

Abstract
Diabetic cardiomyopathy is associated with suppression of cardiac autophagy, and activation of AMP-activated protein kinase (AMPK) restores cardiac autophagy and prevents cardiomyopathy in diabetic mice, albeit by an unknown mechanism. We hypothesized that AMPK-induced autophagy ameliorates diabetic cardiomyopathy by inhibiting cardiomyocyte apoptosis and examined the effects of AMPK on the interaction between Beclin1 and Bcl-2, a switch between autophagy and apoptosis, in diabetic mice and high glucose–treated H9c2 cardiac myoblast cells. Exposure of H9c2 cells to high glucose reduced AMPK activity, inhibited Jun NH2-terminal kinase 1 (JNK1)–B-cell lymphoma 2 (Bcl-2) signaling, and promoted Beclin1 binding to Bcl-2. Conversely, activation of AMPK by metformin stimulated JNK1–Bcl-2 signaling and disrupted the Beclin1–Bcl-2 complex. Activation of AMPK, which normalized cardiac autophagy, attenuated high glucose–induced apoptosis in cultured H9c2 cells. This effect was attenuated by inhibition of autophagy. Finally, chronic administration of metformin in diabetic mice restored cardiac autophagy by activating JNK1–Bcl-2 pathways and dissociating Beclin1 and Bcl-2. The induction of autophagy protected against cardiac apoptosis and improved cardiac structure and function in diabetic mice. We concluded that dissociation of Bcl-2 from Beclin1 may be an important mechanism for preventing diabetic cardiomyopathy via AMPK activation that restores autophagy and protects against cardiac apoptosis.
Diabetic cardiomyopathy, a clinical condition characterized by ventricular dysfunction, develops in many diabetic patients in the absence of coronary artery disease or hypertension (1,2). An increasing number of studies have demonstrated that hyperglycemia is central to the development of diabetic cardiomyopathy, which triggers a series of downstream signals that lead to cardiomyocyte apoptosis, chamber dilation, and cardiac dysfunction (3). In support of this view, diabetes-induced cardiac cell death has been observed in diabetic patients (3) and streptozotocin (STZ)-induced diabetic animals (4). The mechanisms of pathogenesis, however, remain elusive.
Autophagy is a highly conserved process for bulk degradation and recycling of cytoplasmic components in lysosomes (5). In the heart, constitutive autophagy is a homeostatic mechanism for maintaining cardiac structure and function (6). However, excessive induction of autophagy may destroy the cytosol and organelles and release apoptosis-related factors, leading to cell death and cardiac dysfunction (7,8). Thus, autophagy appears to regulate both cell survival and cell death. Emerging evidence suggests that cross-talk occurs between autophagic and apoptotic pathways. For instance, the antiapoptotic protein B-cell lymphoma 2 (Bcl-2) inhibits starvation-induced autophagy by binding to Beclin1, and this binding effectively sequesters Beclin1 away from the core kinase complex formed from Beclin1 and vacuolar sorting protein (VPS34), a class III phosphatidylinositol 3-kinase (PI3K), which is required for the induction of autophagy (9). Recently we demonstrated that in diabetic animals, suppression of autophagy is associated with an increase in cardiac apoptosis (10,11); however, whether the induction of autophagy serves as a protective response in the development of diabetic cardiomyopathy remains unknown.
The AMP-activated protein kinase (AMPK) is a conserved cellular energy sensor that plays an important role in maintaining energy homeostasis (12). In addition, AMPK also regulates many other cellular processes, such as cell growth, protein synthesis (13,14), apoptosis (15,16), and autophagy (17,18). In the heart, AMPK is responsible for activation of glucose uptake and glycolysis during low-flow ischemia and plays an important role in limiting apoptotic activity associated with ischemia and reperfusion (19). Moreover, activation of AMPK by ischemia also stimulates autophagy and protects against ischemic injury (18). Mechanistically, AMPK appears to induce autophagy through phosphorylation and activation of ULK1 (the mammalian homolog of yeast autophagy-related gene 1 [Atg1]) (20,21); however, the molecular mechanism by which AMPK regulates the switch between autophagy and apoptosis in the development of diabetic cardiomyopathy remains to be established.
In this study, we sought to determine whether autophagy plays a role in protection against cell death during the development of diabetic cardiomyopathy and to explore the mechanism by which activation of AMPK regulates the switch between autophagy and apoptosis in this disease. We found that activation of AMPK restores cardiac autophagy by disrupting the interaction between Beclin1 and Bcl-2 and protects against cardiac cell apoptosis, ultimately leading to improvement in cardiac structure and function in diabetic mice.
RESEARCH DESIGN AND METHODS
Animals.
Male Friend virus B (FVB) mice from The Jackson Laboratory (Bar Harbor, ME) were used for the experiments. Eight-week-old mice were rendered diabetic by intraperitoneal injections of STZ (50 mg/kg) on 5 consecutive days, whereas control mice were injected with vehicle (citrate buffer, pH 4.5). One week after the injections, blood glucose was measured by applying tail blood to a glucometer as previously described (22,23). Mice with blood glucose levels >350 mg/dL were considered diabetic. The diabetic mice were randomly assigned to be treated with or without metformin (200 mg/kg/day in drinking water) for 4 months. In addition, 8-week-old control FVB and cardiac-specific transgenic mice that overexpress a dominant-negative (DN) α2 subunit (D157A) of AMPK (DN-AMPKα2; gift of Dr. Rong Tian, University of Washington, Seattle, WA) (24) were treated with STZ and metformin as described above. Four months after the treatment, left ventricular (LV) function was measured using an isolated buffer-perfused heart preparation as described previously (23,25). All animal protocols were reviewed and approved by the University of Oklahoma Institutional Animal Care and Use Committee.
Cell culture and treatments.
H9c2 cardiac myoblast cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and were incubated in a humidified atmosphere of 5% CO2/95% air at 37°C. Upon reaching 50–60% confluence, the cells were incubated with control (5.5 mmol/L) or high-glucose (30 mmol/L) medium for the indicated times as previously described (21,22). Osmotic control groups (to account for medium hyperosmolarity) were exposed to mannitol (24.5 mmol/L) in normal medium containing glucose (5.5 mmol/L).
Immunoprecipitation and Western blotting.
H9c2 cells and hearts were homogenized in lysis buffer, and protein contents were measured using the Bradford assay. Total lysates were immunoprecipitated with specific antibodies. Immunoprecipitates or cell lysates were subjected to Western blotting as described previously (26,27).
Immunohistochemistry and apoptosis assays.
Immunohistochemistry was carried out as previously described (11,28). Apoptosis in heart sections and cultured H9c2 cells was measured by TUNEL staining using a cell death detection assay kit (Roche Applied Science, Indianapolis, IN). To quantify cell apoptosis in cultured H9c2 cells, apoptosis was analyzed using an Annexin-V-FLUOS staining kit (Roche Applied Science). After treatment, H9c2 cells were collected and labeled for 15 min with Alexa 488–conjugated Annexin V and propidium iodide (PI). The apoptotic cells (annexin V+/PI− cells) were detected in the FACScan flow cytometer (BD, San Jose, CA).
AMPK activity assay.
AMPK activity was measured in ∼20 mg of cardiac tissue as described previously (29,30), using the SAMS peptide as a substrate.
Visualization of autophagic vacuoles.
H9c2 cells were plated at 5 × 104 cells per well, allowed to adhere to the plate overnight, and then infected with green fluorescent protein (GFP)–light chain 3 (LC3) adenovirus. After 24 h of transfection, the cells were incubated with chloroquine (CQ; 5 μmol/L) for 16 h in normal or high-glucose media. Fluorescence images were obtained using an inverted fluorescent microscope (Olympus America, Meliville, NY). Autophagy was measured by quantifying the average number of autophagosomes per cell for each sample. A minimum of 100 cells per sample were counted.
RESULTS
High glucose inhibits autophagy in cultured H9c2 cardiac myoblast cells.
We first determined whether high glucose alters autophagy in H9c2 cells. During autophagy, cytosolic microtubule-associated protein LC3-I is lipidated and converted to LC3-II, which is translocated to the autophagosomal membrane (31). Thus, conversion of LC3-I to LC3-II and accumulation of GFP-LC3 provide effective ways to measure the autophagosome (31). H9c2 cells were treated with d-glucose at a final concentration of 20 or 30 mmol/L for 48 h, and cells treated with 5.5 mmol/L d-glucose were used as a control. To exclude the effect of hyperosmolarity, the cells were treated with 5.5 mmol/L d-glucose plus 24.5 mmol/L of mannitol for 48 h, which did not affect AMPK activity and autophagy (data not shown). LC3-II protein levels began to decline at 20 mmol/L and were reduced about threefold at 30 mmol/L (Fig. 1A).
FIG. 1.

Activation of AMPK prevents autophagy inhibition by high glucose (HG) in H9c2 cells. A: H9c2 cells were treated with different concentrations of glucose for 48 h, and cell lysates were analyzed by Western blot using an antibody against LC3B (n = 6; *P < 0.05, †P < 0.01 vs. control [Con]). B: H9c2 cells were exposed to HG (30 mmol/L) for indicated times, and LC3-II protein levels in cell lysates were detected by Western blotting (n = 6; *P < 0.05 vs. Con). C: H9c2 cells were treated with HG for 48 h in the presence or absence of CQ (5 μmol/L), and expression of LC3-II in cell lysates was determined by Western blot (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. CQ). D and E: H9c2 cells were infected with an adenovirus encoding GFP-LC3 for 24 h. The cells were then treated with HG, CQ (5 μmol/L), or HG plus CQ for 48 h. GFP-LC3 was detected using an inverted fluorescence microscope. D: Representative microphotography of GFP-LC3 staining. E: Autophagy was quantified by counting the GFP-LC3 puncta in the cells (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. CQ). F: H9c2 cells were treated with HG, metformin (Met; 1 mmol/L), HG plus Met, or HG/Met/CQ for 48 h. Cell lysates were analyzed by Western blot using antibodies against P-AMPK (Thr172), P-ACC (Ser79), and LC3B (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. HG, ‡P < 0.05 vs. HG/Met). G and H: H9c2 cells transfected with GFP-LC3 were treated with HG, Met, HG plus Met, or HG/Met/CQ for 48 h. GFP-LC3 was detected using an inverted fluorescence microscope. G: Representative microphotography of GFP-LC3 staining. H: Autophagy was quantified by counting the GFP-LC3 puncta in the cells (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. HG, ‡P < 0.05 vs. HG/Met). I: H9c2 cells were transfected with control siRNA (C-siRNA) or AMPK siRNA (AMPK-si) for 48 h. The cells were incubated in HG medium and treated with or without Met (1 mmol/L) for 48 h. Cell lysates were analyzed by Western blot using antibodies against AMPK and LC3B (n = 3; *P < 0.05 vs. C-siRNA, †P < 0.05 vs. HG/C-siRNA, ‡P < 0.05 vs. HG/Met/C-siRNA). J: H9c2 cells infected with GFP or CA-AMPK adenovirus were incubated in HG medium for 48 h. Cell lysates were subjected to Western blot to determine the expression of LC3 (n = 3; *P < 0.05 vs. GFP, †P < 0.05 vs. GFP/HG, ‡P < 0.05 vs. CA-AMPK alone, ♣P < 0.05 vs. GFP/HG/Met, #P < 0.05 vs. CA-AMPK/HG).
We next determined the time course of autophagy suppression in a high-glucose environment. H9c2 cells were exposed to high glucose for 12–48 h. A significant reduction in LC3-II protein levels was observed at 24 h, and this effect persisted throughout the study period (Fig. 1B). To study the effect of high glucose on the autophagic flux, H9c2 cells were treated with high glucose in the presence or absence of CQ, a lysosome and autophagosome fusion inhibitor (32). Administration of CQ increased LC3-II accumulation under normal glucose conditions. This increase was attenuated by high-glucose treatment (Fig. 1C). Immunofluorescence analysis demonstrated that elevated glucose levels reduced the number and distribution of GFP-LC3 punctate staining, suggesting a reduction in autophagosome formation. Under normal glucose conditions, CQ treatment enhanced the number and distribution of the GFP-LC3 spots. Notably, this increase was depressed in a high-glucose environment, indicating that elevated glucose levels reduce autophagic flux (Fig. 1D and E).
Activation of AMPK normalizes autophagic activity in H9c2 cells under high-glucose conditions.
Metformin induces autophagy through activation of AMPK (11). To explore the mechanisms by which this kinase regulates both autophagy and apoptosis during the development of diabetic cardiomyopathy, we first examined whether AMPK activation by metformin induces autophagy. High glucose inhibited AMPK activity, as estimated by decreased phosphorylation of AMPK and acetyl-CoA carboxylase (ACC), and conversion of LC3-I to LC3-II was decreased (Fig. 1F). Administration of metformin activated AMPK and enhanced LC3-II accumulation under normal glucose conditions. The recovered AMPK activity that occurred after metformin treatment restored the autophagic capacity in high glucose–treated H9c2 cells (Fig. 1F). Under this condition, administration of CQ caused a further increase in LC3-II accumulation. Immunofluorescence analysis revealed that high glucose reduced the number and distribution of GFP-LC3 punctate staining, which was reversed after administration of metformin. Addition of both metformin and CQ to cells treated with high glucose caused an additional increase in the number and distribution of GFP-LC3 puncta. Taken together, these data suggest that metformin restores the autophagic flux in a high-glucose environment (Fig. 1G and H). The effect of AMPK on autophagy was further assessed using genetic means. Transfection of cells with AMPK small interfering RNA (siRNA) reduced LC3-II protein levels under basal conditions. Importantly, metformin treatment restored autophagy capacity in the cells transfected with control siRNA but failed to do so in the cells transfected with AMPK siRNA (Fig. 1I). Conversely, transfection of constitutively active AMPK (CA-AMPK) adenovirus in H9c2 cells enhanced LC3-II protein levels. In a high-glucose environment, metformin treatment induced more autophagy in the cells transfected with CA-AMPK adenovirus than in the cells transfected with GFP adenovirus (Fig. 1J). Taken together, our results implicate that AMPK activation by metformin stimulates autophagy.
AMPK activation attenuates high glucose–induced apoptosis in H9c2 cells.
We next investigated whether activation of AMPK prevents high glucose–induced apoptosis in H9c2 cells. As compared with normal glucose and osmotic control conditions, elevated glucose levels increased TUNEL-positive cells (Fig. 2A and B) and apoptotic markers, such as cleavage of caspase-3 and PARP (Fig. 2C). Flow cytometric analysis revealed that high glucose induced more apoptotic cell death (11.7 ± 2.2%) than normal glucose (3.6 ± 1.2%) or osmotic control (3.9 ± 1.1%) (Fig. 2D and E). In the cells exposed to high glucose, metformin treatment prevented the reduction in phosphorylation of AMPK and ACC (Fig. 2F), reduced the cleavage of caspase-3 and PARP (Fig. 2G), and diminished the number of apoptotic cells (Fig. 2H).
FIG. 2.

AMPK activation inhibits high glucose (HG)–induced apoptosis in H9c2 cells. A and B: H9c2 cells were exposed to 5 mmol/L d-glucose (Control), 30 mmol/L d-glucose (HG), or 5.5 mmol/L d-glucose plus 24.5 mmol/L mannitol for 48 h. Then, TUNEL staining was performed (A) and the number of TUNEL-positive cells (B) were quantified (n = 6; *P < 0.05 vs. control or mannitol). C: Cleaved caspase 3 (C-Casp3) and cleaved PARP (C-PARP) were determined by immunoblot analysis (n = 5; *P < 0.05 vs. control). D and E: The apoptotic ratios of H9c2 cells in different groups were determined by flow cytometry using FITC-annexin V/PI double staining. D: Representative diagram of annexin V/PI staining. E: The apoptotic cells were calculated as the ratio of annexin V+/PI− cells to total cells (n = 5; *P < 0.05 vs. control or mannitol). F: H9c2 cells were pretreated with or without metformin (Met; 1 mmol/L) for 1 h and then treated with HG (30 mmol/L) for 48 h. AMPK activation in cell lysates was determined by Western analysis of phosphorylation of AMPK and ACC (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. HG). G: Expression of C-Casp3 and C-PARP in cell lysates was detected by Western blot analysis (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. HG). H: Apoptosis was assessed by flow cytometry using annexin V/PI double staining, and the apoptotic cells were calculated as the ratio of annexin V+/PI− cells to total cells (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. HG).
AMPK activation–attenuated high glucose–induced apoptosis is dependent on autophagy.
To determine the functional role of autophagy in the development of diabetic cardiomyopathy, we assessed apoptosis after manipulation of autophagic activity in H9c2 cells. Under normal glucose conditions, metformin treatment enhanced LC3-II accumulation (Fig. 3A), and the conversion of LC3-I to LC3-II was blocked by the autophagy inhibitor 3-methylademine (3-MA) (33). In a high-glucose environment, metformin prevented the reduced LC3-II protein levels; however, this effect was attenuated by administration of 3-MA (Fig. 3A). In addition, metformin attenuated the enhancement of apoptotic markers (cleavage of caspase-3 and PARP) by high glucose, and this protective effect was abrogated by 3-MA treatment (Fig. 3B).
FIG. 3.
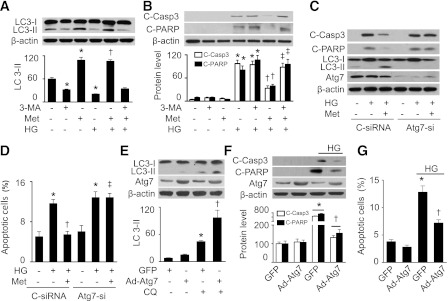
AMPK mitigates high glucose (HG)–induced apoptosis through induction of autophagy. A and B: H9c2 cells were pretreated with 3-MA (10 μmol/L) for 30 min and then treated with HG (30 mmol/L) in the presence or absence of metformin (Met; 1 mmol/L) for 48 h. Cell lysates were analyzed by Western blot to determine the expression of LC3 (A) and apoptosis markers (B) including cleaved caspase-3 (C-Casp3) and cleaved PARP (C-PARP) (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. HG, ‡P < 0.05 vs. HG/Met). C: H9c2 cells were transfected with control siRNA (C-siRNA) or Atg7 siRNA (Atg7-si) for 48 h. The cells were incubated in HG medium and treated with or without Met (1 mmol/L) for 48 h. Cell lysates were analyzed by Western blot using antibodies against C-Casp3, C-PARP, LC3B, and Atg7. D: Apoptosis was assessed by flow cytometry using annexin V/PI double staining, and the apoptotic cells were calculated as the ratio of annexin V+/PI− cells to total cells (n = 5; *P < 0.05 vs. control/C-siRNA, †P < 0.05 vs. C-siRNA/HG, ‡P < 0.05 vs. HG/Met/C-siRNA). E: H9c2 cells were infected with adenovirus encoding GFP or Atg7 for 48 h and then treated with or without CQ (5 μmol/L) for 16 h. Expression of LC3 in cell lysates was determined by Western blot (n = 5; *P < 0.05 vs. GFP, †P < 0.01 vs. CQ/GFP). F: H9c2 cells infected with GFP or Atg7 adenovirus were incubated in HG medium for 48 h. Cell lysates were subjected to Western blot to determine the expression of C-Casp3 and C-PARP (n = 5; *P < 0.05 vs. GFP, †P < 0.05 vs. GFP/HG). G: Apoptosis was assessed by flow cytometry using annexin V/PI double staining, and the apoptotic cells were calculated as the ratio of annexin V+/PI− cells to total cells (n = 5; *P < 0.05 vs. GFP, †P < 0.05 vs. GFP/HG).
To exclude nonspecific effects of 3-MA (34), we suppressed autophagy by gene silencing of Atg7. Transfection of Atg7 siRNA not only prevented the conversion of LC3-I to LC3-II under basal conditions but also inhibited metformin-enhanced LC3-II accumulation (Fig. 3C). Importantly, metformin reduced apoptotic cell death and cleavage of caspase-3 and PARP in the cells transfected with control siRNA, whereas this protective effect was attenuated in the cells transfected with Atg7 siRNA (Fig. 3C and D).
To confirm that autophagy is necessary for H9c2 cell survival, we assessed apoptosis in H9c2 cells that overexpressed Atg7 adenovirus. Overexpression of Atg7 caused an increase in autophagic flux, as the addition of CQ induced more LC3-II accumulation in the cells infected with Atg7 adenovirus than in those infected with GFP adenovirus (Fig. 3E). In addition, overexpression of Atg7 attenuated high glucose–induced apoptotic cell death and cleavage of caspase-3 and PARP (Fig. 3F and G).
Activated AMPK dissociates Beclin1 and Bcl-2 in H9c2 cells.
Reports that starvation and a Bcl-2 homology domain 3 molecular mimetic stimulate autophagy through dissociation of the Beclin1–Bcl-2 complex (9,35) prompted us to investigate the effect of AMPK on the interaction between Beclin1 and Bcl-2. Immunoprecipitation of Beclin1 followed by probing for Bcl-2 (or the reverse experiment) indicated that high glucose enhanced the association between Beclin1 and Bcl-2 and that this interaction was disrupted by metformin treatment (Fig. 4A and B). Together with the findings that elevated glucose levels reduced the autophagic flux and metformin restored autophagy in high glucose–treated H9c2 cells, our data suggest that activation of AMPK may stimulate autophagy by dissociation of the Beclin1–Bcl-2 complex.
FIG. 4.

AMPK activates JNK to regulate the association of Beclin1–Bcl-2, induce autophagy, and reduce apoptotic cell death. A and B: H9c2 cells were pretreated with or without metformin (Met; 1 mmol/L) for 1 h and then incubated in high glucose (HG; 30 mmol/L) medium for 48 h. Beclin1 (A) or Bcl-2 (B) was immunoprecipitated (IP) from cell lysates, and Bcl-2 or Beclin1 in the immunoprecipitates was detected by Western blot (IB) (n = 4; *P < 0.05 vs. control [Con], †P < 0.05 vs. HG). C: H9c2 cells were treated with HG (30 mmol/L) for 24 or 36 h in the presence or absence of metformin (Met; 1 mmol/L). Activation of JNK1 was determined by Western blot using antiphospho-JNK1 (Thr183/Tyr185) antibody (n = 5; *P < 0.05 vs. Con, †P < 0.05 vs. HG). C, right: H9c2 cells were treated with metformin (1 mmol/L) under normal or HG for 36 h. JNK1 phosphorylation was determined by Western blot (n = 3; *P < 0.05 vs. Con, †P < 0.05 vs. HG). D: Cell lysates were immunoprecipitated with anti–Bcl-2 antibody and then subjected to immunoblot using P-Bcl-2 (Ser70) and Bcl-2 antibodies (n = 5; *P < 0.05 vs. Con, †P < 0.05 vs. HG). D, right: H9c2 cells were treated with Met (1 mmol/L) under normal glucose or HG conditions for 36 h. Phosphorylation of Bcl-2 was determined by immunoprecipitation and Western blotting (n = 3; *P < 0.05 vs. Con, †P < 0.05 vs. HG). E: H9c2 cells were pretreated with SP600125 (SP; 50 μmol/L) for 30 min and then incubated in HG (30 mmol/L) medium in the presence or absence of Met (1 mmol/L) for 48 h. Total and phospho-JNK1 were detected by Western blot. In addition, cell lysates were subjected to immunoprecipitation with anti-Beclin1 antibody, and the immunoprecipitates were subjected to Western blotting with anti–Bcl-2 antibody (n = 5; *P < 0.05 vs. Con, †P < 0.05 vs. HG, ‡P < 0.01 vs. HG/Met). F: Expression of LC3 was analyzed by Western blot (n = 5; *P < 0.05 vs. Con, †P < 0.05 vs. HG, ‡P < 0.01 vs. HG/Met). G: H9c2 cells were transfected with LacZ or CA-JNK1 plasmid for 24 h and then incubated in HG medium for 36 h. Expression of JNK1 was detected by Western blot. Cell lysates were subjected to immunoprecipitation with anti-Beclin1 antibody, and then Bcl-2 was detected in the immunoprecipitates by Western blotting (n = 5; *P < 0.05 vs. LAZ/Con, †P < 0.05 vs. HG/LacZ). H: Expression of LC3 was analyzed by Western blot (n = 5; *P < 0.05 vs. LAZ/Con, †P < 0.05 vs. HG/LacZ). I: H9c2 cells were pretreated with SP (50 μmol/L) for 30 min followed by treatment with HG (30 mmol/L) in the presence or absence of Met (1 mmol/L) for 48 h. Cell lysates were subjected to Western blot using anti–C-Casp3 and anti–C-PARP antibodies (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. HG, ‡P < 0.05 vs. HG/Met). J: Apoptosis was assessed by flow cytometry using annexin V-PI double staining, and the apoptotic cells were calculated as the ratio of annexin V+/PI− cells to total cells (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. HG, ‡P < 0.05 vs. HG/Met).
JNK1 regulates AMPK-mediated dissociation of the Beclin1–Bcl-2 complex, induction of autophagy, and suppression of apoptosis.
Since activation of Jun NH2-terminal kinase 1 (JNK1) results in phosphorylation of Bcl-2 (36,37) and disruption of the Beclin1–Bcl-2 complex (38), we first analyzed JNK1 phosphorylation in H9c2 cells. The cells were exposed to high glucose for 24–36 h. Reduced JNK1 phosphorylation was observed at 24 h and reached its minimum at 36 h. This reduction was prevented by metformin treatment (Fig. 4C). In line with the alteration in JNK1 activation, high glucose decreased Bcl-2 phosphorylation at Ser70, and this effect was depressed by administration of metformin (Fig. 4D).
Next, using gain- and loss-of-function approaches, the role of JNK1 in regulating the interaction between Beclin1 and Bcl-2 was determined. Under normal and high-glucose conditions, the JNK1 inhibitor SP600125 reduced JNK1 phosphorylation (Fig. 4E). Coimmunoprecipitation experiments indicated that metformin reduced the association between Beclin1 and Bcl-2 in normal glucose medium, whereas high glucose enhanced the association between these two proteins and this interaction was disrupted by metformin treatment. Administration of SP600125 abrogated the effects of metformin on the Beclin1–Bcl-2 complex, as Beclin1 and Bcl-2 associated under these conditions (Fig. 4E). As a result, metformin-enhanced autophagy was attenuated (Fig. 4F). Transfection of H9c2 cells with CA-JNK1 plasmid disrupted the association between Beclin1 and Bcl-2 and recovered the accumulation of LC3-II under high-glucose conditions (Fig. 4G and H). Importantly, inhibition of JNK1 by SP600125 blocked the reduction of apoptotic cell number (Fig. 4I) and apoptotic markers (i.e., cleavage of caspase-3 and PARP) caused by metformin (Fig. 4J). These results suggest that activation of JNK1–Bcl-2 signaling is critical for AMPK to induce autophagy to protect against apoptotic cell death.
AMPK directly phosphorylates JNK1.
We next determined if AMPK directly caused JNK1 phosphorylation. As depicted in Fig. 5A, metformin increased AMPK phosphorylation in a dose-dependent manner, which was accompanied by an increase in JNK1 phosphorylation. Incubation of purified recombinant AMPK with recombinant JNK1 dose-dependently increased JNK1 phosphorylation (Fig. 5B). Exposure of H9c2 cells to elevated glucose levels, which inhibited JNK1 phosphorylation, disrupted the interaction of AMPK and JNK1. Consistently, the disruption was depressed by metformin treatment, indicating a direct interaction of AMPK with JNK1 (Fig. 5C).
FIG. 5.

AMPK phosphorylates JNK1. A: H9c2 cells were treated with indicated concentrations of metformin (Met) for 36 h. Cells lysates were subjected to Western analysis of P-AMPK (Thr172) and P-JNK1 (Thr183/Tyr185) (n = 3; *P < 0.05 vs. control [Con]). B: Recombinant JNK1 (500 ng) was incubated with indicated concentrations of recombinant AMPK. Phosphorylation of JNK1 was determined by Western blotting (n = 3; *P < 0.05 vs. control). C: H9c2 cells were treated with Met (1 mmol/L) for 36 h under normal glucose and HG. The interaction of AMPK and JNK1 was measured by immunoprecipitation (IP) and Western blotting (IB) (n = 3; *P < 0.05 vs. control, †P < 0.05 vs. HG). D: H9c2 cells were transfected with control siRNA (C-siRNA) or mTOR siRNA (mTOR-si) for 48 h, and then incubated in HG (30 mmol/L) medium for 48 h. The association of Beclin1 and Bcl-2 was determined by immunoprecipitation and Western blotting (n = 3; NS, not significant).
We recently reported that hyperglycemia activated the mammalian target of rapamycin (mTOR) signaling pathway and inhibited cardiac autophagy, and activation of AMPK by metformin was associated with inhibition of mTOR signaling and restoration of autophagy (11). In this experiment, we further determined if mTOR affects the Bcl-2–Beclin1 interaction. As shown in Fig. 5D, inhibition of mTOR did not disrupt the association between Beclin1 and Bcl-2 induced by elevated glucose levels.
Metformin induces autophagy through disruption of the Beclin1–Bcl2 complex in diabetic hearts.
Four months after diabetes induction, diabetic mice exhibited significantly higher blood glucose levels than their control counterparts (469 ± 27 vs. 108 ± 5 mg/dL; P < 0.001, n = 11 per group). Administration of metformin failed to normalize blood glucose (451 ± 28 mg/dL; n = 9) in diabetic mice. In FVB mice, metformin treatment increased AMPK and ACC phosphorylation (Fig. 6A), activated JNK1 and Bcl-2, disrupted the association between Beclin1 and Bcl-2 (Fig. 6B), and elevated LC3-II protein levels (Fig. 6C). Compared with FVB controls, diabetes decreased phosphorylation of AMPK and ACC (Fig. 6A), and the phosphorylation levels were restored with metformin treatment (Fig. 6A). Hyperglycemia reduced JNK1 and Bcl-2 phosphorylation and enhanced the interaction between Beclin1 and Bcl-2. Chronic metformin stimulated JNK1 and Bcl-2, leading to dissociation of Beclin1 and Bcl-2 (Fig. 6B). As a result, administration of metformin enhanced LC3-II protein levels in diabetic mice (Fig. 6C).
FIG. 6.

Activation of AMPK disrupts Bcl-2–Beclin1 association and restores autophagy in diabetic hearts. A: Phosphorylation of AMPK and ACC in cardiac tissues from FVB (control), metformin-treated FVB (Met), STZ-treated (STZ), and metformin-treated STZ (STZ/Met) mice was detected by Western blot using P-AMPK (Thr172) and P-ACC (Ser79) antibodies and quantified by densitometry (n = 6 per group; *P < 0.05 vs. Con, †P < 0.05 vs. STZ). B: Expression of total and phospho-JNK1 in cardiac tissues was analyzed by Western blotting. In addition, cardiac tissues were immunoprecipitated (IP) with an anti–Bcl-2 antibody, and then immunoprecipitates were probed by immunoblot (IB) using P-Bcl-2 (Ser70), Beclin1, and Bcl-2 antibodies. The result shown is representative of three independent experiments. C: Cardiac tissues were subjected to Western blotting to determine the expression of LC3 using a specific antibody (n = 5 in each group; *P < 0.05 vs. Con, †P < 0.05 vs. STZ). D: AMPK activity in cardiac tissues was assayed as described in research design and methods (n = 5; *P < 0.05 vs. WT/Con, †P < 0.05 vs. WT/STZ, ‡P < 0.05 vs. WT/STZ/Met). E: FVB (WT) and DN-AMPKα2 (DN) transgenic mice were treated with or without STZ. LC3-II protein levels in cardiac tissues were determined by Western blotting (n = 5; *P < 0.05 vs. WT). F: Diabetic WT and DN mice were treated with or without metformin (Met), and then heart homogenates from these mice were subjected to Western blot to determine the expression of LC3 (n = 5; *P < 0.05 vs. WT/STZ, †P < 0.05 vs. WT/STZ/Met). G: Phosphorylation of Akt at Ser437 in cardiac tissues from FVB control (Con), STZ, and STZ/Met mice was detected by Western blot and quantified by densitometry (n = 6 per group; *P < 0.05 vs. Con, †P < 0.05 vs. STZ).
To establish the role of AMPK in the regulation of cardiac autophagy in vivo, we induced diabetes in FVB (WT) and DN-AMPKα2 (DN) transgenic mice. Four months after diabetes induction, the diabetic mice had higher blood glucose than their nondiabetic control subjects (WT: 477 ± 21 vs. 113 ± 2 mg/dL, P < 0.001, n = 8 in each group; DN: 483 ± 15 vs. 125 ± 9 mg/dL, P < 0.001, n = 9 in each group). AMPKα2 activity was inhibited by overexpression of DN-AMPKα2 and STZ-induced diabetes. Diabetes did not cause a further reduction of AMPKα2 activity in DN hearts. Chronic administration of metformin restored AMPK activity in WT STZ mice but this effect was absent in DN STZ mice (Fig. 6D). Although DN mice have a normal cardiac phenotype, their hearts exhibited lower LC3-II levels (Fig. 6E), indicating a reduction in cardiac autophagy. The hyperglycemia reduced LC3-II accumulation in both WT and DN mice (Fig. 6E). Although metformin failed to normalize blood glucose levels in diabetic mice, this treatment restored LC3-II protein levels in WT diabetic mice. Notably, overexpression of DN-AMPKα2 in the heart depressed the protective effect of metformin on autophagy (Fig. 6F). We also determined if activation of AMPK by metformin improves insulin signaling. As shown in Fig. 6G, metformin treatment prevented diabetes-reduced Akt phosphorylation.
Activation of AMPK-enhanced autophagy is associated with reduced cardiomyocyte apoptosis in diabetic mice.
We next determined the effect of autophagy induction on cardiac apoptosis in vivo. In FVB mice, metformin treatment had no effect on the number of TUNEL-positive cells and cleavage of caspase-3 and PARP (Fig. 7A–C). TUNEL staining demonstrated that TUNEL-positive cells were seldom identified in control mouse hearts, but numerous TUNEL-positive cells were observed in diabetic mouse hearts. The increase was attenuated by chronic metformin treatment (Fig. 7A and B). Consistently, Western analysis of cardiac homogenates revealed a significant increase in cleavage of caspase-3 and PARP in diabetic mouse hearts, and metformin treatment depressed the observed increase in apoptosis markers in diabetic mice (Fig. 7D). Overexpression of DN-AMPKα2 did not increase these apoptotic markers in nondiabetic mice nor did this mutant exaggerate apoptosis in diabetic DN hearts compared with that in WT hearts (Fig. 7E). Administration of metformin prevented diabetes-enhanced cleavage of caspase-3 and PARP in diabetic WT mice but not in diabetic DN mice (Fig. 7F).
FIG. 7.
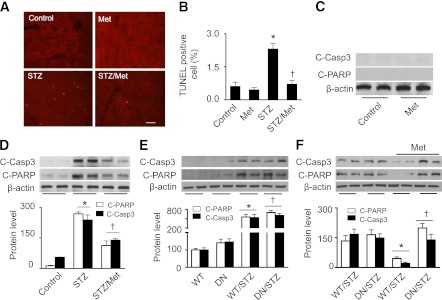
Activation of AMPK protects against apoptosis in diabetic hearts. A: Representative images of TUNEL staining. Apoptotic cells in heart sections from control, metformin-treated FVB (Met), STZ-treated (STZ), and metformin-treated STZ (STZ/Met) mice were labeled by TUNEL staining. B: The number of TUNEL-positive cells is indicated in the bar graph (n = 6 per group; *P < 0.05 vs. control, †P < 0.05 vs. STZ). C and D: Western analysis of cleaved caspase-3 (C-Caspa3) and cleaved PARP (C-PARP) in cardiac homogenates (n = 5; *P < 0.05 vs. control, †P < 0.05 vs. STZ). E: FVB (WT) and DN-AMPKα2 (DN) transgenic mice were treated with or without STZ, and then cardiac tissues were subjected to Western blot analysis to determine the expression of C-Casp3 and C-PARP (n = 5 in each group; *P < 0.05 vs. WT, †P < 0.05 vs. DN). F: Diabetic WT and DN mice were treated with or without metformin (Met), and then heart homogenates were subjected to Western blot to determine the expression of C-Casp3 and C-PARP (n = 5; *P < 0.05 vs. WT/STZ, †P < 0.05 vs. WT/STZ/Met).
Metformin-restored autophagy improves cardiac structure and function in diabetic mice.
Chronic administration of metformin had no effect on collagen I deposition (Fig. 8A and B) and cardiac functions (Fig. 8C–E) in nondiabetic mice. Compared with control hearts, diabetic hearts exhibited increased deposition of collagen I, and this effect was attenuated by metformin treatment (Fig. 8A and B). LV systolic and diastolic functions were determined by analysis of maximal LV-developed pressure, calculated dp/dtmax (maximal change in pressure per unit time), and dp/dt minimum (dp/dtmin). In comparison with FVB control mice, diabetic mice exhibited a significant reduction in LV-developed pressure (Fig. 8C) and dp/dtmax (Fig. 8D). Chronic metformin restored both LV-developed pressure and dp/dtmax (Fig. 8C and D) to wild-type levels. In addition, dp/dtmin, a parameter that reflects LV diastolic function, was impaired in diabetic mice relative to FVB control mice. This impairment was prevented by chronic administration of metformin (Fig. 8E).
FIG. 8.
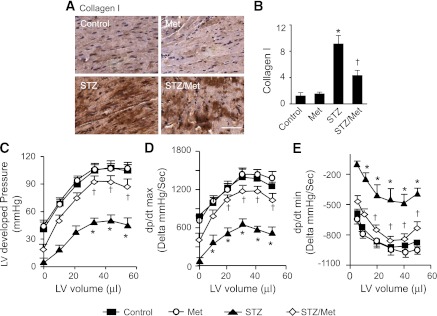
Metformin ameliorates cardiac fibrosis and improves cardiac function in diabetic hearts. A: Immunohistochemical staining for collagen I in cardiac tissues. Heart sections from control, metformin-treated FVB (Met), STZ-treated (STZ), and metformin-treated STZ (Met/STZ) mice were stained with antibody against collagen I, and the nuclei were counterstained with hemotoxylin. B: Quantitative analysis of collagen I–stained areas is shown by the bar graph (n = 6 in each group; *P < 0.05 vs. control, †P < 0.05 vs. STZ). C–E: The relationship between LV-developed pressure and volume in control, STZ, and STZ/Met mice was determined by Langendorff perfusion analysis. The developed pressure was depressed in STZ mice, and this depression was prevented by metformin treatment (C). Metformin improved LV dp/dtmax (D) and dp/dtmin (E) in STZ mice (n = 6 in each group; *P < 0.05 vs. control, †P < 0.05 vs. STZ).
DISCUSSION
In the current study, we demonstrated that hyperglycemia inhibits JNK–Bcl-2 signaling and promotes the interaction between Beclin1 and Bcl-2. Concomitantly, high levels of glucose induce apoptosis and suppress cardiac autophagy. Activation of AMPK stimulates JNK1, which mediates Bcl-2 phosphorylation and subsequent Beclin1–Bcl-2 disassociation, leading to the restoration of cardiac autophagy and protection against cardiac apoptosis. As a result, cardiac structure and function are improved in diabetic mice. Our findings suggest that activation of JNK1–Bcl-2 signaling and subsequent disruption of the Beclin1–Bcl-2 complex may be an essential mechanism for AMPK to regulate the switch between autophagy and apoptotic cell death pathways under diabetic conditions.
Cardiomyopathy in diabetes is characterized by decreased muscle mass, interstitial fibrosis, and impaired ventricular function. Since adult cardiomyocytes rarely proliferate, the loss of cardiomyocytes would eventually lead to compromised cardiac function. In STZ-induced diabetic mice, hyperglycemia enhanced the number of TUNEL-positive cells and cleavage of caspase-3 and PARP, which is associated with increased deposition of collagen I and impaired systolic and diastolic cardiac function. Chronic administration of metformin, a well-known AMPK activator, protected against apoptotic cell death, resulting in reduced cardiac fibrosis and improved cardiac function. More important, cardiac autophagy was suppressed in diabetic mice. Activation of AMPK by chronic metformin treatment prevented diabetes-suppressed cardiac autophagy. These observations provide strong evidence in support of the hypothesis that cardiomyocyte apoptosis is a critical event in the development of diabetic cardiomyopathy. These findings also suggest that the interplay between autophagy and apoptotic cell death pathways is important in the pathogenesis of diabetic cardiomyopathy.
Although in theory autophagy may lead to cell death through excessive self-digestion and degradation of essential cellular constituents, this process has actually been reported to protect cells from apoptosis (39). For example, increased autophagy promotes cell survival under conditions of nutrient deprivation or growth factor withdrawal through inhibition of apoptosis (40,41). However, a recent study demonstrates that high glucose directly inhibits autophagic flux in neonatal rat cardiomyocytes, and in these cells, the reduction of autophagy appears to be an adaptive response that functions to limit high glucose–induced cardiomyocyte injury (42). Neonatal cardiomyocytes have been reported to behave substantially different from adult cardiomyocytes (43). Especially, autophagy is upregulated in the neonatal cardiac tissue during the perinatal period of relative starvation (44). Thus, autophagy could be either protective or detrimental depending on the cell type and cellular environment. In our study, enhanced autophagy by metformin or overexpression of Atg7 protein attenuated hyperglycemia-induced apoptotic cell death, cardiac fibrosis, and cardiac dysfunction, but these protective actions of metformin are absent in mice deficient in AMPKα2. These data indicate that induction of autophagy by activated AMPK constitutes a protective mechanism against hyperglycemic injury to cardiomyocytes.
The interaction between the antiapoptotic protein Bcl-2 and the autophagy protein Beclin1 has been shown to regulate the switch between the autophagic and apoptotic machinery (38). Beclin1 is part of the class III PI3K complex that is required for the formation of the autophagic vesicle, and interference with Beclin1 prevents autophagy induction (45). Beclin1 is an interacting protein of Bcl-2 (46). Binding of Bcl-2 to Beclin1 inhibits Beclin1-mediated autophagy via sequestration of Beclin1 away from class III PI3K (35,47). Here, we observed a strong interaction between Beclin1 and Bcl-2 in H9c2 cells treated with elevated glucose levels and in hearts from diabetic animals. Treatment of cells and animals with metformin to activate AMPK resulted in disruption of the association between Beclin1 and Bcl-2, and the free Beclin1 bound to class III PI3K to form the kinase complex (9), which is essential for the induction of autophagy. The induction of autophagy degrades and recycles cytoplasmic components and selectively removes damaged mitochondria, serving as a cytoprotective mechanism. In addition, phosphorylated Bcl-2 could preserve the integrity of the mitochondrial outer membrane and prevent proapoptotic proteins from releasing into the cytoplasm (48,49), thus protecting against apoptosis. Therefore, dissociation of Bcl-2 from Beclin1 may be an important mechanism by which AMPK activation restores autophagy and protects against apoptosis under diabetic conditions.
In agreement with the finding that disruption of the association between Beclin1 and Bcl-2 by starvation is dependent on JNK1-mediated phosphorylation of Bcl-2 on multiple residues of the protein (38,50), JNK1–Bcl-2 signaling also regulates the interaction between Beclin1 and Bcl-2 in the development of diabetic cardiomyopathy. Since AMPK can directly phosphorylate JNK1, inhibition of AMPK in the presence of hyperglycemia decreases phosphorylation of JNK1 and Bcl-2, enhances the interaction between Beclin1 and Bcl-2, and suppresses autophagic activity. Activation of AMPK by metformin or overexpression of constitutively active JNK1 stimulated the JNK1–Bcl-2 pathway, leading to dissociation of the Beclin1–Bcl-2 complex and restoration of autophagy. These effects were blocked by inhibition of JNK1. More importantly, suppression of autophagy by inhibition of JNK1 prevented the apoptotic cell death induced by high-glucose levels, indicating that JNK1–Bcl-2 signaling is a crucial pathway involved in AMPK promotion of cardiomyocyte survival under diabetic conditions.
In conclusion, we demonstrated that AMPK activation attenuates diabetic cardiomyopathy through regulation of the switch between autophagy and apoptotic machinery. This effect is attributable to JNK-mediated Bcl-2 phosphorylation and subsequent Beclin1–Bcl-2 disassociation. Our results provide new insight into the regulation of autophagy in the development of diabetic cardiomyopathy and suggest that specific modulation of autophagy, perhaps by stimulation of AMPK, may represent a novel approach for the treatment of heart failure in diabetic patients.
ACKNOWLEDGMENTS
This study was supported by funding from the following sources: the National Institutes of Health (HL-079584, HL-080499, HL-074399, HL-089920, HL-096032, and HL-110488 to M.Z. and 1P20-RR-024215-01 to Z.X. and M.Z.), the American Heart Association Scientist Development Grant (Z.X.), and the Oklahoma Center for the Advancement of Science and Technology (Z.X.). M.H.Z. is a recipient of the National Established Investigator Award of the American Heart Association.
No potential conflicts of interest relevant to this article were reported.
C.H. researched data and prepared the manuscript. H.Z. and H.L. researched data. M.-H.Z. conceived the project and wrote the paper. Z.X. designed the experiments, analyzed data, and wrote the manuscript. Z.X. is the guarantor of this work, had full access to all the data, and takes full responsibility for the integrity of data and the accuracy of data analysis.
The authors thank Dr. Rong Tian (University of Washington, Seattle, WA) for providing DN-AMPKα2 cardiac-specific transgenic mice.
REFERENCES
- 1.Francis GS. Diabetic cardiomyopathy: fact or fiction? Heart 2001;85:247–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Picano E. Diabetic cardiomyopathy. the importance of being earliest. J Am Coll Cardiol 2003;42:454–457 [DOI] [PubMed] [Google Scholar]
- 3.Frustaci A, Kajstura J, Chimenti C, et al. Myocardial cell death in human diabetes. Circ Res 2000;87:1123–1132 [DOI] [PubMed] [Google Scholar]
- 4.Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 2002;51:1938–1948 [DOI] [PubMed] [Google Scholar]
- 5.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007;8:931–937 [DOI] [PubMed] [Google Scholar]
- 6.Martinet W, Knaapen MW, Kockx MM, De Meyer GR. Autophagy in cardiovascular disease. Trends Mol Med 2007;13:482–491 [DOI] [PubMed] [Google Scholar]
- 7.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007;8:741–752 [DOI] [PubMed] [Google Scholar]
- 8.Nishida K, Yamaguchi O, Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circ Res 2008;103:343–351 [DOI] [PubMed] [Google Scholar]
- 9.Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007;26:2527–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 2011;7:1254–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Z, Lau K, Eby B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011;60:1770–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 2003;144:5179–5183 [DOI] [PubMed] [Google Scholar]
- 13.Tian R, Musi N, D’Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation 2001;104:1664–1669 [DOI] [PubMed] [Google Scholar]
- 14.Shibata R, Ouchi N, Ito M, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med 2004;10:1384–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shibata R, Ouchi N, Kihara S, Sato K, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis in response to tissue ischemia through stimulation of amp-activated protein kinase signaling. J Biol Chem 2004;279:28670–28674 [DOI] [PubMed] [Google Scholar]
- 16.Hickson-Bick DL, Buja LM, McMillin JB. Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J Mol Cell Cardiol 2000;32:511–519 [DOI] [PubMed] [Google Scholar]
- 17.Meijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy 2007;3:238–240 [DOI] [PubMed] [Google Scholar]
- 18.Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007;100:914–922 [DOI] [PubMed] [Google Scholar]
- 19.Russell RR, 3rd, Li J, Coven DL, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest 2004;114:495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011;331:456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie Z, Zhang J, Wu J, Viollet B, Zou MH. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes 2008;57:3222–3230 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Xie Z, Singh M, Singh K. Osteopontin modulates myocardial hypertrophy in response to chronic pressure overload in mice. Hypertension 2004;44:826–831 [DOI] [PubMed] [Google Scholar]
- 24.Xing Y, Musi N, Fujii N, et al. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem 2003;278:28372–28377 [DOI] [PubMed] [Google Scholar]
- 25.Eberli FR, Sam F, Ngoy S, Apstein CS, Colucci WS. Left-ventricular structural and functional remodeling in the mouse after myocardial infarction: assessment with the isovolumetrically-contracting Langendorff heart. J Mol Cell Cardiol 1998;30:1443–1447 [DOI] [PubMed] [Google Scholar]
- 26.Zou MH, Li H, He C, Lin M, Lyons TJ, Xie Z. Tyrosine nitration of prostacyclin synthase is associated with enhanced retinal cell apoptosis in diabetes. Am J Pathol 2011;179:2835–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie Z, Singh M, Siwik DA, Joyner WL, Singh K. Osteopontin inhibits interleukin-1beta-stimulated increases in matrix metalloproteinase activity in adult rat cardiac fibroblasts: role of protein kinase C-zeta. J Biol Chem 2003;278:48546–48552 [DOI] [PubMed] [Google Scholar]
- 28.He C, Choi HC, Xie Z. Enhanced tyrosine nitration of prostacyclin synthase is associated with increased inflammation in atherosclerotic carotid arteries from type 2 diabetic patients. Am J Pathol 2010;176:2542–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie Z, Dong Y, Zhang J, Scholz R, Neumann D, Zou MH. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol Cell Biol 2009;29:3582–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 2008;117:952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010;140:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwai-Kanai E, Yuan H, Huang C, et al. A method to measure cardiac autophagic flux in vivo. Autophagy 2008;4:322–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stroikin Y, Dalen H, Lööf S, Terman A. Inhibition of autophagy with 3-methyladenine results in impaired turnover of lysosomes and accumulation of lipofuscin-like material. Eur J Cell Biol 2004;83:583–590 [DOI] [PubMed] [Google Scholar]
- 34.Caro LH, Plomp PJ, Wolvetang EJ, Kerkhof C, Meijer AJ. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur J Biochem 1988;175:325–329 [DOI] [PubMed] [Google Scholar]
- 35.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005;122:927–939 [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 1999;19:8469–8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blagosklonny MV. Unwinding the loop of Bcl-2 phosphorylation. Leukemia 2001;15:869–874 [DOI] [PubMed] [Google Scholar]
- 38.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008;30:678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis 2008;13:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boya P, González-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005;25:1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lum JJ, Bauer DE, Kong M, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005;120:237–248 [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi S, Xu X, Chen K, Liang Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy 2012;8:577–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothen-Rutishauser BM, Ehler E, Perriard E, Messerli JM, Perriard JC. Different behaviour of the non-sarcomeric cytoskeleton in neonatal and adult rat cardiomyocytes. J Mol Cell Cardiol 1998;30:19–31 [DOI] [PubMed] [Google Scholar]
- 44.Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature 2004;432:1032–1036 [DOI] [PubMed] [Google Scholar]
- 45.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005;115:2679–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999;402:672–676 [DOI] [PubMed] [Google Scholar]
- 47.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008;4:600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997;275:1129–1132 [DOI] [PubMed] [Google Scholar]
- 49.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997;275:1132–1136 [DOI] [PubMed] [Google Scholar]
- 50.Saeki K, Yuo A, Okuma E, et al. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ 2000;7:1263–1269 [DOI] [PubMed] [Google Scholar]