycin-treated β-cells and their matched controls. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
FIG. 7.

Rapamycin (Rapa.) augments the generation of autolysosomes in wild-type and Akita β-cells. Autolysosome content was assessed using live-cell imaging of P62-mCherry and LAMP-1–GFP reporters. Wild-type and Akita β-cells were cotransfected with LAMP-1–GFP and P62-mCherry and then treated without (UT) and with rapamycin (R) for 16 h. Starved cells treated with the lysosome inhibitor bafilomycin A (B) were used as positive controls. Autolysosomes appear as P62-mCherry+/LAMP-1–GFP+ puncta (A). Quantification of autolysosomes, expressed as percentage of all P62-mCherry+ puncta, is shown (B) (n = 10–13 cells per each treatment). Wild-type cells, white bars; Akita cells, black bars. C: Quantification of autolysosome size (n = 10–13 cells per each treatment, at least 225 autolysosomes analyzed per treatment). *P < 0.05, **P < 0.01, #P < 0.001 for the difference between the indicated groups. Scale bar, 5 μm.
Thus, basal autophagy is increased in Akita β-cells and islets. In both WT and Akita β-cells, rapamycin stimulated autophagy by increasing autophagosome generation and autophagosome–lysosome fusion.
Torin1 mimics rapamycin effects on autophagy and cellular stress.
Rapamycin inhibits mTORC1 via direct binding to FK506-binding protein 12, which is required for raptor binding to mTOR, without altering the mTOR intrinsic catalytic activity (33). To confirm that rapamycin effects on autophagy and ER stress are mediated via mTOR, we used Torin1, a selective ATP-competitive inhibitor of the mTOR serine/threonine kinase (34). WT and Akita β-cells were treated without or with Torin1 for 16 h in the presence or absence of bafilomycin A1 during the last 4 h of treatment. Torin1, like rapamycin, inhibited mTORC1, shown by decreased S6 phosphorylation (Supplementary Fig. 5A). Torin1 inhibits both mTORC1 and mTORC2, the latter regulating Akt/PKB activity (34). Consequently, it decreased Akt/PKB phosphorylation in WT and Akita β-cells (Supplementary Fig. 5A). Similar to rapamycin, Torin1 stimulated autophagy, as evidenced by decreased P62/SQSTM1 (without affecting LC3-II levels), which in Akita β-cells was associated with decreased cellular stress and apoptosis (Supplementary Fig. 5). Of note, Torin1 stimulation of autophagic flux was not abolished by 4 h treatment with bafilomycin A1, indicating that Torin1 is a potent stimulator of autophagy. Treatment with bafilomycin A1 increased cellular stress and apoptosis in WT and Akita β-cells, which were attenuated by Torin1 (Supplementary Fig. 5).
Regulation of β-cell survival by ER stress-autophagy cross talk.
We studied whether increased autophagy in Akita β-cells results from ER stress. β-Cells were treated with the chemical chaperones 4-phenyl butyrate or tauroursodeoxycholic acid for 16 h in presence or absence of bafilomycin A1 during the last 4 h of treatment. As expected, chaperones inhibited cellular stress (CHOP expression) in Akita β-cells. This markedly decreased the accumulation of LC3-II in presence of bafilomycin A1, indicating that ER stress indeed stimulates autophagy in β-cells (Supplementary Fig. 6).
Next, we examined whether stimulation of autophagy by rapamycin and reduction of ER stress are causally related. We treated WT and Akita β-cells together with rapamycin and the lysosome inhibitor chloroquine for 16 h and studied the impact on stress and apoptosis (Fig. 8). Pharmacological inhibition of autophagy by chloroquine increased cellular stress and apoptosis. Inhibition of autophagy abolished the rapamycin effect on JNK phosphorylation and CHOP expression (Fig. 8). Moreover, β-cell apoptosis was markedly increased in the presence, as well as absence of rapamycin. Thus, autophagy is an adaptive response to ER stress and mediates the rapamycin protection of stressed β-cells.
FIG. 8.

Inhibition of autophagy induces stress and apoptosis in β-cells and abolishes the beneficial effects of rapamycin in Akita β-cells. Wild-type and Akita β-cells were treated without and with rapamycin (50 nmol/L) and the lysosomal enzyme inhibitor chloroquine (50 μmol/L) for 16 h, followed by analysis for different markers of ER stress and apoptosis by Western blot (A). S6 phosphorylation was used as a marker for mTORC1 inhibition by rapamycin. Accumulation of LC3-ΙΙ in response to treatment with chloroquine indicates inhibition of autophagic flux (B). JNK phosphorylation, CHOP expression, and cleaved caspase 3 were used to assess stress and apoptosis (C). Quantification of apoptosis was performed by ELISA for cytosolic oligonucleosomes (D). Representative blots and quantifications are shown. Results are means ± SE of three to eight individual experiments. *P < 0.05, **P < 0.01, #P < 0.001 for the difference between the indicated groups. Cl., cleaved; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
DISCUSSION
The Akita mouse is an excellent model of ER stress-induced diabetes, in which β-cell dysfunction results from retention of misfolded protein in the ER, leading to stress, insulin deficiency, and apoptosis. Deletion of the proapoptotic transcription factor CHOP delayed the onset of diabetes in Akita mice, emphasizing that ER stress is at center stage in the pathophysiology of diabetes in this model (35).
We found that the mutant Akita proinsulin is accumulated in the ER and stimulates the UPR death effectors JNK/c-JUN and CHOP. Interestingly, the IRE1α pathway was inhibited, evident by decreased IRE1α expression and Xbp-1 splicing. Consistent with this, a previous report showed that chronic ER stress attenuated IRE1α activity, whereas PERK signaling was maintained (36). Inhibition of IRE1α activity under chronic stress increased susceptibility for apoptosis. Thus, insufficient UPR activation under chronic stress might contribute to β-cell apoptosis. Insulin signaling was also attenuated in Akita compared with WT cells, including decreased IRS2 expression and PKB/Akt and S6 phosphorylation. This also might trigger β-cell dysfunction and apoptosis, considering the important role of the canonical insulin-signaling pathway for β-cell survival (37). However, rapamycin did not modify the activities of insulin and IRE1α signaling pathways, despite markedly improving β-cell well being; we therefore assume that their role is less important in the present context.
Rapamycin decreased stress-induced mediators of cell death including JNK, c-JUN, and CHOP. Consistent with this, we previously showed that rapamycin decreased stress in β-cells exposed to glucolipotoxicity via inhibition of JNK (38). How rapamycin attenuates ER stress is not clear. Rapamycin increased rather than decreasing proinsulin biosynthesis in WT and Akita β-cells, nor did it affect proinsulin biosynthesis under glucolipotoxicty (38). Thus, rapamycin does not reduce proinsulin mRNA translation, which makes us conclude that rapamycin inhibition of ER stress is not mediated via reduction of the proinsulin load. Rapamycin does not inhibit all mTORC1 functions and may have dissimilar effects on the translation of different mRNAs (39). Further studies are required to clarify how rapamycin increased proinsulin biosynthesis in Akita β-cells and whether this simply reflects functional improvement in response to alleviation of cellular stress.
We hypothesized that mTORC1 inhibitors may attenuate ER stress through stimulation of autophagy. Indeed, rapamycin markedly increased autophagosome and autolysosome generation. Notably, stimulation of autophagy by rapamycin or by Torin1 did not lead to the expected accumulation of LC3-II, despite marked reduction in P62/SQSTM1 expression. Our findings strongly suggest that stimulated autophagosome formation was coupled to autophagosome–lysosome fusion, leading to robust increase in autolysosome number and size, therefore preventing LC3-II accumulation. These findings are consistent with a recent report suggesting that in addition to regulating the initiation step of autophagy, mTORC1 also regulates autophagosome–lysosome fusion in starvation-induced autophagy (40).
The impact of diabetes on autophagy is controversial. Some in vivo and in vitro studies suggested that autophagy is increased in diabetes (17,41), whereas others showed impaired autophagic turnover in β-cells exposed to lipotoxicity (42) and possibly also in β-cells of diabetic patients (43). Similarly, high-fat diet and obesity impaired autophagy in the liver, resulting in ER stress and insulin resistance (44). Our triple-reporter system (LC3-II–GFP, P62-mCherry, and LAMP-1–GFP), together with measurement of P62/SQSTM1 and LC3-II expression in presence and absence of lysosome enzyme inhibitors, allows reliable assessment of autophagic flux (32). The comprehensive analysis performed in this study clearly showed that both basal and rapamycin-stimulated autophagic flux were increased in Akita β-cells, suggesting that proinsulin misfolding/ER stress stimulates autophagy. Consistent with this, attenuation of ER stress by chaperones in Akita β-cells decreased autophagy. In remarkable symmetry, inhibition of autophagy increased ER stress and apoptosis, more so in Akita β-cells; furthermore, it abolished the protective effects of rapamycin. Collectively, these findings suggest that in β-cells, autophagy is activated by ER stress as an essential compensatory mechanism for cell survival. Additional stimulation of autophagy by mTORC1 inhibitors may further decrease stress and protect from apoptosis. Intriguingly, Torin1 alleviated stress in Akita β-cells despite its inhibition of mTORC2–Akt/PKB signaling, expected to impair cell survival. This emphasizes the dominating role of autophagy in β-cell adaptation to stress, which can override the potential toxicity of deranging other survival mechanisms. The recent demonstration that impairment of autophagy diminishes UPR activation and increases β-cell susceptibility for ER stress and exacerbates diabetes in ob/ob mice (45) supports this paradigm. Altogether, our results bring to focus the intimate UPR–autophagy cross talk that governs β-cell function and survival under ER stress.
The striking finding of this study is that rapamycin ameliorated diabetes under severe ER stress induced by proinsulin misfolding in vivo. Male Akita mice were extremely hyperglycemic near weaning and rapamycin did not affect blood glucose (not shown); therefore, we studied female Akita mice, in which diabetes is milder but definitive with marked hyperglycemia, hypoinsulinemia, and increased β-cell apoptosis, although β-cell mass was not decreased at 6–10 weeks. Rapamycin ameliorated diabetes, attenuated hypoinsulinemia, and prevented β-cell apoptosis. This was accompanied by lower β-cell proliferation, hence reduced overall β-cell turnover, without affecting β-cell mass. This implies that amelioration of diabetes was mainly due to improved β-cell function. This protective effect of rapamycin is striking, as rapamycin has pleiotropic effects, mostly deleterious in diabetes. We found previously that rapamycin increased insulin resistance in diabetic Psammomys obesus and induced severe dyslipidemia, β-cell apoptosis, and marked metabolic exacerbation (46). Others showed that rapamycin decreases β-cell proliferation, preventing recovery of β-cell mass following injury (47). Indeed, mTORC1 inhibitors can induce insulin resistance and diabetes (48,49). We suggest that the response to rapamycin is determined by the specific pathophysiology of diabetes. In diabetes induced by marked ER stress, as in Akita mice, β-cell function and survival strongly rely on autophagy. Therefore, in this context, stimulation of autophagy by rapamycin overrides the deleterious effects of the drug (e.g., increased insulin resistance as also noted in this study), and diabetes is improved by promoting β-cell adaptation to stress. In this regard, the finding that feeding elderly mice with rapamycin may extend life span (50) is relevant and may support the hypothesis that the beneficial versus deleterious outcomes of rapamycin treatment depend on the genetic, environmental, and metabolic milieu of the recipient. Unfortunately, no genetic means are available to stimulate autophagy in β-cells in vivo; therefore, whether rapamycin effects outside of autophagy regulation also impact the phenotype cannot be ruled out at this stage.
Rapamycin is obviously unsuitable for diabetes treatment due to its multiple side effects. However, our study is a proof of concept that autophagy is a key adaptive mechanism in β-cell stress. Improvement of proinsulin folding capacity and more selective stimulation of autophagy could become attractive therapeutic approaches for mutant INS gene–induced diabetes of youth and type 2 diabetes in general.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a grant from the Israel Science Foundation (to G.L.).
No potential conflicts of interest relevant to this article were reported.
E.B.-W. researched data and was involved in writing the manuscript. J.D.W. researched data and edited the manuscript. Y.A. researched data. B.T., N.K., and E.C. contributed to discussion and reviewed and edited the manuscript. G.L. wrote the manuscript. G.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank A. Koizumi (Kyoto University) and S. Ramanadham (University of Alabama) for providing the Akita and WT control β-cell lines. The LAMP-1–eGFP construct was kindly provided by S. Di Pietro (Colorado State University), GFP-LC3 and mCherry-P62 were both kindly provided by T. Johansen (Tromsoe University), the WT–insulin-GFP and Akita insulin-GFP constructs were kindly provided by L. Philipson (University of Chicago), and the RFP-ER construct was kindly provided by F. Pimentel Muinos (Consejo Superior de Investigaciones Cientificas-University of Salamanca). The authors also thank Tomer Nir, Judith Furth-Lavi, and Sharona Tornovsky-Babay (all from The Hebrew University of Jerusalem) for technical help.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1474/-/DC1.
REFERENCES
- 1.Van Lommel L, Janssens K, Quintens R, et al. Probe-independent and direct quantification of insulin mRNA and growth hormone mRNA in enriched cell preparations. Diabetes 2006;55:3214–3220 [DOI] [PubMed] [Google Scholar]
- 2.Schuit FC, In’t Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci USA 1988;85:3865–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donath MY, Ehses JA, Maedler K, et al. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005;54(Suppl. 2):S108–S113 [DOI] [PubMed] [Google Scholar]
- 4.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008;29:42–61 [DOI] [PubMed] [Google Scholar]
- 5.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006;313:1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev 2008;29:317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunha DA, Hekerman P, Ladrière L, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci 2008;121:2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss MA. Proinsulin and the genetics of diabetes mellitus. J Biol Chem 2009;284:19159–19163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu M, Haataja L, Wright J, et al. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS ONE 2010;5:e13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodish I, Liu M, Rajpal G, et al. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J Biol Chem 2010;285:685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci USA 2007;104:15841–15846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M, Li Y, Cavener D, Arvan P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J Biol Chem 2005;280:13209–13212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hodish I, Absood A, Liu L, et al. In vivo misfolding of proinsulin below the threshold of frank diabetes. Diabetes 2011;60:2092–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006;26:9220–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujitani Y, Ueno T, Watada H. Autophagy in health and disease. 4. The role of pancreatic beta-cell autophagy in health and diabetes. Am J Physiol Cell Physiol 2010;299:C1–C6 [DOI] [PubMed] [Google Scholar]
- 16.Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 2008;8:325–332 [DOI] [PubMed] [Google Scholar]
- 17.Jung HS, Chung KW, Won Kim J, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008;8:318–324 [DOI] [PubMed] [Google Scholar]
- 18.Tooze SA, Jefferies HB, Kalie E, et al. Trafficking and signaling in mammalian autophagy. IUBMB Life 2010;62:503–508 [DOI] [PubMed] [Google Scholar]
- 19.Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell 2011;147:724–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012;32:2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009;20:1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lei X, Zhang S, Barbour SE, et al. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. J Biol Chem 2010;285:6693–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falcón-Pérez JM, Nazarian R, Sabatti C, Dell’Angelica EC. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J Cell Sci 2005;118:5243–5255 [DOI] [PubMed] [Google Scholar]
- 24.Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005;171:603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajan S, Eames SC, Park SY, et al. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am J Physiol Endocrinol Metab 2010;298:E403–E410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klee M, Pimentel-Muiños FX. Bcl-X(L) specifically activates Bak to induce swelling and restructuring of the endoplasmic reticulum. J Cell Biol 2005;168:723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attali V, Parnes M, Ariav Y, Cerasi E, Kaiser N, Leibowitz G. Regulation of insulin secretion and proinsulin biosynthesis by succinate. Endocrinology 2006;147:5110–5118 [DOI] [PubMed] [Google Scholar]
- 28.Barlow AD, Xie J, Moore CE, et al. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2). Diabetologia 2012;55:1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briaud I, Dickson LM, Lingohr MK, McCuaig JF, Lawrence JC, Rhodes CJ. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in beta-cells. J Biol Chem 2005;280:2282–2293 [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Beugnet A, Murakami M, Yamanaka S, Proud CG. Distinct signaling events downstream of mTOR cooperate to mediate the effects of amino acids and insulin on initiation factor 4E-binding proteins. Mol Cell Biol 2005;25:2558–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bravo R, Vicencio JM, Parra V, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 2011;124:2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008;4:151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oshiro N, Yoshinko K, Hidayat S, et al. Dissociation of raptor from mTOR is a mechanism of rapamycin induced inhibition of mTOR function. Genes Cells 2004;9:359–366 [DOI] [PubMed] [Google Scholar]
- 34.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal 2009;2:pe24. [DOI] [PubMed] [Google Scholar]
- 35.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 2002;109:525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007;318:944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulkarni RN. New insights into the roles of insulin/IGF-I in the development and maintenance of beta-cell mass. Rev Endocr Metab Disord 2005;6:199–210 [DOI] [PubMed] [Google Scholar]
- 38.Bachar E, Ariav Y, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G. Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS ONE 2009;4:e4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huo Y, Iadevaia V, Yao Z, et al. Stable isotope-labelling analysis of the impact of inhibition of the mammalian target of rapamycin on protein synthesis. Biochem J 2012;444:141–151 [DOI] [PubMed] [Google Scholar]
- 40.Korolchuk VI, Saiki S, Lichtenberg M, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 2011;13:453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han D, Yang B, Olson LK, et al. Activation of autophagy through modulation of 5′-AMP-activated protein kinase protects pancreatic beta-cells from high glucose. Biochem J 2010;425:541–551 [DOI] [PubMed] [Google Scholar]
- 42.Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in β-cells. J Biol Chem 2011;286:42534–42544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masini M, Bugliani M, Lupi R, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009;52:1083–1086 [DOI] [PubMed] [Google Scholar]
- 44.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010;11:467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quan W, Hur KY, Lim Y, et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012;55:392–403 [DOI] [PubMed] [Google Scholar]
- 46.Fraenkel M, Ketzinel-Gilad M, Ariav Y, et al. mTOR inhibition by rapamycin prevents β-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 2008;57:945–957 [DOI] [PubMed] [Google Scholar]
- 47.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest 2007;117:2553–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pereira MJ, Palming J, Rizell M, et al. mTOR inhibition with rapamycin causes impaired insulin signalling and glucose uptake in human subcutaneous and omental adipocytes. Mol Cell Endocrinol 2012;355:96–105 [DOI] [PubMed] [Google Scholar]
- 49.Lamming DW, Ye L, Katajisto P, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012;335:1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009;460:392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.