

Abstract
Maternal nutrient reduction (MNR) during fetal development may predispose offspring to chronic disease later in life. Increased regeneration of active glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) in metabolic tissues is fundamental to the developmental programming of metabolic syndrome, but underlying mechanisms are unknown. Hexose-6-phosphate dehydrogenase (H6PD) generates NADPH, the cofactor for 11β-HSD1 reductase activity. CCAAT/enhancer binding proteins (C/EBPs) and the glucocorticoid receptor (GR) regulate 11β-HSD1 expression. We hypothesize that MNR increases expression of fetal C/EBPs, GR, and H6PD, thereby increasing expression of 11β-HSD1 and reductase activity in fetal liver and adipose tissues. Pregnant MNR baboons ate 70% of what controls ate from 0.16 to 0.9 gestation (term, 184 days). Cortisol levels in maternal and fetal circulations increased in MNR pregnancies at 0.9 gestation. MNR increased expression of 11β-HSD1; H6PD; C/EBPα, -β, -γ; and GR in female but not male perirenal adipose tissue and in male but not female liver at 0.9 gestation. Local cortisol level and its targets PEPCK1 and PPARγ increased correspondingly in adipose and liver tissues. C/EBPα and GR were found to be bound to the 11β-HSD1 promoter. In conclusion, sex- and tissue-specific increases of 11β-HSD1, H6PD, GR, and C/EBPs may contribute to sexual dimorphism in the programming of exaggerated cortisol regeneration in liver and adipose tissues and offsprings’ susceptibility to metabolic syndrome.
Although central obesity, genetic susceptibility, aging, sedentary lifestyle, and stress are recognized as important contributing factors to development of diabetes and metabolic syndrome, it is now understood that an adverse intrauterine environment resulting from poor maternal nutrition, stress, or overexposure to glucocorticoids can lead to developmental programming of these conditions in later life (1). Maternal nutrient reduction (MNR) affects a significant portion of the population in the developing world, but it is much less an issue in developed countries where maternal smoking, pregnancy-induced hypertension, and placental insufficiency or other placental pathologies may all result in restriction of intrauterine growth restriction. To survive these adverse conditions, the fetus has to make adaptive alterations in the expression patterns of genes involved in glucose and insulin metabolism. Permanent changes to these genes may predispose the fetus to the development of metabolic syndrome in later life, an adaptation often referred to as the “thrifty phenotype hypothesis” (2–7).
Excess cortisol is associated with metabolic syndrome (8). Increased glucocorticoid activity in these tissues also is proposed as a key mechanism underlying the developmental programming of metabolic syndrome (9). Increased expression of glucocorticoid receptor (GR) expression is observed in hepatic, renal, and adipose tissues after developmental programming in several animal species (9,10), although other reports indicate no change (11). Although circulating levels of glucocorticoids are not changed in patients with obesity and metabolic syndrome (12,13), increased local regeneration of biologically active glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) in the major metabolic tissues has been suggested to play a pivotal role in the pathogeneses of metabolic syndrome (14). The reductase activity of 11β-HSD1 regenerates cortisol (humans)/corticosterone (rodents) from their biologically inactive metabolites cortisone/11-dehydrocorticosterone, thus amplifying the actions of glucocorticoids (15). Such local hypercortisolism is believed to cause metabolic and other complications in metabolic syndrome (8,16,17). A key role for 11β-HSD1 in metabolic syndrome is supported by findings that 11β-HSD1−/− mice are protected from developing metabolic syndrome (18,19), whereas selective overexpression of 11β-HSD1 in adipose or liver tissue, the two major tissues expressing 11β-HSD1 in the body, leads to a metabolic syndrome phenotype (20,21). MNR has been shown to cause a persistent increase in 11β-HSD1 expression in the visceral adipose or liver tissues in a number of animal models (9,22). However, evidence of overexpression of 11β-HSD1 in developing fetal liver and adipose tissue in response to the challenge of MNR in the fetal primate is lacking, although a persistent postnatal increase of 11β-HSD1 expression was observed in the liver and subcutaneous adipose tissues in the common marmoset after prenatal exposure to dexamethasone (23). Because the baboon shares 96% genomic homology with humans (24), characterizing the effect of MNR on 11β-HSD1 expression in the liver and adipose tissues in a fetal primate model would improve understanding of human development and aid translation of developmental programming during human development.
Despite considerable evidence of a role for 11β-HSD1 in the pathogenesis of developmental programming of metabolic syndrome, the mechanism that increases 11β-HSD1 expression during developmental challenges is unknown. 11β-HSD1 is among the target genes that are regulated by the CCAAT/enhancer binding proteins (C/EBPs) and GR (25,26). We hypothesize that the challenge of nutrient restriction to the developing fetus results in a persistent increase of C/EBP and GR expression in the major glucocorticoid-sensitive tissues, such as liver and fat, and as a consequence, downstream genes including 11β-HSD1 are up-regulated, leading to a feed-forward production of cortisol and expression of glucocorticoid-target genes in these cells. 11β-HSD1 is a bidirectional enzyme capable of both reductase and oxidase activity (15). The reductase activity of 11β-HSD1 regenerating cortisol/corticosterone depends on the availability of the cofactor NADPH, derived from the enzymatic activity of hexose-6-phosphate dehydrogenase (H6PD) (27). Thus parallel increases of H6PD and 11β-HSD1 may be a prerequisite for the reductase activity of 11β-HSD1.
To address these hypotheses, we studied the effect of 70% maternal ad libitum food intake from 0.16 gestation (term, 184 days) to 0.9 gestation on the expression of 11β-HSD1, H6PD, GR, and C/EBP, which are central to the peripheral generation of cortisol in fetal adipose tissue, as well as the mineralocorticoid receptor (MR) that may also mediate the effects of cortisol (28) and its targets phosphoenolpyruvate carboxykinase 1 (PEPCK1) and peroxisome proliferator-activated receptor γ (PPARγ), which are essential for hepatic energy metabolism and adipocyte differentiation (29,30).
RESEARCH DESIGN AND METHODS
Animal care.
Baboon (Papio species) singleton pregnancies were studied at the Southwest National Primate Research Center at the Texas Biomedical Research Institute. Healthy female baboons with similar body weights (10–15 kg) were randomly assigned to one of two outdoor group cages and were maintained in social groups of 10–16 with a vasectomized male. At the end of the acclimation period (30 days), a fertile male was placed in each breeding cage. Pregnancy was dated initially by timing of ovulation and changes in sex skin color and was confirmed at 30 days of gestation (term, ∼184 days) by ultrasonography. Details about housing have been published elsewhere (31). All procedures were approved by the University of Texas Health Science Center and the Texas Biomedical Research Institute Institutional Animal Care and Use Committees and was performed in facilities approved by the Centers for Disease Control and Prevention, and the Association for Assessment and Accreditation of Laboratory Animal Care.
Study design.
The animals were individually fed between either 0700 and 0900 h or 1100 and 1300 h, as described in detail elsewhere (31). At feeding time, all baboons exited their group cage and passed along a chute into individual feeding cages. Water was available continuously in each feeding cage and the animals were fed Purina Monkey Diet 5038 (Purina, St. Louis, MO). At the start of the feeding period, control baboons, which were fed ad libitum, were given 60 biscuits in their individual cage feeding tray. At the end of the 2-h feeding period, the remaining biscuits were counted. From 0.16 gestation, 13 mothers were in a group fed 70% of the control diet on a per kilogram basis. Food consumption of the animals and their weight and health status were recorded daily. Fetuses were delivered at 0.9 gestation by caesarean sections under general anesthesia, as previously described (32). After hysterectomy, the umbilical cord was identified and elevated to the surgical opening to exsanguinate the fetus while still under general anesthesia. This method is approved by the American Veterinary Medical Association. Previous study has demonstrated fewer tissue artifacts after exsanguination than after administration of euthanasia solutions (33). Maternal and fetal blood was collected from the femoral artery and umbilical vein, respectively. Postoperative maternal analgesia was provided (buprenorphine hydrochloride, 0.015 mg · kg−1 · day−1) for 3 days.
Morphometric measurements, including fetal weight, length, and waist and hip circumferences, were performed. After complete pathological evaluation and recording the liver weight, the right lobe of the liver and perirenal adipose tissue were dissected. The perirenal adipose tissue was collected as representative of visceral adipose tissue because this fat compartment represents the majority of total fetal fat stores. It should be noted that the perirenal adipose tissue in most mammals, including humans, has been reported to contain varying amount of brown adipose tissue, particularly in the early stage of life (34).
Cortisol assay.
Cortisol was measured in fetal liver and adipose tissues as well as maternal and fetal plasma using an enzyme immunoassay (Cayman, MI) after homogenization and extraction with ethyl acetate.
Quantitative real-time PCR.
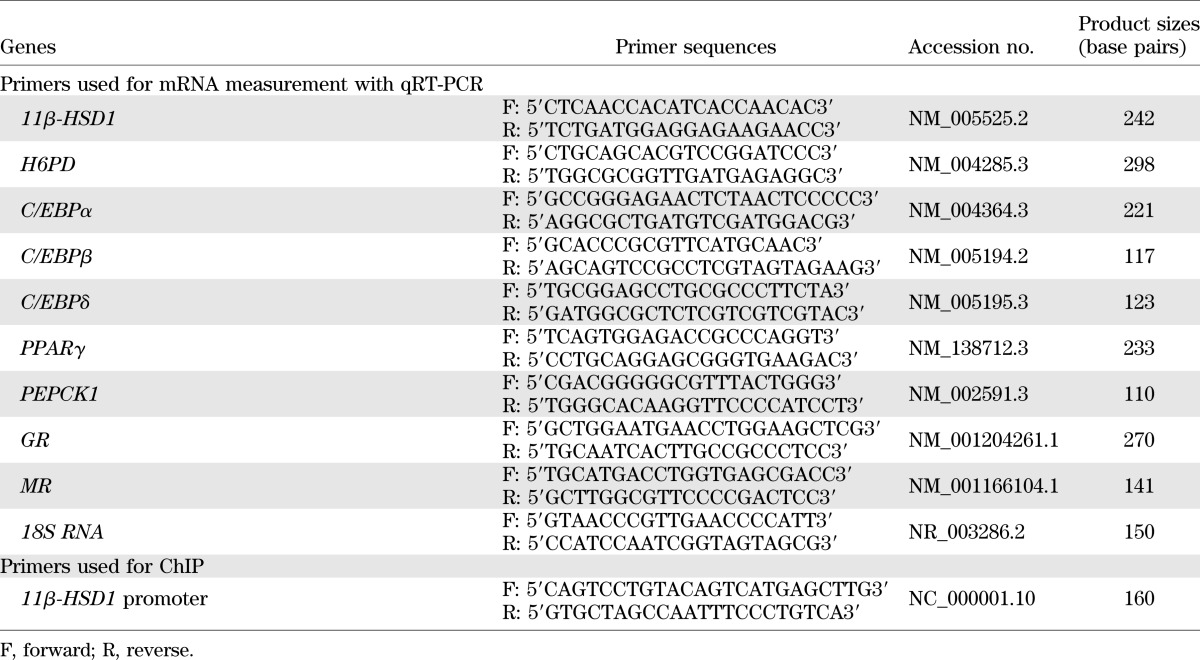
Total RNA was isolated from the homogenized fetal liver and perirenal adipose tissues using the RNeasy Mini Kit (QIAGEN, Valencia, CA). After reverse transcription, the cDNA was used for subsequent measurement of 11β-HSD1; H6PD; C/EBPα, -β, -δ; GR; MR; PEPCK1; and PPARγ mRNA levels with quantitative real-time PCR (qRT-PCR). The absolute mRNA levels in each sample were calculated according to a standard curve set up using serial dilutions of known amounts of specific templates against corresponding cycle threshold values. To control for sampling errors, qRT-PCR for the housekeeping gene 18S RNA was performed on each sample. The ratio of the target genes to 18S RNA in each sample was obtained to normalize the expression of the target gene. The primers used for PCR are illustrated in Table 1.
TABLE 1.
Primers used for mRNA measurement with qRT-PCR and for ChIP assay
Western blotting.
Total protein from fetal liver and perirenal adipose tissues was extracted in RIPA lysis buffer (Millipore, Billerica, MA) containing 1:1,000 protease cocktail (Sigma, St. Louis, MO) and phosphatase inhibitor (Active Motif, Carlsbad, CA). Protein samples (35 μg) were electrophoresed on 4–20% Tris–hydrogen chloride gels and electroblotted onto 0.45-μm nitrocellulose membranes (BioRad, Hercules, CA). After blocking with nonfat milk, the blotted membranes were incubated with primary antibodies overnight at 4°C. The primary antibody information is as follows: 1:500 11β-HSD1 (Cayman); 1:1,000 H6PD (Abgent, San Diego, CA); 1:1,000 C/EBPα (Cell Signaling, Danvers, MA); 1:500 C/EBPβ (Santa Cruz Biotechnology, Santa Cruz, CA); 1:500 C/EBPδ (Santa Cruz Biotechnology); 1:500 GR (Santa Cruz Biotechnology); 1:500 MR (Santa Cruz Biotechnology); 1:5,000 PPARγ (Santa Cruz Biotechnology); 1:2,000 PEPCK1 (Abcam, Cambridge, MA); 1:5,000 GADPH (Sigma); or 1:5,000 β-actin (Sigma). After washing, a secondary horseradish peroxidase-conjugated antibody was applied. Specific protein bands were developed using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and visualized using a G Box iChemi Chemiluminescence image capture system (SynGene, Cambridge, UK). The ratio of band intensities of target protein to the housekeeping genes β-actin in the liver or GAPDH in the adipose was obtained. The expression level of β-actin or GAPDH in the liver or adipose tissue did not differ between the experimental groups or by sex.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) assay was conducted to examine the binding of C/EBPα, GR, and MR to the 11β-HSD1 promoter, using a kit from Upstate Biotechnology (Millipore). The frozen chopped liver and adipose tissue were fixed with 1% formaldehyde on ice to cross-link the proteins bound to the chromatin DNA. After washing, the tissue was homogenized and the chromatin DNA was sheared by sonication to produce DNA fragments of around 500–1,000 base pairs. The same amounts of sheared DNA were used for immunoprecipitation with antibodies against human C/EBPα (Cell Signaling), GR (Santa Cruz Biotechnology), and MR (Santa Cruz Biotechnology) or an equal amount of preimmune rabbit IgG (Millipore). The immunoprecipitate then was incubated with protein A agarose/salmon sperm DNA (Millipore), and the antibody/protein/DNA/agarose complex was collected for subsequent reverse cross-linking. The same amount of sheared DNA without antibody precipitation was processed for reverse cross-linking and served as input control. DNA recovered from reverse cross-linking was used for qRT-PCR. The primers for qRT-PCR are illustrated in Table 1. DNA precipitated with antibody against acetylated histone 3 lysine 9 was used as a positive control. The amount of DNA precipitated was calculated from the threshold cycle number of the amplification curve of qRT-PCR. The percentage of antibody-precipitated and preimmune IgG-precipitated DNA to input DNA was compared.
Statistical analysis.
All data are reported as mean ± SEM. Unpaired Student’s t test or two-way ANOVA followed by the post hoc Student-Newman-Keuls test were performed when appropriate. Bivariate correlation analysis was performed to assess the correlation between the expression levels of the two genes concerned. Significance was set at P < 0.05. Statistical analyses were undertaken using SPSS software version 12.0.
RESULTS
Effect of MNR on fetal morphometrics.
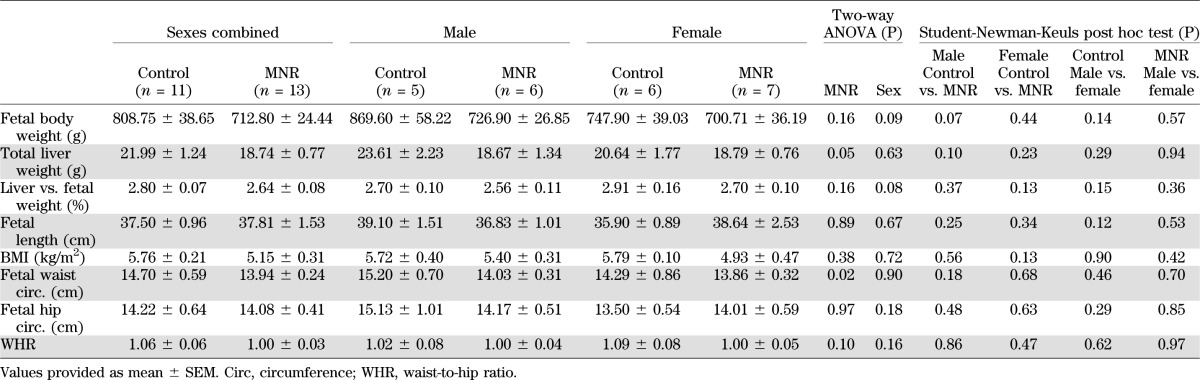
Control mothers (fed ad libitum) gained 11.6 ± 1.8% of body weight during pregnancy, whereas MNR mothers lost 3.7 ± 2.8% of their prepregnancy weight (P < 0.05). Analysis by two-way ANOVA revealed that fetal liver weight and waist circumference were significantly decreased in fetuses of MNR mothers, but the fetal weight, length, hip circumference and waist-to-hip ratio of MNR fetuses were not significantly changed when sexes were combined (Table 2). When sexes were separated, there were no statistically significant differences in any the indices measured, but the reduction of body weight in the male MNR fetuses reached marginal significance (P = 0.07) (Table 2). These data indicate that MNR (70% of food intake) has a significant influence on the body weight gain in mother but less influence on the fetus, and the male fetus is more likely than the female fetus to be affected by MNR in the mother.
TABLE 2.
Fetal morphometrics
Fetal and maternal cortisol and glucose levels.
Circulating cortisol concentrations in control mothers fed ad libitum were about twofold higher than in their offspring at 0.9 gestation. Proportional significant increases in cortisol were observed in both maternal and fetal plasma in MNR baboons, thereby maintaining a similar maternal-to-fetal circulating cortisol gradient in control and MNR pregnancies (Fig. 1A). Cortisol levels in the perirenal adipose tissue were increased significantly in fetuses of MNR mothers, but this increase was only statistically significant in female fetuses (Fig. 1B). Cortisol level in the liver of fetuses of MNR mothers was also higher than that of the fetuses of the control mothers, but the difference did not reach statistical significance when sexes were combined or separated (Fig. 1C).
FIG. 1.
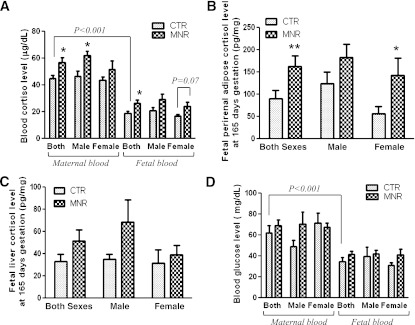
Cortisol and glucose levels in maternal and fetal plasma and cortisol levels in the fetal liver and adipose tissues of control (CTR) and MNR baboons at 0.9 gestation. A: Cortisol levels in maternal and fetal plasma of control and MNR baboons. B: Local cortisol levels in fetal perirenal adipose tissue of control and MNR baboons. C: Local cortisol levels in fetal liver tissue of control and MNR baboons. CTR groups in A–C included 5 male and 5 or 6 female baboons; MNR groups included 4–6 male and 4–7 female baboons. D: Glucose levels in maternal and fetal plasma of CTR baboons (male, n = 3; female, n = 4) and MNR baboons (male, n = 3; female, n = 3). *P < 0.05, **P < 0.01, vs. respective controls.
Plasma glucose level in control mothers fed ad libitum was significantly higher than that of their fetuses at 0.9 gestation (Fig. 1D). However, no difference was observed in glucose levels between control mothers fed ad libitum and MNR mothers or their offspring when sexes were either combined or separated (Fig. 1D).
Expression of 11β-HSD1, H6PD, GR, and MR in the fetal liver and perirenal adipose tissues.
There were no significant differences in 11β-HSD1, H6PD, GR, and MR mRNA levels between sexes of the control group either in the liver or adipose tissue. 11β-HSD1 and H6PD mRNA and protein were increased significantly in MNR female fetal perirenal adipose tissue but not in MNR male tissue (Fig. 2A and B). In contrast, fetal liver 11β-HSD1 and H6PD mRNA and protein levels were increased significantly only in the MNR male fetuses and not in MNR female fetuses (Fig. 2C and D). A similar pattern of changes in GR and MR mRNA also was observed in MNR female fetal perirenal adipose and MNR male fetal liver (Fig. 3A and C). Western blotting showed that the splice variant MR (MRΔ5,6), lacking exons 5 and 6 rather than the full-length MR, predominated in both fetal liver and adipose tissues. In addition to the splice variant form of MR, the full-length MR was detected in the fetal liver tissue but not in the adipose tissue. Variable levels of GR protein were observed in the control and MNR groups at either tissue site, but the MRΔ5,6 and full-length MR proteins were increased significantly in MNR female fetal adipose tissue and male fetal liver tissue, respectively (Fig. 3B and D).
FIG. 2.

11β-HSD1 and H6PD mRNA and protein levels in fetal perirenal adipose and liver tissues of control (CTR) and MNR baboons at 0.9 gestation. 11β-HSD1 and H6PD mRNA levels in fetal perirenal adipose (A) and liver (C) tissues and 11β-HSD1 and H6PD protein levels in fetal perirenal adipose (B) and liver (D) tissues. Top and bottom panels of each figure are images of Western blots and the mean protein levels, respectively. CTR groups included 5 male and 5 or 6 female baboons; MNR groups included 4–6 male and 4–7 female baboons. *P < 0.05, **P < 0.01, vs. male CTR; #P < 0.05, ##P < 0.01, vs. female CTR.
FIG. 3.

GR and MR mRNA and protein levels in fetal perirenal adipose and liver tissues of control (CTR) and MNR baboons at 0.9 gestation. GR and MR mRNA levels in fetal perirenal adipose (A) and liver (C) tissues and GR and MR protein levels in fetal perirenal adipose (B) and liver (D) tissues. Top and bottom panels of each figure are images of Western blots and the mean protein levels, respectively. CTR groups included 5 male and 5 or 6 female baboons; MNR groups included 4–6 male and 4–7 female baboons. *P < 0.05, **P < 0.01, vs. male CTR; #P < 0.05, vs. female CTR. MR, full-length mineralocorticoid receptor; MRΔ5,6, splice variant of MR lacking exons 5 and 6.
Expression of C/EBP in the fetal liver and perirenal adipose tissues.
There were no significant differences in C/EBPα, -β, and -δ mRNA levels between sexes of the control group in either liver or adipose tissue (Fig. 4). C/EBPα, -β, and -δ mRNA and protein were increased in MNR female fetal perirenal adipose tissue but not in MNR male fetuses. In contrast, significant increases of C/EBPα, -β, and -δ mRNA or protein were observed in MNR male but not female fetal liver (Fig. 4A–D). Thus, these sexually dimorphic, MNR-induced changes followed the same pattern of changes of 11β-HSD1 and H6PD in the fetal perirenal adipose and liver tissues described above.
FIG. 4.

C/EBPα, -β, and -δ mRNA and protein levels in fetal perirenal adipose and liver tissues of control (CTR) and MNR baboons at 0.9 gestation. mRNA levels of C/EBPα, -β, and -δ in fetal perirenal adipose (A) and liver (C) tissues. C/EBPα, -β, and -δ protein levels in fetal perirenal adipose (B) and liver (D) tissues. Top and bottom panels of each figure are images of Western blots and the mean protein levels, respectively. CTR groups included 5 male and 5 or 6 female baboons; MNR groups included 4–6 male and 4–7 female baboons. * P < 0.05, **P < 0.01, vs. male CTR; #P < 0.05, ##P < 0.01, vs. female CTR.
Expression of PEPCK1 and PPARγ in the fetal liver and perirenal adipose tissues.
There were no significant differences in PEPCK1 and PPARγ mRNA levels between sexes of the control group in either liver or adipose tissue. PPARγ mRNA and protein were increased significantly only in MNR female fetal perirenal adipose tissue, not in male MNR fetuses (Fig. 5A and B). MNR-induced changes in PEPCK1 mRNA and protein in fetal liver tissue were observed only in MNR male fetuses, not female fetuses (Fig. 5C and D). This increased expression of PEPCK in the fetal liver, due to an increased gluconeogenetic response to the MNR insult at this stage, may account for the unchanged fetal plasma glucose level.
FIG. 5.

PPARγ and PEPCK1 mRNA and protein levels in fetal perirenal adipose or liver tissue of control (CTR) and MNR baboons at 0.9gestation. A and B: mRNA and protein levels of PPARγ in fetal perirenal adipose tissue. C and D: mRNA and protein levels of PEPCK1 in fetal liver tissue. Top and bottom panels of B and D are images of Western blots and the mean protein levels, respectively. CTR groups included 5 male and 5 or 6 female baboons; MNR groups included 4–6 male and 4–7 female baboons. *P < 0.05, **P < 0.01, vs. male CTR; #P < 0.05, ##P < 0.01, vs. female CTR.
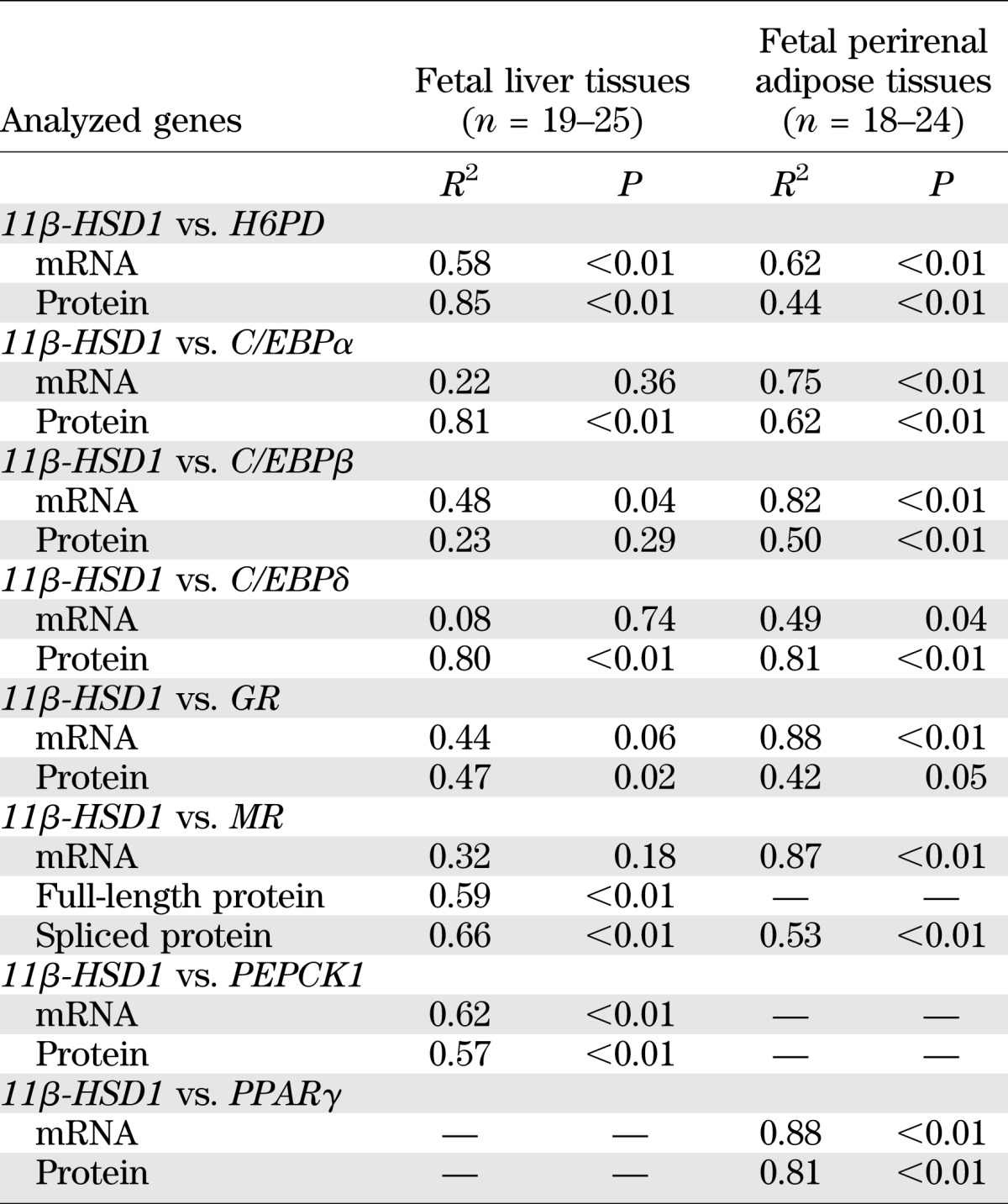
Correlation analysis of expression levels of the genes involved in cortisol regeneration and glucocorticoid-target genes in the fetal liver and perirenal adipose tissues.
ChIP assay revealed that there were significantly more 11β-HSD1 promoter DNA fragments precipitated by the C/EBPα, GR, and acetylated H3K9 antibodies than by the preimmune IgG in the liver tissue and by the GR, MR, and acetylated H3K9 antibodies in the adipose tissue (Fig. 6). The amount of 11β-HSD1 promoter DNA fragments precipitated by MR antibody and C/EBPα antibodies also reached marginal statistical significance in the liver (P = 0.07) and adipose tissue (P = 0.05), respectively. Consistent with the ChIP data, bivariate correlation analysis revealed that the expression level of 11β-HSD1 in both adipose and liver tissue was significantly correlated with C/EBPα, GR, and MR at either the mRNA or protein level (Table 3). In addition, the expression level of 11β-HSD1 was also significantly correlated with C/EBPβ and -δ, H6PD, PPARγ, and PEPCK1 at either the mRNA or protein level in both adipose and liver tissue (Table 3).
FIG. 6.

Chromatin immunoprecipitation assay demonstrating the binding of C/EBPα, GR, and MR and the enrichment of acetylated histone3 lysine 9 (H3K9ac) to the 11β-HSD1 promoter in fetal baboon liver and adipose tissues. The top panel illustrates the C/EBP and GR binding sites on the 11β-HSD1 promoter (−1,000 base pairs) based on the human 11β-HSD1 promoter sequence, and the position of the primers designed to amplify the 160-base pair PCR product spanning the glucocorticoid response element (GRE), C/EBP binding site, and transcription start site (TSS). The representative amplification curves of real-time PCR are shown above each bar graph of the mean data showing the amounts of 11β-HSD1 promoter DNA fragments precipitated by C/EBPα, GR, MR, and H3K9ac antibodies and preimmune IgG. n = 3–12. *P < 0.05, **P < 0.01, vs. preimmune IgG.
TABLE 3.
Bivariate correlation analyses of the correlation of 11β-HSD1 with H6PD, C/EBPα/-β/-δ, GR, MR, and PEPCK1 or PPARγ
DISCUSSION
Epidemiologic studies provide compelling evidence for an association between low birth weight and obesity, ischemic heart disease, and type 2 diabetes in later life (35,36). The effect of MNR on birth weight and subsequent disease during adulthood is well demonstrated in studies of exposure to famine, most notably the Dutch Hunger Winter (36). This developmental programming of adult disease by MNR has been supported extensively by studies of a variety of animal models (37). However, the molecular mechanism underlying the developmental programming of disease may differ between sexes. Maternal malnutrition during early but not mid and late gestation is associated with higher BMI and waist circumference later in life only in female (not male) offspring at 50 years of age (36), whereas MNR seems to affect the vascular and nephronic function mainly in male offspring, leading to the development of hypertension in adulthood (38,39). Although metabolic syndrome may manifest in offspring of both sexes exposed to MNR, glucose intolerance in male offspring has been ascribed to insulin resistance, whereas, in female offspring, glucose intolerance is reported to be due to insulin deficiency, further suggesting possible sex-specific mechanisms (40). These observations suggest that a disturbance of endocrine regulatory systems established during early gestation may contribute to sexually dimorphic development of metabolic syndrome in later life.
Among the endocrine disturbances, a chronic increase in either systemic or local glucocorticoids in the metabolic tissues is well recognized to play a central role in the development of metabolic syndrome (8). In situations of increased demand for glucocorticoid actions, glucocorticoids seem to have evolved to mobilize stored peripheral energy, directing it to central depots that serve the substrate needs of life-saving hepatic gluconeogenesis. However, excessive glucocorticoid activity is considered a major cause of metabolic syndrome (8,14,41). Increased expression of 11β-HSD1 in the liver and adipose tissues has been demonstrated in a number of animal models of developmental programming (9,22,23,42). In this study we demonstrated, for the first time, increased expression of 11β-HSD1 in the fetal liver and perirenal adipose tissues in a sex-specific manner in a primate model of MNR. Baboons share more than 96% genomic homology with human and thus are an excellent model for translation to the human situation with regard to developmental programming. Consistent with the sex-specific changes in the expression of 11β-HSD1 in the liver and adipose tissues, we also observed sexually dimorphic changes of H6PD, the enzyme that provides NADPH for the reductase activity of 11β-HSD1 (27). The role of H6PD in 11β-HSD1 activity is well demonstrated by a study showing H6PD knock-out mice lack 11β-HSD1-mediated generation of glucocorticoids (27). The finding of parallel increases of H6PD and 11β-HSD1 in the male fetal liver and the female fetal perirenal adipose tissue suggest that this parallel up-regulation of H6PD may be a fundamental feature in the increase in reductase activity of 11β-HSD1.
Sequence analysis of the 11β-HSD1 gene promoter reveals multiple C/EBP and GR binding sites (25). The C/EBP family and GR have been demonstrated to regulate basal and induced expression of 11β-HSD1 in a number of studies (25,26,43,44). In this study, C/EBPα and GR were found bound to the 11β-HSD1 gene promoter in baboon liver and adipose tissue, suggesting the transcriptional expression of 11β-HSD1 may also be under the regulation of C/EBPα and GR in baboons.
C/EBPs are transcription factors that are enriched by liver and adipose tissue and control numerous genes involved in energy metabolism and adipocyte differentiation. Knocking out or attenuating the genes expressing C/EBPα, -β, and -δ causes hypoglycemia and blocks adipocyte differentiation and lipid accumulation, suggesting a crucial role for these C/EBPs in energy metabolism, adipocyte differentiation, and obesity (45,46). Thus the findings of increased expression of C/EBP in the liver and perirenal adipose tissues may account not only for the increased 11β-HSD1 in these tissues but also for the disturbance in energy metabolism in the liver of male offspring and exaggerated adipocyte differentiation in female offspring programmed by MNR.
In addition to GR, we also observed a sexually dimorphic change of MR in the liver and adipose tissues in the offspring of MNR mothers. MR is suggested to mediate some of the specific actions or regulate the actions of glucocorticoids via either homodimerization or heterodimerization with GR (28,47) and contribute significantly to glucocorticoid-induced adipogenesis (28). The sexually dimorphic changes of MR may thus play an important role in the developmental programming of metabolic syndrome by glucocorticoids. Of interest, we found the splice variant MRΔ5,6, which lacks the ligand binding domain (47), predominated in both liver and adipose tissues. At this time we have no explanation for the dominant expression of MRΔ5,6 in these tissues of the baboon.
Expression of PEPCK1 and PPARγ was increased in the liver of male offspring and in the perirenal adipose tissue of the female offspring, respectively. Because both PEPCK1 and PPARγ are important glucocorticoid target proteins in liver and adipose tissues (29,30), the increased expression of PEPCK1 and PPARγ may be the consequence of increased regeneration of cortisol by 11β-HSD1 in these tissues or increased circulating cortisol in the fetal blood. We found that MNR significantly increased the cortisol level in both fetal and maternal circulation as well as the local level of cortisol in the perirenal adipose tissue of female fetuses, although the increase in cortisol level in the male MNR fetal liver tissue did not reach significance. Because the expression of 11β-HSD1 is induced in a feed-forward manner by glucocorticoids via GR and C/EBPs (26,43), we postulate that increased circulating cortisol resulting from MNR induces the expression of 11β-HSD1 in the major metabolic tissues, including the liver and adipose tissues, thus forming a feed-forward cycle in terms of cortisol regeneration and the expression of glucocorticoid-target genes involved in energy metabolism and adipocyte differentiation. This positive feedback cycle could be detrimental by exposing the fetus to glucocorticoid levels higher than are appropriate for the current stage of maturation, thereby predisposing the fetus to the development of hyperglycemia, insulin resistance, and obesity in later life.
Epigenetic modulation of these genes—particularly PEPCK1, H6PD, GR, and C/EBPs that carry multiple CpG islands in their promoters—by excessive cortisol may also provide a mechanism for their increased expression in later life (4,32,48); glucocorticoid-induced gene demethylation has been shown to contribute to establishing a “memory” or “marker” of the developmental nutritional challenge (49,50).
Conclusions.
We have demonstrated that moderately reduced nutrient availability during pregnancy increased circulating cortisol levels in the fetus. This increased cortisol level potentially triggers a cycle of positive feed-forward induction of 11β-HSD1 via C/EBPs, GR, and MR in a sex-specific manner. The parallel increase of H6PD would ensure an enhanced reductase activity of 11β-HSD1, thus increasing the local cortisol level, with subsequent increased expression of glucocorticoid-target genes PEPCK1 and PPARγ in major metabolic tissues such as liver and adipose tissues. These findings may explain sexual dimorphism in programming of the susceptibility to metabolic syndrome.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (HD-31514), National Institute of Child Health and Human Development (HD-21350), and the National Key Basic Research Program of China (2011CB944403).
No potential conflicts of interest relevant to this article were reported.
C.G. and K.S. designed the study. C.G., C.L., and K.S. researched data. L.M. and P.W.N. developed the animal model and contributed to the writing and editing of the manuscript. K.S. wrote the manuscript. C.G. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of data and the accuracy of the data analysis.
The authors thank Dr. Mark Nijland, University of Texas Health Science Center at San Antonio, for his contribution to the development of the animal model.
REFERENCES
- 1.Harding JE. The nutritional basis of the fetal origins of adult disease. Int J Epidemiol 2001;30:15–23 [DOI] [PubMed] [Google Scholar]
- 2.Desai M, Crowther NJ, Lucas A, Hales CN. Organ-selective growth in the offspring of protein-restricted mothers. Br J Nutr 1996;76:591–603 [DOI] [PubMed] [Google Scholar]
- 3.Choi GY, Tosh DN, Garg A, Mansano R, Ross MG, Desai M. Gender-specific programmed hepatic lipid dysregulation in intrauterine growth-restricted offspring. Am J Obstet Gynecol 2007;196:477.e1–477.e7 [DOI] [PubMed] [Google Scholar]
- 4.Zheng S, Rollet M, Pan YX. Maternal protein restriction during pregnancy induces CCAAT/enhancer-binding protein (C/EBPβ) expression through the regulation of histone modification at its promoter region in female offspring rat skeletal muscle. Epigenetics 2011;6:161–170 [DOI] [PubMed] [Google Scholar]
- 5.Choi J, Li C, McDonald TJ, Comuzzie A, Mattern V, Nathanielsz PW. Emergence of insulin resistance in juvenile baboon offspring of mothers exposed to moderate maternal nutrient reduction. Am J Physiol Regul Integr Comp Physiol 2011;301:R757–R762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 2005;85:571–633 [DOI] [PubMed] [Google Scholar]
- 7.Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull 2001;60:5–20 [DOI] [PubMed] [Google Scholar]
- 8.Dallman MF, Akana SF, Pecoraro NC, Warne JP, la Fleur SE, Foster MT. Glucocorticoids, the etiology of obesity and the metabolic syndrome. Curr Alzheimer Res 2007;4:199–204 [DOI] [PubMed] [Google Scholar]
- 9.Whorwood CB, Firth KM, Budge H, Symonds ME. Maternal undernutrition during early to midgestation programs tissue-specific alterations in the expression of the glucocorticoid receptor, 11beta-hydroxysteroid dehydrogenase isoforms, and type 1 angiotensin ii receptor in neonatal sheep. Endocrinology 2001;142:2854–2864 [DOI] [PubMed] [Google Scholar]
- 10.Bertram C, Trowern AR, Copin N, Jackson AA, Whorwood CB. The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11beta-hydroxysteroid dehydrogenase: potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology 2001;142:2841–2853 [DOI] [PubMed] [Google Scholar]
- 11.Wake DJ, Rask E, Livingstone DE, Söderberg S, Olsson T, Walker BR. Local and systemic impact of transcriptional up-regulation of 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue in human obesity. J Clin Endocrinol Metab 2003;88:3983–3988 [DOI] [PubMed] [Google Scholar]
- 12.Rask E, Olsson T, Söderberg S, et al. Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab 2001;86:1418–1421 [DOI] [PubMed] [Google Scholar]
- 13.Strain GW, Zumoff B, Strain JJ, Levin J, Fukushima DK. Cortisol production in obesity. Metabolism 1980;29:980–985 [DOI] [PubMed] [Google Scholar]
- 14.Walker BR, Andrew R. Tissue production of cortisol by 11beta-hydroxysteroid dehydrogenase type 1 and metabolic disease. Ann N Y Acad Sci 2006;1083:165–184 [DOI] [PubMed] [Google Scholar]
- 15.Tomlinson JW, Walker EA, Bujalska IJ, et al. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev 2004;25:831–866 [DOI] [PubMed] [Google Scholar]
- 16.Gaillard D, Wabitsch M, Pipy B, Négrel R. Control of terminal differentiation of adipose precursor cells by glucocorticoids. J Lipid Res 1991;32:569–579 [PubMed] [Google Scholar]
- 17.Chrousos GP. Is 11beta-hydroxysteroid dehydrogenase type 1 a good therapeutic target for blockade of glucocorticoid actions? Proc Natl Acad Sci U S A 2004;101:6329–6330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kotelevtsev Y, Holmes MC, Burchell A, et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci U S A 1997;94:14924–14929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morton NM, Holmes MC, Fiévet C, et al. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem 2001;276:41293–41300 [DOI] [PubMed] [Google Scholar]
- 20.Masuzaki H, Paterson J, Shinyama H, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001;294:2166–2170 [DOI] [PubMed] [Google Scholar]
- 21.Paterson JM, Morton NM, Fievet C, et al. Metabolic syndrome without obesity: Hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci U S A 2004;101:7088–7093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McMillen IC, Warnes KE, Adams MB, Robinson JS, Owens JA, Coulter CL. Impact of restriction of placental and fetal growth on expression of 11beta-hydroxysteroid dehydrogenase type 1 and type 2 messenger ribonucleic acid in the liver, kidney, and adrenal of the sheep fetus. Endocrinology 2000;141:539–543 [DOI] [PubMed] [Google Scholar]
- 23.Nyirenda MJ, Carter R, Tang JI, et al. Prenatal programming of metabolic syndrome in the common marmoset is associated with increased expression of 11beta-hydroxysteroid dehydrogenase type 1. Diabetes 2009;58:2873–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.VandeBerg JL, Williams-Blangero S. Advantages and limitations of nonhuman primates as animal models in genetic research on complex diseases. J Med Primatol 1997;26:113–119 [DOI] [PubMed] [Google Scholar]
- 25.Williams LJ, Lyons V, MacLeod I, et al. C/EBP regulates hepatic transcription of 11beta -hydroxysteroid dehydrogenase type 1. A novel mechanism for cross-talk between the C/EBP and glucocorticoid signaling pathways. J Biol Chem 2000;275:30232–30239 [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, Guo C, Zhu P, Li W, Myatt L, Sun K. Role of glucocorticoid receptor and CCAAT/enhancer-binding protein alpha in the feed-forward induction of 11beta-hydroxysteroid dehydrogenase type 1 expression by cortisol in human amnion fibroblasts. J Endocrinol 2007;195:241–253 [DOI] [PubMed] [Google Scholar]
- 27.Lavery GG, Walker EA, Draper N, et al. Hexose-6-phosphate dehydrogenase knock-out mice lack 11 beta-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J Biol Chem 2006;281:6546–6551 [DOI] [PubMed] [Google Scholar]
- 28.Marzolla V, Armani A, Zennaro MC, et al. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol Cell Endocrinol 2012;350:281–288 [DOI] [PubMed] [Google Scholar]
- 29.Vidal-Puig AJ, Considine RV, Jimenez-Liñan M, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest 1997;99:2416–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cassuto H, Kochan K, Chakravarty K, et al. Glucocorticoids regulate transcription of the gene for phosphoenolpyruvate carboxykinase in the liver via an extended glucocorticoid regulatory unit. J Biol Chem 2005;280:33873–33884 [DOI] [PubMed] [Google Scholar]
- 31.Schlabritz-Loutsevitch NE, Howell K, Rice K, et al. Development of a system for individual feeding of baboons maintained in an outdoor group social environment. J Med Primatol 2004;33:117–126 [DOI] [PubMed] [Google Scholar]
- 32.Nijland MJ, Mitsuya K, Li C, et al. Epigenetic modification of fetal baboon hepatic phosphoenolpyruvate carboxykinase following exposure to moderately reduced nutrient availability. J Physiol 2010;588:1349–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grieves JL, Dick EJ, Jr, Schlabritz-Loutsevich NE, et al. Barbiturate euthanasia solution-induced tissue artifact in nonhuman primates. J Med Primatol 2008;37:154–161 [DOI] [PubMed] [Google Scholar]
- 34.Tanuma Y, Tamamoto M, Ito T, Yokochi C. The occurrence of brown adipose tissue in perirenal fat in Japanese. Arch Histol Jpn 1975;38:43–70 [DOI] [PubMed] [Google Scholar]
- 35.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet 1989;2:577–580 [DOI] [PubMed] [Google Scholar]
- 36.Ravelli AC, van Der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr 1999;70:811–816 [DOI] [PubMed] [Google Scholar]
- 37.Barnes SK, Ozanne SE. Pathways linking the early environment to long-term health and lifespan. Prog Biophys Mol Biol 2011;106:323–336 [DOI] [PubMed] [Google Scholar]
- 38.Brawley L, Itoh S, Torrens C, et al. Dietary protein restriction in pregnancy induces hypertension and vascular defects in rat male offspring. Pediatr Res 2003;54:83–90 [DOI] [PubMed] [Google Scholar]
- 39.Bagby SP. Maternal nutrition, low nephron number, and hypertension in later life: pathways of nutritional programming. J Nutr 2007;137:1066–1072 [DOI] [PubMed] [Google Scholar]
- 40.Shepherd PR, Crowther NJ, Desai M, Hales CN, Ozanne SE. Altered adipocyte properties in the offspring of protein malnourished rats. Br J Nutr 1997;78:121–129 [DOI] [PubMed] [Google Scholar]
- 41.Wamil M, Seckl JR. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 as a promising therapeutic target. Drug Discov Today 2007;12:504–520 [DOI] [PubMed] [Google Scholar]
- 42.Nammi S, Dembele K, Nyomba BL. Increased 11beta-hydroxysteroid dehydrogenase type-1 and hexose-6-phosphate dehydrogenase in liver and adipose tissue of rat offspring exposed to alcohol in utero. Am J Physiol Regul Integr Comp Physiol 2007;292:R1101–R1109 [DOI] [PubMed] [Google Scholar]
- 43.Yang Z, Zhu P, Guo C, Zhu X, Sun K. Expression of 11beta-hydroxysteroid dehydrogenase type 1 in human fetal lung and regulation of its expression by interleukin-1beta and cortisol. J Clin Endocrinol Metab 2009;94:306–313 [DOI] [PubMed] [Google Scholar]
- 44.Ignatova ID, Kostadinova RM, Goldring CE, Nawrocki AR, Frey FJ, Frey BM. Tumor necrosis factor-alpha upregulates 11beta-hydroxysteroid dehydrogenase type 1 expression by CCAAT/enhancer binding protein-beta in HepG2 cells. Am J Physiol Endocrinol Metab 2009;296:E367–E377 [DOI] [PubMed] [Google Scholar]
- 45.Tanaka T, Yoshida N, Kishimoto T, Akira S. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J 1997;16:7432–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang ND, Finegold MJ, Bradley A, et al. Impaired energy homeostasis in C/EBP alpha knockout mice. Science 1995;269:1108–1112 [DOI] [PubMed] [Google Scholar]
- 47.Zennaro MC, Souque A, Viengchareun S, Poisson E, Lombès M. A new human MR splice variant is a ligand-independent transactivator modulating corticosteroid action. Mol Endocrinol 2001;15:1586–1598 [DOI] [PubMed] [Google Scholar]
- 48.Turner JD, Pelascini LP, Macedo JA, Muller CP. Highly individual methylation patterns of alternative glucocorticoid receptor promoters suggest individualized epigenetic regulatory mechanisms. Nucleic Acids Res 2008;36:7207–7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomassin H, Flavin M, Espinás ML, Grange T. Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J 2001;20:1974–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wyrwoll CS, Mark PJ, Waddell BJ. Developmental programming of renal glucocorticoid sensitivity and the renin-angiotensin system. Hypertension 2007;50:579–584 [DOI] [PubMed] [Google Scholar]