

Abstract
Glucagon is a critical regulator of glucose homeostasis; however, mechanisms regulating glucagon action and α-cell function and number are incompletely understood. To elucidate the role of the hepatic glucagon receptor (Gcgr) in glucagon action, we generated mice with hepatocyte-specific deletion of the glucagon receptor. GcgrHep−/− mice exhibited reductions in fasting blood glucose and improvements in insulin sensitivity and glucose tolerance compared with wild-type controls, similar in magnitude to changes observed in Gcgr−/− mice. Despite preservation of islet Gcgr signaling, GcgrHep−/− mice developed hyperglucagonemia and α-cell hyperplasia. To investigate mechanisms by which signaling through the Gcgr regulates α-cell mass, wild-type islets were transplanted into Gcgr−/− or GcgrHep−/− mice. Wild-type islets beneath the renal capsule of Gcgr−/− or GcgrHep−/− mice exhibited an increased rate of α-cell proliferation and expansion of α-cell area, consistent with changes exhibited by endogenous α-cells in Gcgr−/− and GcgrHep−/− pancreata. These results suggest that a circulating factor generated after disruption of hepatic Gcgr signaling can increase α-cell proliferation independent of direct pancreatic input. Identification of novel factors regulating α-cell proliferation and mass may facilitate the generation and expansion of α-cells for transdifferentiation into β-cells and the treatment of diabetes.
The islets of Langerhans comprise distinct populations of differentiated endocrine cells whose functions are critical for maintenance of metabolic homeostasis. Considerable progress has been made in understanding the control of β-cell growth, function, and survival (1). Moreover, advances in understanding the developmental and adaptive control of β-cell formation coupled with insights gleaned from studies of adult and embryonic pancreatic endocrine stem cells have yielded new information regarding the cellular origin and formation of differentiated adult β-cells.
In contrast, much less is known about mechanisms governing the development, growth, and survival of the glucagon-secreting α-cell (2). Glucagon secretion is stimulated by exercise or hypoglycemia; conversely, glucagon secretion is suppressed during conditions of fuel abundance. However, development of diabetes is often associated with failure to suppress glucagon secretion in the fed state (3,4); hence, therapeutic efforts to suppress α-cell function for the treatment of type 2 diabetes are ongoing (4). Moreover, α-cell mass appears dynamic in the context of diabetes, with expansion of α-cell mass noted in the diabetic primate (5) and human pancreas (6).
The observation that functionally differentiated β-cells can arise from α-cell precursors (7–9) has engendered additional interest in the control of α-cell growth. α-Cell hyperplasia is frequently observed in settings of partial or complete glucagon deficiency (10,11) or resistance to glucagon action (12). Mice with targeted disruption of Pcsk2 exhibit impaired generation of bioactive glucagon, mild hypoglycemia, and marked α-cell proliferation, findings that are rapidly reversed by glucagon administration (13). Similarly, transgenic expression of Pax4 in pancreatic endocrine cells results in relative glucagon deficiency and compensatory α-islet cell proliferation; exogenous glucagon administration in this setting also reduces α-cell proliferation (7). Both transient genetic reduction of glucagon receptor (Gcgr) expression in normoglycemic or diabetic mice using antisense oligonucleotides or complete genetic germline disruption of GCGR signaling are associated with α-cell hyperplasia (10,14). Collectively, these findings raise the possibility that α-cell transdifferentiation toward a β-cell phenotype may represent an alternative strategy for replenishment of β-cell mass in vivo.
Despite evidence linking reduction in GCGR signaling to α-cell hyperplasia, the precise tissues and signals important for stimulation of α-cell proliferation remain unknown. Because levels of GLP-1, a potent stimulator of islet cell proliferation, are extremely high in mice with partial or complete attenuation of Gcgr signaling (10,14), we analyzed α-cell mass in Gcgr−/−:Glp1r−/− mice (15). Although elimination of the Glp1r in Gcgr−/− mice reversed improvements in β-cell function, fasting glycemia, and inhibition of gastric emptying, Gcgr−/−:Glp1r−/− mice continued to exhibit marked islet and α-cell hyperplasia. Similarly, mice with liver-specific disruption of Gsα exhibit α-cell hyperplasia despite genetic elimination of the Glp1r (16). Hence, although GLP-1 controls α-cell function, the Glp1r is not required for development of α-cell hyperplasia in Gcgr−/− mice.
To elucidate mechanisms and tissues through which reduction in GCGR signaling promotes α-cell hyperplasia, we have assessed the importance of the liver and the tissue environment. Surprisingly, selective elimination of the hepatic Gcgr in GcgrHep−/− mice was sufficient to recapitulate the phenotype of α-cell hyperplasia in the endogenous pancreas, suggesting that reduction of GCGR signaling in liver originates one or more signals that promote α-cell proliferation. Remarkably, transplantation of Gcgr+/+ islets under the kidney capsule of Gcgr−/− or GcgrHep−/− mice resulted in stimulation of α-cell proliferation and hyperplasia in transplanted islets, implying the existence of one or more circulating factors capable of promoting α-cell hyperplasia independent of the normal pancreatic microenvironment.
RESEARCH DESIGN AND METHODS
Animals.
Gcgr−/− mice on a C57BL/6 background were maintained as previously reported and generated by heterozygous–heterozygous breeding (10). Albumin-Cre (stock 003574) (17) and FLPe (stock 005703) transgenic mice were from Jackson Laboratory. GcgrFlox chimeric mice carrying one loxP site between exons 5 and 6 and two loxP sites in a neomycin cassette inserted between exons 12 and 13 of the Gcgr gene were generated in the C57Bl/6 background, and the neomycin cassette was removed using the FLPe-FRT system. GcgrHep−/− mice were generated by breeding GcgrFlox mice and Albumin-Cre mice. All animals were maintained on a standard rodent chow under a normal 12-h light/12-h dark cycle. All wild-type Gcgr+/+ control groups used were littermates; 1% cholic acid diet was obtained from Harlan. Experiments were conducted according to protocols and guidelines approved by the Mount Sinai Hospital Animal Care Committee or the Vanderbilt University Institutional Animal Care and Use Committee. The metabolic phenotype observed in GcgrHep−/− mice was similar in males and females.
Glucose challenge and measurement of plasma metabolites.
Glucose tolerance tests were performed as described (18,19). For plasma insulin and glucagon determinations, blood samples (100 μL) were drawn from the tail vein during the 0-min and 15-min time periods after glucose administration in a heparinized tube. Plasma insulin was measured using a mouse insulin ELISA kit (Alpco), and plasma levels of glucagon, GIP, GLP-1, and interleukin-6 were measured using a mouse Milliplex assay (Millipore). SDF-1 levels were measured using the mouse CXCL12/SDF-1 α Quantikine ELISA Kit from R&D system. Plasma levels of total bile acids were measured with a total bile acids test kit (Enzyme Cycling) from Diazyme.
Glucagon and insulin tolerance tests.
For glucagon challenge, mice were fasted for 5 h and then injected intraperitoneally with glucagon (16 mg/kg body weight). To test the effect of insulin on plasma glucose (insulin tolerance test), mice fasted for 5 h were administered 0.7 units/kg of human insulin (Novolin GE; Novo Nordisk) by intraperitoneal injection.
Hyperinsulinemic euglycemic clamp.
Hyperinsulinemic euglycemic clamps were performed in conscious unrestrained mice as described, 5–6 days after catheter implantation (21).
Histology.
Pancreata were weighed, fixed in 10% neutral buffered formalin solution for 48 h, and then embedded in paraffin or 4% paraformaldehyde/0.1 mol/L PBS as described (20,22). For assessment of α-cell and β-cell mass, pancreatic sections were immunostained for insulin and/or glucagon, followed by scanning using the ScanScope CS system (Aperio Technologies) at ×20 magnification. Digital images were analyzed with ScanScope software (Aperio Technologies). Immunofluorescent imaging was performed using an Olympus Epifluorescent Microscope or a ScanScope FL Digital Slide Scanner (Aperio, Vista, CA). Cell counts were performed using Meta Imaging Series 7.1 (Metamorph) software. The α-cell and β-cell mass analyses were performed with Spectrum Analysis software (Aperio) using mass analyses algorithm normalizing glucagon or insulin staining area to total amylase staining area as an approximation of total pancreas area.
RNA preparation and real-time PCR.
RNA was extracted from liver using Trizol Reagent (Sigma-Aldrich, St. Louis, MO). Real-time quantitative PCR reactions were performed using the TaqMan Gene Expression Assay Universal PCR Master Mix (Applied Biosystems, Foster City, CA) and an ABI Prism 7900 Sequence Detection System (Applied Biosystems). Values for mRNA transcripts were normalized to the levels of PPia, which were not different between groups.
Islet transplantation experiments.
Pancreatic islets were isolated from wild-type and Gcgr−/− mice for transplantion beneath the renal capsule as described (20,22). Isolated hand-picked islets were incubated overnight in Roswell Park Memorial Institute medium (RPMI) 5.6 mmol/L glucose with 10% FBS before transplantation. Pancreatic islets isolated from 14 week-old wild-type or Gcgr−/− animals were transplanted under the renal capsule of 14-week-old recipients (wild-type, Gcgr−/− GcgrFlox, and GcgrHep−/− mice). After 1, 4, or 8 weeks, the kidneys containing the islet grafts were removed and processed as described. Thin sections (5 µm) were stained for glucagon, insulin, and Ki67 as described (22).
Hormone content in pancreas, isolated islets, and transplanted islet grafts.
Whole pancreata were dissected in 0.1 mol/L PBS and weighed. For isolated islets, islets were isolated and 60 size-matched islets were hand-picked. For graft measurements, islet grafts were dissected from the kidney parenchyma. Tissues were homogenized with a Tissue Tearor or small hand-held homogenizer in 0.14 N HCL/95% ethanol. Aliquots were measured for insulin, C-peptide, and glucagon using radioimmunoassay or for GLP-1 using a mouse Milliplex assay (all from Millipore) by the Vanderbilt Hormone and Analytical Core.
Statistical analysis.
Statistical significance was assessed by one-way or two-way ANOVA using Bonferroni multiple comparison posttest and, when appropriate, by unpaired Student t test using GraphPad Prism 5 (GraphPad Software; San Diego, CA). A P < 0.05 was considered to be statistically significant.
RESULTS
Generation of mice with hepatocyte-specific deletion of the glucagon receptor.
To generate GcgrHep−/− mice, GcgrFlox mice were mated with Alb-Cre mice (Supplementary Fig. 1A). Gcgr expression in the liver was abundant and comparable in wild-type and GcgrFlox mice and was virtually absent in GcgrHep−/− mice (Supplementary Fig. 1B). In contrast, Gcgr mRNA transcripts were expressed at normal levels in other tissues, including pancreas, kidney, and jejunum (Supplementary Fig. 1C–E).
Deletion of the Gcgr in hepatocytes leads to mild hypoglycemia and abrogates the hyperglycemic response to acute glucagon challenge.
GcgrHep−/− mice exhibited normal body weight compared with littermate controls (Supplementary Fig. 2A) and reductions in fasting (Fig. 1A) and random glucose levels (Supplementary Fig. 2B), similar to Gcgr−/− mice (11,16). Plasma glucose levels increased briskly in response to exogenous glucagon in control mice; however, GcgrHep−/− mice failed to exhibit a glycemic response after acute glucagon challenge (Supplementary Fig. 2C), findings that also were reported for Gcgr−/− mice (10).
FIG. 1.
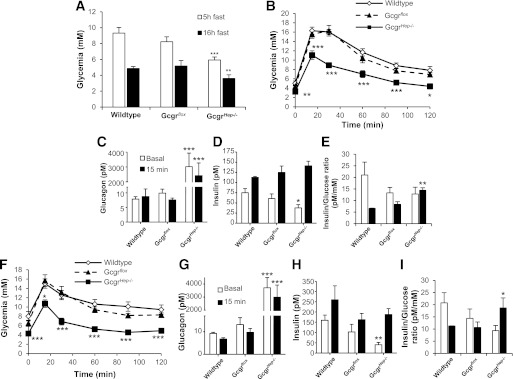
Ablation of the hepatocyte Gcgr leads to hypoglycemia and improved glucose tolerance. A: Glycemia in 12-week-old male GcgrHep−/− and littermate controls fasted for 5 h or 16 h (n = 10–12 mice per group). B: Intraperitoneal (IP) glucose challenge in 9-week-old male GcgrHep−/− and littermate controls fasted for 16 h (n = 8–10 mice). Plasma glucagon (C) and insulin (D) levels measured before and 15 min after IP glucose injection (n = 8–10 mice per group). E: Insulin-to-glucose ratio before and 15 min after IP glucose injection. F: Oral glucose challenge in 8-week-old GcgrHep−/− males and littermate controls fasted for 16 h (n = 10–12 mice per group). Plasma glucagon (G) and insulin (H) levels measured before and 15 min after oral glucose administration (n = 10–12 mice per group). I: Insulin-to-glucose ratio before and 15 min after oral glucose injection. Data are mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. wild-type mice.
Both Gcgr−/− and GcgrHep−/− mice display improved glucose tolerance, decreased fasting insulin, and increased plasma glucagon levels.
Intraperitoneal (Fig. 1B) and oral (Fig. 1F) glucose tolerance were improved in GcgrHep−/− mice compared with littermate controls, findings that are similar to those described for Gcgr−/− mice (10). Plasma glucagon levels were markedly elevated in GcgrHep−/− mice (Fig. 1C, G) and similar to levels reported for Gcgr−/− mice (10,11). Furthermore, plasma insulin levels were reduced in the fasting state, but were not significantly different during glucose challenge in GcgrHep−/− mice compared with wild-type and GcgrFlox littermate controls (Fig. 1D, H), as reported in Gcgr−/− mice (10). Nevertheless, fasting glucose and insulin were lower, and insulin-to-glucose ratios were significantly increased in GcgrHep−/− versus control mice after glucose challenge (Fig. 1B, D–F, H, I). The similarity of the metabolic phenotypes in Gcgr−/− and GcgrHep−/− mice reveals a critical role for the hepatic Gcgr in the phenotype of Gcgr−/− mice.
GcgrHep−/− mice exhibit increased insulin sensitivity.
Insulin challenge resulted in a greater initial decline in blood glucose in GcgrHep−/− mice (Fig. 2A) but no difference in overall glucose excursion (Fig. 2B), as previously shown in Gcgr−/− mice (10). Furthermore, Gcgr−/− mice displayed increased insulin sensitivity compared with Gcgr+/+ littermate controls (Fig. 2C, D). Insulin sensitivity was similarly increased in GcgrHep−/− mice (Fig. 2E, F). The comparable phenotypes of Gcgr−/− versus GcgrHep−/− mice indicate that the impact of glucagon on glucose tolerance and insulin sensitivity is largely mediated by hepatic glucagon receptors.
FIG. 2.

Ablation of the Gcgr in hepatocytes increases insulin sensitivity. A: Intraperitoneal (IP) insulin tolerance test in 13-week-old male GcgrHep−/− and littermate controls fasted for 5 h (n = 8–10 mice). B: Area under the curve (AUC) glucose from the IP insulin tolerance test shown in A. Hyperinsulinemic euglycemic clamp performed in conscious Gcgr−/− (C, E) or GcgrHep−/− (E, F) males and littermate controls fasted for 5 h (n = 5–7 mice). C and E: Glycemic excursion during stabilization and steady-state phase. D and F: Glucose infusion rates during steady state to maintain euglycemia. Data are mean ± SEM. *P < 0.05, **P < 0.01 vs. wild-type mice.
Disruption of GCGR signaling in the liver results in increased pancreas weight, α-cell proliferation, and α-cell hyperplasia.
Gcgr−/− mice exhibit markedly increased plasma glucagon levels, increased mass of the entire pancreas and islet, and α-cell hyperplasia (Fig. 3A, B) (10,11). Although total β-cell mass is increased in Gcgr−/− and GcgrHep−/− mice (Supplementary Fig. 3A, B), this increase is secondary to the enlargement of the pancreas itself, because β-cell mass per gram of pancreas is similar in 20-week-old Gcgr−/− and wild-type littermate controls (Fig. 3C). Additionally, glucagon and GLP-1 content in the pancreas of Gcgr−/− mice is markedly increased, whereas insulin and C-peptide content is unchanged when corrected for changes in pancreatic mass (Supplementary Fig. 2D). The increase in α-cell mass (Fig. 3D) is accompanied by a significant increase in α-cell proliferation (Fig. 3E–G). Despite preservation of Gcgr expression in the pancreas (Supplementary Fig. 1C), GcgrHep−/− mice exhibited markedly elevated plasma GLP-1 and glucagon levels (Supplementary Fig. 4A and Fig. 1C, G), increased pancreas weight (Fig. 4B), and absolute β-cell mass (Supplementary Fig. 3B), but normal β-cell mass per gram of pancreas (Fig. 4C), comparable with the phenotype of Gcgr−/− mice (Fig. 3 and Supplementary Fig. 3A, B). Although GIP is produced in α-cells (23), plasma GIP levels were normal in Gcgr−/− and GcgrHep−/− mice (Supplementary Fig. 4C, D). Despite preservation of pancreatic Gcgr expression, GcgrHep−/− mice exhibited increased α-cell proliferation with islet and α-cell hyperplasia (Fig. 4A, D–G), indicating that this process is mediated by loss of hepatic GCGR signaling.
FIG. 3.
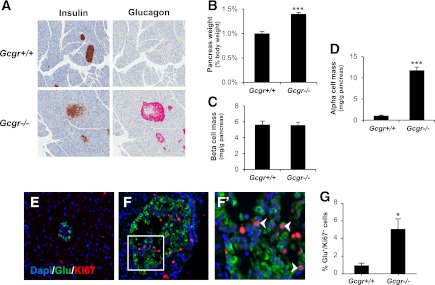
Whole-body ablation of the Gcgr increases pancreatic weight, α-cell mass, and α-cell proliferation. A: Histological sections stained for insulin (left panels) or glucagon (right panels). B: Pancreas weight of 20-week-old Gcgr−/− males and littermate controls corrected for body weight (n = 8–10 mice). Pancreatic β-cell mass (C) and α-cell mass (D) per gram of pancreas. Data are mean ± SEM. ***P < 0.001 vs. Gcgr+/+. Representative immunofluorescent sections stained for dapi (blue), glucagon (green), and Ki67 (red) from 22-week-old Gcgr−/− mice (F) and littermate controls (E). F′: Higher magnification of Ki67-positive nuclei from F. White arrows indicate Ki67-positive nuclei of α-cells. G: The α-cell proliferation rate (percentage of glucagon+/Ki67+ cells per total glucagon+ cells, n = 4 per group; 3 depths/pancreas). Data are mean ± SEM. *P < 0.05 vs. Gcgr+/+.
FIG. 4.
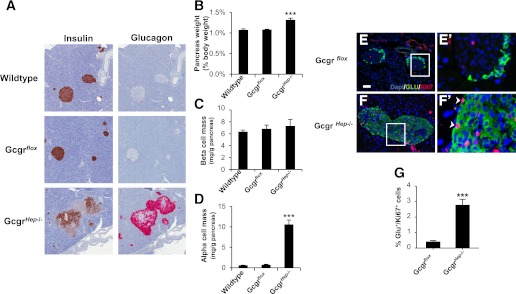
Ablation of the Gcgr in hepatocytes results in increased pancreas weight and α-cell hyperplasia. A: Representative histological sections stained for insulin (left panels) or glucagon (right panels). B: Pancreas weight of 20-week-old GcgrHep−/− males and littermate controls corrected for body weight (n = 8–10 mice). Pancreatic β-cell mass (C) and α-cell mass (D) per gram of pancreas. Data are mean ± SEM. ***P < 0.001 vs. wild type. Representative immunofluorescent sections stained for dapi (blue), glucagon (green), and Ki67 (red) from 22-week-old GcgrHep−/− mice (F) and littermate controls (E). E′ and F′: Higher magnification of sections containing Ki67-positive nuclei, denoted by white arrows, in α-cells. Insets are magnifications of selected Ki67-positive nuclei of α-cells (white box). White scale bar represents 50 μm. G: The α-cell proliferation rate (percentage of glucagon+/Ki67+ cells per total glucagon+ cells, n = 4 per group; 3 depths/pancreas). Data are mean ± SEM. ***P < 0.001 vs. Gcgrflox.
Increased α-cell proliferation in islets transplanted into Gcgr−/− mice.
The findings of islet and α-cell hyperplasia arising in GcgrHep−/− mice implies that disruption of GCGR signaling in the liver initiates signals that are, in turn, communicated to the pancreas, perhaps through neural pathways or circulating factors, promoting α-cell proliferation. To determine if the proliferative signals are dependent on the pancreatic location, we transplanted 14-week-old Gcgr+/+ (wild-type) islets into 14-week-old Gcgr+/+ and Gcgr−/− mice and allowed them to engraft for 8 weeks (Fig. 5A). We also transplanted Gcgr−/− islets into the contralateral kidney of the same Gcgr+/+ and Gcgr−/− mice. In this approach, the endogenous pancreatic islets in the transplant recipient serve as a control. Isolated islets from 14-week-old Gcgr−/− mice before transplantation had higher glucagon and GLP-1 content, but lower insulin and C-peptide content when compared with Gcgr+/+ islets (Fig. 5B). Remarkably, transplantation of Gcgr+/+ islets under the kidney capsule of Gcgr−/− mice resulted in a striking increase in glucagon and GLP-1 content of the grafts (Fig. 5C). A small increase in insulin also was observed in Gcgr+/+ grafts in Gcgr−/− recipient mice. The area of glucagon-positive cells in the transplanted Gcgr+/+ islets also was increased in Gcgr−/− recipients (Fig. 5E, F, I). Furthermore, Gcgr+/+ islets transplanted into Gcgr−/− mice exhibited significantly increased α-cell proliferation compared with Gcgr+/+ recipients, similar to the rate of α-cell proliferation in the Gcgr−/− pancreas (Figs. 3G and 6B, E). In contrast, α-cell proliferation was extremely low after transplantation of Gcgr+/+ islets into Gcgr+/+ mice (Fig. 6A, E). Conversely, the grafts of Gcgr−/− islets transplanted into Gcgr+/+ mice had reduced glucagon and GLP-1 content (Fig. 5D) and reduced α-cell proliferation (Fig. 6C, F) when compared with Gcgr−/− recipients, whereas α-cell area was not significantly different (Fig. 5G, J).
FIG. 5.

Gcgr+/+ islets transplanted into Gcgr−/− mice have development of α-cell hyperplasia. A: Experimental schematic showing islets from 14-week-old Gcgr+/+ or Gcgr−/− mice transplanted beneath the right and left kidney capsule of 14-week-old Gcgr+/+ or Gcgr−/− mice, respectively. The kidneys containing the islet graft were removed for analysis 8 weeks later. B: Hormone content of freshly isolated islets of 14-week-old Gcgr+/+ (open bars) and Gcgr−/− (closed bars) mice. Data are mean ± SEM. *P < 0.05, ***P < 0.001 vs. Gcgr+/+ islets. C: Hormone content of Gcgr+/+ grafts removed from 22-week-old mice Gcgr+/+ (open bars) and Gcgr−/− (light grey bars) recipient mice 8 weeks after transplant. Data are mean ± SEM. *P < 0.05, **P < 0.01 vs. Gcgr+/+ recipients. D: Hormone content of Gcgr−/− grafts removed from 22-week-old mice Gcgr+/+ (closed bars) and Gcgr−/− (dark grey bars) recipient mice 8 weeks after transplantation. Data are mean ± SEM. *P < 0.05, ***P < 0.001 vs. Gcgr−/− recipients. Representative islet graft sections from Gcgr+/+ (donor) to Gcgr+/+ (recipient; E) and Gcgr+/+ (donor) to Gcgr−/− (recipient; F) mice stained for insulin (green) and glucagon (red). E′ and F′: Insets of E and F (white box). Representative islet graft sections from Gcgr−/− (donor) to Gcgr+/+ (recipient; G) and Gcgr−/− (donor) to Gcgr−/− (recipient; H) mice stained for insulin (green) and glucagon (red). G′ and H′: Insets of G and H (white box). White scale bar represents 50 μm. Percentage of islet grafts that stained positive for glucagon for Gcgr+/+ donor (I) and Gcgr−/− donor islets (J) into Gcgr+/+ and Gcgr−/− mice (n = 3/mice, 3 sections/islet graft). Percent glucagon area is defined as the percentage of glucagon area/total insulin + glucagon area. Data are mean ± SEM. ***P < 0.001 vs. Gcgr+/+ recipient grafts.
FIG. 6.
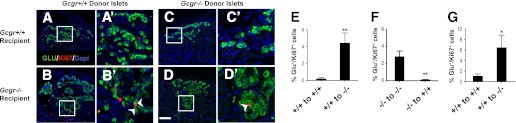
Gcgr+/+ islets transplanted into Gcgr−/− recipients exhibit increased α-cell proliferation. Islet graft sections from Gcgr+/+ (donor) to Gcgr+/+ (recipient; A) and Gcgr+/+ (donor) to Gcgr−/− (recipient; B) mice stained for Ki67 (red), glucagon (green), and dapi (blue). A′ and B′: Insets of A and B (white box from A, B). White arrows indicate Ki67-positive α-cells. Islet graft sections from Gcgr−/− (donor) to Gcgr+/+ (recipient; C) and Gcgr−/− (donor) to Gcgr−/− (recipient; D) mice stained for Ki67 (red), glucagon (green), and dapi (blue). C′ and D′: Insets of C and D (white box from C, D). White arrows indicate Ki67-positive α-cells. White scale bar represents 50 μm. Proliferation rate of α-cells in grafts of islets from 14-week-old Gcgr+/+ (E) or Gcgr−/− (F) mice (donors) transplanted beneath the right and left kidney capsule, respectively, of 14-week-old Gcgr+/+ (recipient) or Gcgr−/− (recipient) mice. The kidneys containing the islet graft were removed for analysis 8 weeks later (n = 3/mice, 3 sections/islet graft). Data are mean ± SEM. **P < 0.01 vs. control grafts (donor and recipient of same genotype). G: Proliferation rate of α-cells in grafts of islets from 14-week-old Gcgr+/+ mice transplanted beneath the right and left kidney capsule of 14-week-old Gcgr+/+ or Gcgr−/− mice. The kidney containing the islet graft was removed for analysis 1 week later (n = 4–6/mice, 3 sections/graft). Data are mean ± SEM. *P < 0.05 vs. Gcgr+/+ recipient grafts.
To investigate how quickly α-cells respond to the proliferative stimulus and whether innervation is involved in the increased α-cell proliferation, 14-week-old wild-type islet grafts were removed 1 week after transplantation into Gcgr−/− mice, a time point before reinnervation occurs or revascularization is complete (20,24). We observed a significant increase in α-cell proliferation in Gcgr+/+ islet grafts in Gcgr−/− recipients 1 week after transplantation (Fig. 6G).
Because α-cell proliferation and hyperplasia occurs in the pancreas of both Gcgr−/− and GcgrHep−/−mice, we tested whether wild-type islets transplanted into GcgrHep−/− recipients recapitulated the phenotype observed in Gcgr−/− recipients. Gcgr+/+ islets transplanted into GcgrHep−/− littermates had an increase in α-cell area and α-cell proliferation 1 month after transplantation compared with those transplanted into GcgrFlox mice (Fig. 7A–F). The magnitude of increase in glucagon area in Gcgr+/+ islets transplanted into GcgrHep−/− recipients after 4 weeks was less than after transplantation of Gcgr+/+ islets into Gcgr−/− mice after 8 weeks.
FIG. 7.
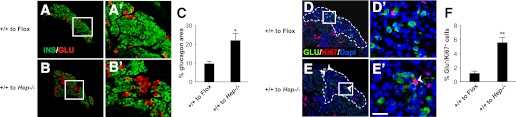
Wild-type islets transplanted into GcgrHep−/− recipients for 4 weeks have increased α-cell area and proliferation. Representative islet graft sections from Gcgr+/+ (donor) to Gcgrflox (recipient; A) and Gcgr+/+ (donor) to GcgrHep−/− (recipient; B) mice stained for insulin (green) and glucagon (red). A′ and B′: Insets of A and B (white box). C: Percentage of islet grafts that stained positive for glucagon for Gcgr+/+ donor islets into Gcgrflox (recipient, open bars) and GcgrHep−/− (recipient, closed bars) mice (n = 4 mice, 3 sections/islet graft). Percent glucagon area is defined as the percentage of glucagon area/total insulin + glucagon area. Data are mean ± SEM. *P < 0.05 vs. Gcgrflox recipient grafts. Islet grafts sections from Gcgr+/+ (donor) to Gcgrflox (recipient; D) and Gcgr+/+ (donor) to GcgrHep−/− (recipient; E) mice stained for Ki67 (red), glucagon (green), and dapi (blue). D′ and E′: Insets of D and E (white box). White arrows indicate the Ki67-positive α-cells. White scale bar represents 25 μm. F: Proliferation rate of α-cells in Gcgr+/+ islet graft (donor) transplanted into Gcgrflox (recipient, open bars) or GcgrHep−/− (recipient, closed bars) mice (n = 4 mice, 3 sections/islet graft). Data are mean ± SEM. **P < 0.01 vs. Gcgrflox recipient grafts.
Analysis of potential mechanisms contributing to α-cell hyperplasia.
Because β-cell hyperplasia arising as a result of liver insulin resistance has been linked to increased ERK1/2 phosphorylation through a liver–pancreas vagal signal, we assessed whether loss of hepatic glucagon signaling leads to enhanced ERK1/2 phosphorylation (25). Western blot analysis of hepatic extracts from wild-type, GcgrFlox, and GcgrHep−/− mice revealed similar levels of ERK1/2 phosphorylation across genotypes (Supplementary Fig. 5).
Plasma levels of interleukin-6, associated with α-cell hyperplasia (26), were similar in control, Gcgr−/−, and GcgrHep−/− mice (Supplementary Fig. 6A, B). Plasma levels of SDF-1, known to induce expression of prohormone convertase-1, GLP-1, and islet cell proliferation (27), were similar in Gcgr−/−, GcgrHep−/−, and littermate controls (Supplementary Fig. 6C, D). Total bile acid levels were significantly increased in plasma from GcgrHep−/− and Gcgr−/− mice (Supplementary Fig. 6E, F), consistent with recent observations (28).
Because bile acids increase the secretion of proglucagon-derived peptides and promote cell proliferation, we fed wild-type mice a diet enriched with 1% cholic acid and assessed α-cell mass. Plasma bile acid levels were significantly increased after cholic acid feeding, in association with reduced levels of glucose and insulin (Supplementary Fig. 6G–I); however, pancreas weight (Supplementary Fig. 6J) and plasma glucagon levels (Supplementary Fig. 6K) were not increased. The α-cell mass was modestly increased, whereas β-cell mass was unchanged in cholic acid–fed mice (Supplementary Fig. 6L, M). Therefore, it seems unlikely that increased bile acids produce the phenotypes of hyperglucagonemia and increased α-cell mass in Gcgr−/− mice. Taken together, the findings from GcgrHep−/− mice and transplantation experiments support the existence of one or more circulating factors that arise after elimination of liver GCGR signaling, which promote α-cell proliferation independent of the normal pancreatic location.
DISCUSSION
By comparing mice with whole-body and hepatocyte-specific inactivation of the Gcgr, we found that the predominant phenotypes arising in Gcgr−/− mice are recapitulated by selective loss of the Gcgr in hepatocytes. Both Gcgr−/− mice and GcgrHep−/− mice had similar reductions in fasting glucose and improvements in oral and intraperitoneal glucose tolerance and insulin sensitivity. Gcgr−/− mice and GcgrHep−/− mice also had development of similar increases in pancreatic and α-cell mass. Notably, normal islets transplanted into either Gcgr−/− or GcgrHep−/− mice displayed a marked increase in α-cell proliferation. This increased rate of α-cell proliferation at an extrapancreatic site establishes the liver as a critical organ for initiation of signals promoting the proliferation of islet α-cells after reduction or elimination of GCGR signaling.
Reduced or absent glucagon receptor signaling produced by several complementary approaches (inactivation or a reduction in glucagon and/or its receptor by genetic manipulation, immunoblockade, or antisense oligonucleotides) leads to α-cell expansion (10,14,29). Conversely, increased glucagon receptor signaling by administration of glucagon several times daily for up to 20 days produces marked atrophy of α-cells in the endocrine pancreas (30). Experimental hyperglucagonemia in rodents with transplantable glucagonomas also produces rapid diminution of islet and α-cell mass, apoptosis of α-cells, and depletion of proglucagon mRNA expression and immunoreactivity of glucagon in the pancreas (31,32). In the transplantation studies presented here, α-cell proliferation and glucagon content are reduced when Gcgr−/− islets are transplanted into Gcgr+/+ recipients versus Gcgr−/−recipients (Figs. 5 and 6). This is similar to the reversal of α-cell hyperplasia observed in Pcsk2−/− mice rescued with glucagon supplementation and GCGR antibody-treated mice on removal of treatment and restoration of glucagon signaling.
Our findings of robust α-cell hyperplasia in GcgrHep−/− mice are consistent with reports of islet and α-cell hyperplasia arising in LGsKO mice with liver-specific inactivation of the gene encoding the Gsα subunit (12,16). In contrast, disruption of Gsα in the entire pancreas (33) or selectively in β-cells (34) does not produce α-cell hyperplasia. Furthermore, direct attenuation of GCGR signaling in α-cells does not result in increased rates of cell proliferation (35). In agreement with findings from LGsKO mice, our data establish the liver as critical for generation of signals promoting α-cell hyperplasia pursuant to disruption of GCGR-dependent signal transduction pathways.
An increase in α-cell proliferation has been reported in the pancreas of Gcgr−/− embryos, raising the possibility that the signal triggering expansion of α-cell mass is attributable to the developmental loss of GCGR signaling (11). However, even partial reduction of GCGR signaling in adult mice (14,29,36) induces α-cell hyperplasia, and adult islets rapidly exhibit increased α-cell proliferation after transplantation into Gcgr−/− recipients. Because albumin-Cre-mediated recombination in hepatocytes occurs largely postnatally (17,37), GcgrHep−/− mice would not likely undergo significant elimination of Gcgr expression during embryonic development. Hence, the available evidence strongly suggests that signals triggering α-cell expansion do not require disruption of GCGR-regulated signaling pathways during liver or islet development.
Possible pathways by which reduced or absent hepatic glucagon receptor signaling leads to α-cell hyperplasia include, among others, a stimulus transmitted from liver via intraislet nerves; however, 1-week transplantation data suggest that islet innervation is not necessary for the α-cell proliferation. Slightly lower blood glucose could stimulate α-cell hyperplasia; however, previous studies show that mild hypoglycemia arising in some murine models of disrupted glucagon action is not required or sufficient to stimulate α-cell hyperplasia (14,29,31,38–40). Impaired glucagon signaling in β-cells could stimulate α-cell hyperplasia by intraislet communication; however, GcgrHep−/− mice retain intact GCGR function in β-cells yet have development of severe hyperglucagonemia. Excessive insulin signaling in α-cells in the absence of adequate glucagon signaling promotes α-cell hyperplasia; a recent report suggests that insulin can induce α-cell proliferation, and Kawamori et al. (41) have shown that interruption of insulin receptor signaling in α-cells alone can reduce α-cell mass. However, interruption of insulin receptor signaling in α-cells is not sufficient to avert α-cell hyperplasia observed in mice with global interruption of GCGR signaling (41). A circulating factor released from the liver or released from other tissues after loss of liver GCGR signaling could be responsible for regulating α-cell mass. The current results strongly support the latter possibility, namely the existence of one or more circulating factors regulating α-cell mass. Because α-cell proliferation increased to a greater extent in islets transplanted into Gcgr−/− recipients for 8 weeks, relative to findings in Gcgr+/+ islets engrafted in GcgrHep−/− mice for 4 weeks, interpretation of quantitative differences in the magnitude of changes observed in islets transplanted into Gcgr−/− versus GcgrHep−/− mice will require additional experimentation.
Imai et al. (25) demonstrated that increased activity of a hepatic extracellular regulated kinase triggered β-cell proliferation in murine models of insulin resistance or deficiency, and pancreatic afferent vagotomy abolished this stimulation of β-cell proliferation and increased ERK1/2 phosphorylation through a liver–pancreas vagal signal. We did not find a change in ERK1/2 phosphorylation or in plasma levels of interleukin-6, a circulating factor that promotes α-cell proliferation in experimental models of diabetes (26). Similarly, plasma levels of SDF-1, a chemokine known to promote islet GLP-1 production and islet cell proliferation, were normal. Although plasma levels of bile acids are markedly elevated in Gcgr−/− mice, α-cell mass appears only modestly increased and glucagon levels are not increased in cholic acid–fed mice. Hence, the mechanisms responsible for induction of α-cell proliferation remain elusive and require further investigation.
Insulin-resistant states also have been shown to trigger β-cell proliferation in transplanted islets. Transplantation of murine islets under the kidney capsule of lean or obese normoglycemic insulin-resistant mice resulted in stimulation of β-cell proliferation and expansion of islet mass in transplanted islets (42). The findings in the current report are similar in that islet cell proliferation at an extrapancreatic site appears to be induced by a circulating factor. Gu et al. (43) transplanted human islets underneath the kidney capsule of streptozotocin-treated mice, and then administered a single course of a monoclonal antibody directed against the glucagon receptor. Although the GCGR antibody improved glucose tolerance and increased plasma levels of glucagon and GLP-1, the extent, if any, of α-cell proliferation in the islet transplant was not examined (43). Because of efforts to develop therapies targeting the GCGR for the treatment of type 2 diabetes, it is important to establish whether α-cell proliferation and hyperplasia can occur in adult human islets in response to acute interruption of GCGR signaling.
The α-cell hyperplasia arising pursuant to chronic reduction or elimination of GCGR signaling also has been reported in humans. Zhou et al. (44) reported a 60-year-old woman presenting with a pancreatic mass, extremely elevated levels of glucagon, and persistent hyperglucagonemia; marked α-cell hyperplasia was detected after surgical resection of the pancreatic mass, and sequencing of genomic DNA revealed a homozygous missense mutation in the Gcgr gene, resulting in markedly reduced receptor affinity. Furthermore α-cell hyperplasia, adenomatosis, and hyperglucagonemia have been reported in patients without histological or clinical evidence of malignancy, some of whom may harbor loss-of-function mutations in the Gcgr (45).
Recent evidence for enhanced proliferation of α-cells in the pancreas from patients with newly diagnosed type 1 diabetes (46), coupled with reports demonstrating conversion or transdifferentiation of α-cells to β-cells (7–9), have rekindled therapeutic interest in understanding the signals and mechanisms regulating α-cell proliferation. Our data combining disruption of the Gcgr and islet transplantation establish a new model for analysis of the control of α-cell hyperplasia. Together with recent efforts directed at promoting generation of functional β-cells from α-cells (7,8), identification of this circulating factor may provide new opportunities for regeneration of functional β-cell mass for the treatment of diabetes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant MOP93739 from the Canadian Institutes of Health Research to D.J.D., by a Canada Research Chair in Regulatory Peptides and the Banting and Best Diabetes Centre–Novo Nordisk Chair in Incretin Biology to D.J.D., the Juvenile Diabetes Research Foundation International, the VA Research Service, the National Institutes of Health (DK-66636, DK-69603), the Beta Cell Biology Consortium (DK72473, DK89572), the Vanderbilt Mouse Metabolic Phenotyping Center (DK59637), and the Vanderbilt Diabetes Research and Training Center (DK20593, Islet Procurement and Analysis Core, Hormone Assay and Analytical Services Core, and Cell Imaging Shared Resource).
No potential conflicts of interest relevant to this article were reported.
C.L., A.M.R., E.D.D., C.D., S.A., V.d.C., and I.M. designed and carried out experiments and wrote and reviewed the manuscript. P.M.V., M.J.C., A.C.P., and D.J.D. designed experiments, reviewed results, and wrote the manuscript. A.C.P. and D.J.D. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
The authors give special thanks to Courtney Thompson and Greg Poffenberger (Vanderbilt University) for technical assistance.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-1605/-/DC1.
REFERENCES
- 1.Seymour PA, Sander M. Historical perspective: beginnings of the beta-cell: current perspectives in beta-cell development. Diabetes 2011;60:364–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bramswig NC, Kaestner KH. Transcriptional regulation of α-cell differentiation. Diabetes Obes Metab 2011;13(Suppl. 1):13–20 [DOI] [PubMed] [Google Scholar]
- 3.Müller WA, Faloona GR, Aguilar-Parada E, Unger RH. Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med 1970;283:109–115 [DOI] [PubMed] [Google Scholar]
- 4.Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev 2007;28:253–283 [DOI] [PubMed] [Google Scholar]
- 5.Dufrane D, Maillart JF, Aouassar N, Goebbels RM, Guiot Y, Gianello P. Native pancreatic alpha-cell adaptation in streptozotocin-induced diabetic primates: importance for pig islet xenotransplantation. Xenotransplantation 2009;16:152–163 [DOI] [PubMed] [Google Scholar]
- 6.Yoon KH, Ko SH, Cho JH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003;88:2300–2308 [DOI] [PubMed] [Google Scholar]
- 7.Collombat P, Xu X, Ravassard P, et al. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009;138:449–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thorel F, Népote V, Avril I, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010;464:1149–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung CH, Hao E, Piran R, Keinan E, Levine F. Pancreatic β-cell neogenesis by direct conversion from mature α-cells. Stem Cells 2010;28:1630–1638 [DOI] [PubMed] [Google Scholar]
- 10.Gelling RW, Du XQ, Dichmann DS, et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA 2003;100:1438–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vuguin PM, Kedees MH, Cui L, et al. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology 2006;147:3995–4006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen M, Gavrilova O, Zhao WQ, et al. Increased glucose tolerance and reduced adiposity in the absence of fasting hypoglycemia in mice with liver-specific Gs alpha deficiency. J Clin Invest 2005;115:3217–3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webb GC, Akbar MS, Zhao C, Swift HH, Steiner DF. Glucagon replacement via micro-osmotic pump corrects hypoglycemia and alpha-cell hyperplasia in prohormone convertase 2 knockout mice. Diabetes 2002;51:398–405 [DOI] [PubMed] [Google Scholar]
- 14.Sloop KW, Cao JX, Siesky AM, et al. Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest 2004;113:1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ali S, Lamont BJ, Charron MJ, Drucker DJ. Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. J Clin Invest 2011;121:1917–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen M, Mema E, Kelleher J, et al. Absence of the glucagon-like peptide-1 receptor does not affect the metabolic phenotype of mice with liver-specific G(s)α deficiency. Endocrinology 2011;152:3343–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000;26:149–150 [DOI] [PubMed] [Google Scholar]
- 18.Flock G, Baggio LL, Longuet C, Drucker DJ. Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes 2007;56:3006–3013 [DOI] [PubMed] [Google Scholar]
- 19.Maida A, Hansotia T, Longuet C, Seino Y, Drucker DJ. Differential importance of glucose-dependent insulinotropic polypeptide vs glucagon-like peptide 1 receptor signaling for beta cell survival in mice. Gastroenterology 2009;137:2146–2157 [DOI] [PubMed] [Google Scholar]
- 20.Brissova M, Shostak A, Shiota M, et al. Pancreatic islet production of vascular endothelial growth factor—a is essential for islet vascularization, revascularization, and function. Diabetes 2006;55:2974–2985 [DOI] [PubMed] [Google Scholar]
- 21.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 2006;55:390–397 [DOI] [PubMed] [Google Scholar]
- 22.Brissova M, Fowler M, Wiebe P, et al. Intraislet endothelial cells contribute to revascularization of transplanted pancreatic islets. Diabetes 2004;53:1318–1325 [DOI] [PubMed] [Google Scholar]
- 23.Fujita Y, Wideman RD, Asadi A, et al. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology 2010;138:1966–1975 [DOI] [PubMed] [Google Scholar]
- 24.Korsgren O, Andersson A, Jansson L, Sundler F. Reinnervation of syngeneic mouse pancreatic islets transplanted into renal subcapsular space. Diabetes 1992;41:130–135 [DOI] [PubMed] [Google Scholar]
- 25.Imai J, Katagiri H, Yamada T, et al. Regulation of pancreatic beta cell mass by neuronal signals from the liver. Science 2008;322:1250–1254 [DOI] [PubMed] [Google Scholar]
- 26.Ellingsgaard H, Ehses JA, Hammar EB, et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci USA 2008;105:13163–13168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Stanojevic V, Avadhani S, Yano T, Habener JF. Stromal cell-derived factor-1 (SDF-1)/chemokine (C-X-C motif) receptor 4 (CXCR4) axis activation induces intra-islet glucagon-like peptide-1 (GLP-1) production and enhances beta cell survival. Diabetologia 2011;54:2067–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, MacDougall ML, McDowell MT, et al. Polyomic profiling reveals significant hepatic metabolic alterations in glucagon-receptor (GCGR) knockout mice: implications on anti-glucagon therapies for diabetes. BMC Genomics 2011;12:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu W, Yan H, Winters KA, et al. Long-term inhibition of the glucagon receptor with a monoclonal antibody in mice causes sustained improvement in glycemic control, with reversible alpha-cell hyperplasia and hyperglucagonemia. J Pharmacol Exp Ther 2009;331:871–881 [DOI] [PubMed] [Google Scholar]
- 30.Petersson B, Hellman B. Effects of long term administration of glucagon on the pancreatic islet tissue of rats and guinea-pigs. Acta Endocrinol (Copenh) 1963;44:139–149 [DOI] [PubMed] [Google Scholar]
- 31.Blume N, Skouv J, Larsson LI, Holst JJ, Madsen OD. Potent inhibitory effects of transplantable rat glucagonomas and insulinomas on the respective endogenous islet cells are associated with pancreatic apoptosis. J Clin Invest 1995;96:2227–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ehrlich P, Tucker D, Asa SL, Brubaker PL, Drucker DJ. Inhibition of pancreatic proglucagon gene expression in mice bearing subcutaneous endocrine tumors. Am J Physiol 1994;267:E662–E671 [DOI] [PubMed] [Google Scholar]
- 33.Xie T, Chen M, Weinstein LS. Pancreas-specific Gsalpha deficiency has divergent effects on pancreatic alpha- and beta-cell proliferation. J Endocrinol 2010;206:261–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie T, Chen M, Zhang QH, Ma Z, Weinstein LS. Beta cell-specific deficiency of the stimulatory G protein alpha-subunit Gsalpha leads to reduced beta cell mass and insulin-deficient diabetes. Proc Natl Acad Sci USA 2007;104:19601–19606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, Kim W, Chen Z, et al. Insulin and glucagon regulate pancreatic α-cell proliferation. PLoS ONE 2011;6:e16096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winzell MS, Brand CL, Wierup N, et al. Glucagon receptor antagonism improves islet function in mice with insulin resistance induced by a high-fat diet. Diabetologia 2007;50:1453–1462 [DOI] [PubMed] [Google Scholar]
- 37.Kellendonk C, Opherk C, Anlag K, Schütz G, Tronche F. Hepatocyte-specific expression of Cre recombinase. Genesis 2000;26:151–153 [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Komiya I, Inman L, et al. Effects of hypoglycemia and prolonged fasting on insulin and glucagon gene expression. Studies with in situ hybridization. J Clin Invest 1989;84:711–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shigeyama Y, Kobayashi T, Kido Y, et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol 2008;28:2971–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sund NJ, Vatamaniuk MZ, Casey M, et al. Tissue-specific deletion of Foxa2 in pancreatic beta cells results in hyperinsulinemic hypoglycemia. Genes Dev 2001;15:1706–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawamori D, Akiyama M, Hu J, Hambro B, Kulkarni RN. Growth factor signalling in the regulation of α-cell fate. Diabetes Obes Metab 2011;13(Suppl. 1):21–30 [DOI] [PubMed] [Google Scholar]
- 42.Flier SN, Kulkarni RN, Kahn CR. Evidence for a circulating islet cell growth factor in insulin-resistant states. Proc Natl Acad Sci USA 2001;98:7475–7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu W, Winters KA, Motani AS, et al. Glucagon receptor antagonist-mediated improvements in glycemic control are dependent on functional pancreatic GLP-1 receptor. Am J Physiol Endocrinol Metab 2010;299:E624–E632 [DOI] [PubMed] [Google Scholar]
- 44.Zhou C, Dhall D, Nissen NN, Chen CR, Yu R. Homozygous P86S mutation of the human glucagon receptor is associated with hyperglucagonemia, alpha cell hyperplasia, and islet cell tumor. Pancreas 2009;38:941–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henopp T, Anlauf M, Schmitt A, et al. Glucagon cell adenomatosis: a newly recognized disease of the endocrine pancreas. J Clin Endocrinol Metab 2009;94:213–217 [DOI] [PubMed] [Google Scholar]
- 46.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Evidence of increased islet cell proliferation in patients with recent-onset type 1 diabetes. Diabetologia 2010;53:2020–2028 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.