

Abstract
Activation of protein kinase C (PKC) has been implicated in the pathogenesis of diabetic nephropathy with proteinuria and peritubular extracellular matrix production. We have previously shown that the PKC isoforms α and β mediate different cellular effects. PKC-β contributes to hyperglycemia-induced renal matrix production, whereby PKC-α is involved in the development of albuminuria. We further tested this hypothesis by deletion of both isoforms and used a PKC inhibitor. We analyzed the phenotype of nondiabetic and streptozotocin (STZ)-induced diabetic homozygous PKC-α/β double-knockout mice (PKC-α/β−/−). After 8 weeks of diabetes mellitus, the high-glucose–induced renal and glomerular hypertrophy as well as transforming growth factor-β1) and extracellular matrix production were diminished in the PKC-α/β−/− mice compared with wild-type controls. Urinary albumin/creatinine ratio also was significantly reduced, however, it was not completely abolished in diabetic PKC-α/β−/− mice. Treatment with CGP41252, which inhibits PKC-α and PKC-β, is able to prevent the development of albuminuria and to reduce existing albuminuria in type 1 (STZ model) or type 2 (db/db model) diabetic mice. These results support our hypothesis that PKC-α and PKC-β contribute to the pathogenesis of diabetic nephropathy, and that dual inhibition of the classical PKC isoforms is a suitable therapeutic strategy in the prevention and treatment of diabetic nephropathy.
Diabetic nephropathy is the most common cause of progressive chronic kidney disease and end-stage renal disease in the Western world (1). It is postulated that diabetic nephropathy may result from a local interplay of metabolic and hemodynamic factors either through direct effects of high-glucose levels or autocrine and paracrine actions of various vasoactive substances in the diabetic kidney (2). More than 20 years ago, it was described that activation of the protein kinase C (PKC) system by hyperglycemia may represent an important mediator of glucotoxicity in diabetic nephropathy (3,4). PKC constitutes a family of homologous serine/threonine kinases that are involved in many signaling events (5). In mammals, a gene family of nine independent gene loci is distributed over the whole genome (6). Because of biochemical properties and sequence homologies, the PKC family is divided into classical (α, β I, β II, γ), novel (δ, ɛ, η, θ), and atypical (ζ, ι/λ) isoforms. The functional role of distinct PKC isoforms in the development of diabetic nephropathy has recently been further elucidated by means of single isoform-specific knockout mice (7–13). We and others have revealed that activation of the PKC-β isoform contributes to high-glucose–induced, transforming growth factor (TGF)-β1–mediated renal hypertrophy and extracellular matrix expansion (9,14), whereas perlecan and vascular endothelial growth factor (VEGF) as well as nephrin expression are regulated by a PKC-α–dependent signaling pathway leading to diabetic albuminuria (7,8).
To further understand the role of PKC isoforms in the development of diabetic nephropathy, we characterized the renal phenotype of homozygous PKC-α/β double knockout (KO) mice and tested the hypothesis that deletion of both classical PKC isoforms, PKC-α and PKC-β, is able to completely abolish the development of experimental diabetic nephropathy in the streptozotocin (STZ)-induced diabetic stress model. Furthermore, we also tested if pharmacological inhibition of the classical PKC isoforms with the classical PKC inhibitor CGP41251, the N-Benzoyl derivative of the naturally occurring alkaloid staurosporine, is safely achievable and beneficial in type 1 (STZ model) and type 2 diabetic (db/db mice) animal models. CGP41251 previously has been used in several phase I–III cancer trials, showing an IC50 for the classical PKC isoforms of ∼20–30 nmol/L and for the novel isoforms between 160 and 1,250 nmol/L (15).
RESEARCH DESIGN AND METHODS
Animal studies.
Experiments were performed with male 129/SV wild-type (WT) and PKC-α/β−/− KO mice. The latter were generated by breeding PKC-α−/− and PKC-β−/−, which have the same 129/SV background strain (7,9). The heterozygous PKC-α−/− and PKC-β−/− mice from the F1 generation were paired and double KO mice were identified by genetic testing. The breeding was difficult because a maximum of one double KO mouse was found per litter. Although mice homozygotes for both mutations are viable and have normal life expectancy, they were not fertile. In consequence, no direct breeding of PKC-α/β−/− KO mice was possible to build a colony, which significantly limited our experimental setting for this study. A head-to-head comparison with the single KO PKC-α−/− and PKC-β−/− KO mouse strain was not performed; instead, historical controls were used. Pharmacological studies with CGP41251 were performed in 8- to 10-week-old male 129/SV (Harlan) or 8-week-old db/db mice (Charles River). The animals received a standard diet with free access to tap water. All procedures were performed according to guidelines from the Federation of European Laboratory Animal Science Associations and were approved by local authorities. The 8- to 10-week-old mice received either 125 mg/kg body weight STZ (Sigma-Aldrich) in 50 mmol/L sodium citrate (pH 4.5) or sodium citrate buffer intraperitoneally on days 1 and 4. Glucose levels from tail blood were measured with the glucometer Elite (Bayer, Leverkusen, Germany) every other day. Animals with glucose levels ≥16 mmol/L on two consecutive measurements were regarded as hyperglycemic and glucose measurements were controlled every 2 weeks. The mice received no insulin.
Pharmacological treatment.
CGP41251 was synthesized by LC Laboratories with a purity of 99% and mixed into the standard chow. In a pilot study, blood concentration of CGP41251 was measured by mass spectroscopy (LC laboratories). In the STZ-induced diabetic 129/SV mice, treatment with CGP41251 (1 mg/kg) was started immediately after the mice became diabetic, respectively, after 4 weeks and 12 weeks of diabetes. In the db/db mouse model, the pharmacological treatment was started immediately. ProQinase GmbH (Freiburg, Germany) determined the IC50 for the different PKC isoforms (α, β 1, β 2, γ, δ, ε, θ, and ζ) by a radiometric filter-binding kinase assay.
Clinical chemistry.
In animals participating in the pharmacological intervention, 24-h urine was collected using metabolic cages (Tecniplast). Albumin concentration was measured with a commercially available competitive ELISA following the instructions of the manufacturer (Exocell, Philadelphia, PA) and was normalized to urine creatinine defined as the albumin/creatinine ratio. In the KO mice, urine was collected by puncturing the bladder with a 23-gauge needle during organ removal. Albumin was quantified as described and creatinine was measured by means of the chemistry analyzer Olympus AU400 (Olympus) using standard reagents. The albumin/creatinine ratio was then calculated.
Histological assessment.
We obtained the animal kidneys for further morphological and immunohistochemical analyses. For this purpose, animals were euthanized according to the following protocol: after anesthesia with isofluran via a nasal mask (2%) was administered, a laparotomy was performed. The organs were perfused with lactated Ringer solution via the left ventricle and the left kidney was removed after clipping of the renal artery and immediately frozen in liquid nitrogen for Western blotting. Subsequently, the right kidney was perfused with 3% paraformaldehyde, removed, and fixed for an additional 20 h in 3% paraformaldehyde, and then embedded in paraffin, sectioned at 2 µm. For morphological evaluation, paraffin sections were stained with trichrome Masson-Goldner staining and Sirius red staining following a standard protocol.
Immunohistochemistry was performed using the following primary antibodies: perlecan (RDI, Flanders, NJ), nephrin (Progen, Heidelberg, Germany), VEGF (Santa Cruz Biotechnology, Santa Cruz, CA), nitrotyrosine, fibronectin (Abcam, Cambridge, U.K.), type IV collagen (Southern Biotech, Birmingham, AL), Wilms tumor-1, and TGF-β1 (Santa Cruz Biotechnology). For indirect immunofluorescence, nonspecific binding sites were blocked with 10% normal donkey serum (Jackson ImmunoResearch Laboratory) for 30 min. Then, sections were incubated with the primary antibody for 1 h. All incubations were performed in a humid chamber at room temperature. For fluorescent visualization of bound primary antibodies, sections were further incubated with Cy3 conjugated secondary antibodies (Jackson ImmunoResearch Laboratory) for 1 h. Specimens were analyzed using a Zeiss Axioplan-2 imaging microscope with the computer program AxioVision 4.8 (Zeiss, Jena, Germany). Semi-quantitative analyses for the various target proteins were performed mainly by counting the numbers of glomeruli with distinct expression.
Western blot.
Kidney fragments were homogenized in modified RIPA lysis buffer (50 mmol/L Tris-HCl, pH 7.4; 1% Triton X-100; 0.25% Na-deoxycholate; 150 mmol/L NaCl; 1 mmol/L EDTA) supplemented with 1 mmol/L PMSF, 1 µg/mL aprotinin, 1 µg/mL leupeptin, 1 mmol/L Na3VO4, and 1 mmol/L NaF. Homogenates were clarified by centrifugation at 12,800 rpm for 10 min and were used for electrophoresis. Semi-dry Western blotting of PVDF membrane (Roche Applied Science) was performed using Bio-Rad Mini-PROTEAN system. VEGF and TGF-β1 were quantified using the same antibody as used for immunochemistry. Chemiluminescent Western blotting images were captured using VersaDoc-3000 and were quantified using Quantity One software (Bio-Rad Laboratories).
Statistics.
Data are shown as mean ± SD (if not otherwise indicated). The data were compared by ANOVA with Bonferroni as post hoc test. Significant differences were accepted with P < 0.05. Data analysis was performed using Prism 5.0 software (GraphPad Software).
RESULTS
First, we performed our investigation of PKC-α/β−/− double KO mice in nondiabetic and diabetic states to analyze early changes of the renal phenotype in comparison with appropriate WT littermates. Hyperglycemia (nonfasted values) persisted in both WT (37.7 ± 4.8 mmol/L) and KO (40.3 ± 5.6 mmol/L) animals 8 weeks after STZ injection as compared with 8.6 ± 1.1 and 10.4 ± 1.2 mmol/L, respectively, in the appropriate sham-injected control mice (Table 1).
TABLE 1.
Blood glucose levels and body/kidney ratio as well as glomerular changes and proteinuria levels in WT and PKC-α/β−/− mice after 8 weeks of diabetes
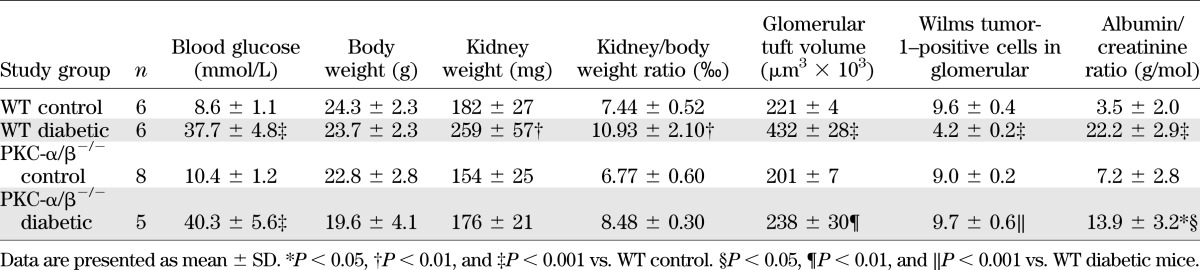
Because we previously have shown that PKC-α−/− mice are protected against the development of albuminuria in the diabetic state (7,8), we then tested albuminuria in our PKC-α/β−/− mice (Table 1). After 8 weeks of diabetes, the albumin/creatinine ratio was significantly increased in the diabetic WT mice with a median of 22.0 ± 3.1 g/mol compared with 3.1 ± 1.9 g/mol in the sham-injected control WT animals (P < 0.001). Notably, the albumin/creatinine ratio was significantly diminished in hyperglycemic PKC-α/β−/− mice compared with diabetic WT animals (13.9 ± 3.2 g/mol; P < 0.05). However, there was still a notable amount of albuminuria when compared with nondiabetic KO controls (7.2 ± 2.8 g/mol; P < 0.05).
We previously have suggested that the prevention of albuminuria in STZ-induced diabetic PKC-α−/− mice is possibly related to diabetes-induced expression changes of the heparan sulfate proteoglycan perlecan (7) or the slit membrane protein nephrin (8). Furthermore, we have shown that suppression of the VEGF system contributes to the prevention of albuminuria in diabetic PKC-α−/− mice (7). We observed only a weak expression level of VEGF, mainly located in podocytes, in nondiabetic study mice (Fig. 1A–E). Under hyperglycemic conditions, a significant increase of VEGF was observed in WT animals. Notably, this increase was significantly reduced in diabetic PKC-α/β−/− mice. This finding was confirmed by Western blotting (data not shown). The expressions of perlecan (Fig. 1F–J) and nephrin (Fig. 1K–O) were reduced in the glomeruli of diabetic WT animals, whereas the diabetes-induced loss was prevented in PKC-α/β−/− mice, as previously described in diabetic PKC-α−/− mice (7).
FIG. 1.

Expression levels of VEGF (A–E), perlecan (F–J), nephrin (K–O), and nitrotyrosine (P–T) in the glomerulus/renal cortex of nondiabetic and diabetic WT mice and diabetic PKC-α/β−/− (dKO) mice, and of diabetic mice treated with CGP41251 (1 mg/kg) after 8 weeks of hyperglycemia. *P < 0.01 vs. WT; **P < 0.05 vs. WT. (A high-quality color representation of this figure is available in the online issue.)
We next studied renal and glomerular hypertrophy as well as extracellular matrix expansion in our PKC-α/β−/− double KO model because we previously proved protection from fibrotic events in diabetic PKC-β−/− single KO mice (9). In this study, the kidney weight in the diabetic WT group was significantly augmented, indicating renal hypertrophy in the early phase of diabetic nephropathy (Table 1). Interestingly, the kidney weight did not increase significantly in the diabetic double KO group, suggesting prevention of renal hypertrophy in addition to albuminuria in the diabetic PKC-α/β−/− mice. When calculating the kidney/body weight ratio, we found a significant increase in the diabetic WT animals, whereas deletion of both PKC-α and PKC-β isoforms prevented renal hypertrophy during hyperglycemia (Table 1). Furthermore, we then analyzed glomerular tuft volume in our PKC-α/β−/− mouse model compared with appropriate controls. We revealed that in diabetic WT mice, glomerular tuft volume is significantly increased, whereas PKC-α/β−/− mice were protected from such changes under hyperglycemic conditions (Table 1). Additionally, the number of Wilms tumor-1–positive podocytes was reduced in diabetic WT mice, but not in diabetic KO mice (Table 1).
To study early structural changes in the kidneys from PKC-α/β−/− mice, we next performed Masson-Goldner trichrome staining of 8-week-old animals of all four study groups. The structural analysis revealed glomerular and extracellular matrix expansion in the WT diabetic mice, whereas in PKC-α/β−/− mice less extracellular matrix was detectable in the diabetic state (data not shown). A possible explanation for these findings is the observation that the profibrotic cytokine TGF-β1 expression was significantly upregulated in hyperglycemic WT animals, whereas diabetic PKC-α/β−/− mice showed a significantly reduced TGF-β1 protein expression (Fig. 2A–E). This finding was confirmed by Western blotting (data not shown). Consistent with the histological alterations and effects on TGF-β1 upregulation, we found less type IV collagen (Fig. 2F–J) and fibronectin (Fig. 2K–O) expression in diabetic PKC-α/β−/− mice, and Sirius red staining (Fig. 2P–T) revealed that the diabetic PKC-α/β−/− mice had less extracellular matrix between the tubuli than the WT mice.
FIG. 2.

Expression levels of TGF-β1 (A–E), type IV collagen (F–J), and fibronectin (K–O), as well as Sirius red staining (P–T), in the glomerulus/renal cortex of nondiabetic and diabetic WT mice, and of diabetic PKC-α/β−/− (dKO) mice and diabetic mice treated with CGP41251 (1 mg/kg) after 8 weeks of hyperglycemia. *P < 0.01 vs. WT; **P < 0.05 vs. WT.
One mechanism involved in diabetic nephropathy is oxidative stress, and we analyzed nitrotyrosine as a marker. Under diabetic conditions, nitrotyrosine was markedly upregulated, but it was significantly reduced in the PKC-α/β−/− mice, suggesting that some of the beneficial effects observed might be attributable to a reduction of oxidative stress (Fig. 1P–T).
To analyze if a pharmacological interventional approach shows a similar benefit as observed in the diabetic PKC-α/β−/− mice, we also performed experimental studies in two independent diabetic rodent models using the known classical PKC inhibitor CGP41251, which blocks PKC-α and PKC-β isoforms. as well as the PKC-γ isoform (which is, however, only present in neurons). First, we performed initial in vivo experiments to determine the dose to be administered to achieve sufficient blood concentration. The blood concentration was 100–300 nmol/L after 2–9 weeks of treatment with 1 mg/kg of CGP41251, whereas the administration of 3 mg/kg was associated with blood levels of 2,000–4,000 nmol/L, resulting in increased mortality in this group. Therefore, we decided to further study administration of 1 mg/kg CGP41251 to prevent the development of diabetic nephropathy in the STZ model (Table 2). Eight weeks of treatment did diminish the occurrence of albuminuria (Table 2) and had a positive effect on the nephrin and perlecan expression (Fig. 1). VEGF was only slightly modified. We observed no significant effect on renal hypertrophy, and TGF-β1 expression was only reduced slightly (Table 2 and Fig. 2).
TABLE 2.
Blood glucose levels and body and kidney weight in untreated and treated 129SV and db/m and db/db mice with diabetes
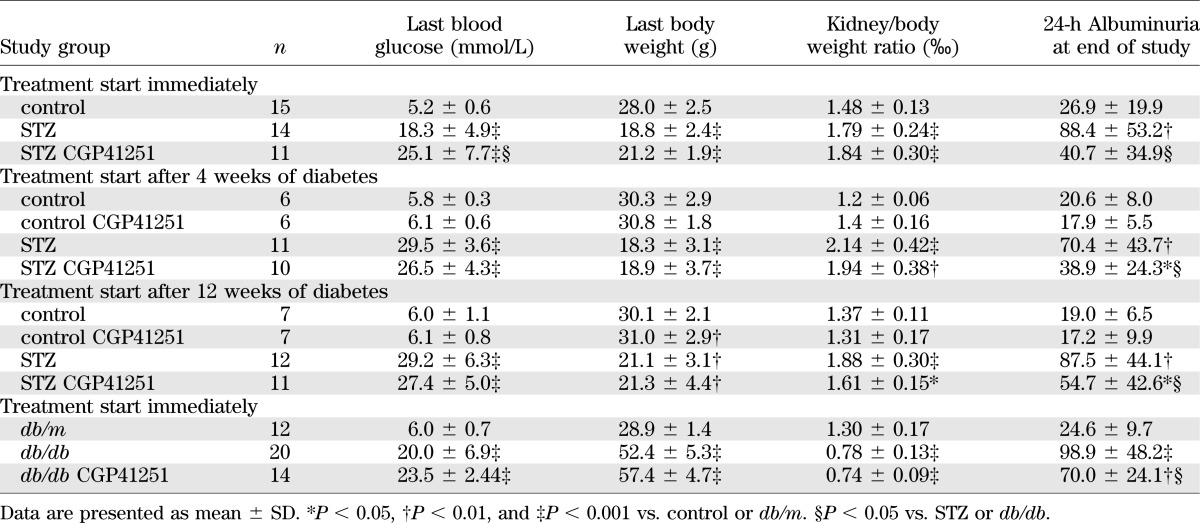
To determine if treatment with CGP41251 could improve already existing diabetic kidney damage, we started treatment after 4 and 12 weeks of diabetes. During the 8-week treatment period, we observed a significant improvement of albuminuria (Table 2 and Fig. 3A and B), whereas the effect on renal hypertrophy was only mild.
FIG. 3.
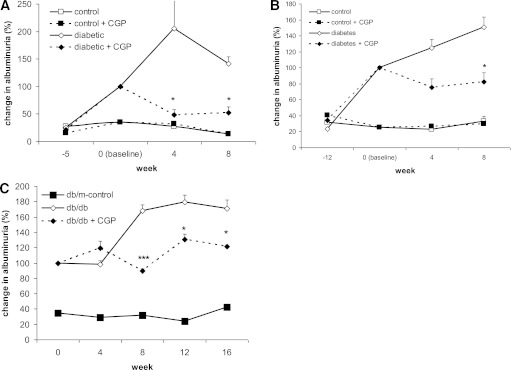
Change in albuminuria in 129/SV and db/db mice treated with CGP41251 (1 mg/kg). Treatment was started at time point 0. Albuminuria levels in the diabetic mice at time point 0 (baseline) were set at 100% for better comparison. A: Treatment was started after 4 weeks of diabetes in 129/SV mice with STZ-induced hyperglycemia. B: Treatment was started after 12 weeks of diabetes in 129/SV mice with STZ-induced hyperglycemia. C: Treatment was commenced in 8-week-old db/db mice. *P < 0.01, ***P < 0.001 vs. diabetic control.
Finally, we also tested if CGP41251 also would have an effect in a type 2 diabetic mouse model (db/db mice). We treated db/db mice for a total of 16 weeks. Treatment improved albuminuria but, again, had only little effect on renal hypertrophy (Table 2 and Fig. 3C).
Because the renal hypertrophy was only mildly affected, we wondered if CGP41251 is a specific classical PKC inhibitor. Therefore, we determined the IC50 of CGP41251 for the distinct PKC isoforms. Contrary to the published data, we found that the IC50 for classical PKC isoforms was between 40 and 60 nmol/L, and for novel PKC isoforms it was between 50 and 100 nmol/L, indicating that CGP41251 can be considered as a pan-PKC inhibitor.
DISCUSSION
We demonstrate that removal of the two classical PKC-α and PKC-β isoforms has a synergistic effect on experimental diabetic nephropathy models. Blocking both isoforms has a beneficial effect on the development of renal hypertrophy and on albuminuria under diabetic conditions. Additionally, we show that pharmaceutical inhibition of these two isoforms has a positive effect on albuminuria. These data extend our own findings from various PKC KO models that two important features of diabetic nephropathy, albuminuria and glomerular hypertrophy, are differentially regulated via PKC isoform–selective signaling events (Table 3) (7–12,16).
TABLE 3.
Expression of different proteins in diabetic WT, PKCα−/−, PKCβ−/−, and PKCα/β−/−
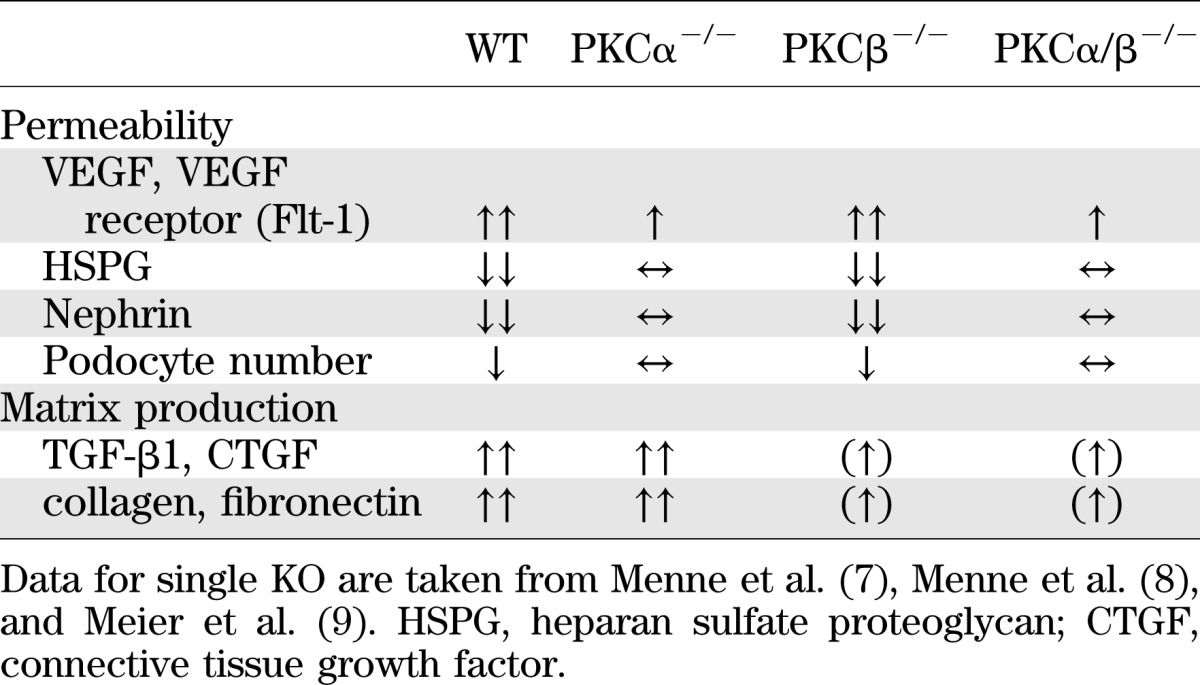
We previously have shown that diabetic PKC-β−/− mice demonstrate reduced renal hypertrophy and diminished high-glucose–induced expression of extracellular matrix (ECM) proteins and of the profibrotic cytokine TGF-β1 (9), which previously had been demonstrated to be unaltered in diabetic PKC-α−/− mice (7,8). However, we also had shown that diabetic PKC-α−/− mice are protected against the development of albuminuria, possibly because of effects on VEGF expression, a diminished loss of negatively charged heparan sulfate proteoglycans or the slit diaphragm protein nephrin (7,8). Furthermore, we recently characterized the molecular mechanism of the latter as the endocytosis of nephrin is PKC-α–dependent (17). These previous studies indicate that a cell-type–specific expression pattern of the various PKC isoforms is pivotal, mainly in such a complex cellular structure as the kidney. Therefore, we suggested that the PKC-β isoform is more important in the upregulation of TGF-β1 and for the development of renal hypertrophy and extracellular matrix production under hyperglycemic conditions, whereas PKC-α isoform seems to play a critical role in the development of albuminuria by perpetuating the glomerular filtration barrier. Notably, the breakdown of the glomerular filtration barrier with development of albuminuria in the diabetic PKC-β−/− mice did occur, although inhibition of TGF-β1 was obtained by the deletion of the PKC-β isoform.
In addition to the described molecular effects of the two classical PKC isoforms α and β, it is noteworthy that both are involved in many intracellular molecular pathways important for the development of diabetes complications. Several authors previously have demonstrated that PKC-α and PKC-β activations are associated with increased NADPH activity and NADPH-dependent superoxide production (14,18). Because excessive production of reactive oxygen species has been implicated in the pathogenesis of diabetic nephropathy, this observation might be clinically relevant, and we found that oxidative stress is reduced in PKC-α/β−/− mice. It also has been demonstrated that advanced glycation end products (AGEs) mediate at least some of their damaging effects via PKC-α signaling (18).
Our experimental data now suggest that it is more promising to inhibit PKC-α and PKC-β simultaneously in the treatment of diabetic nephropathy; otherwise, important components of long-term kidney damage (albuminuria, interstitial structural changes, or reactive oxygen stress) would not be sufficiently inhibited. Notably, the data obtained from the double KO underline this concept. Albuminuria, however, was not completely abolished. The reason for this observation remains unclear. One possible explanation could be an altered VEGF balance, which may be deleterious to the maintenance of the glomerular filtration barrier, regardless of whether it is up or down (19).
Generating PKC isoform–selective inhibitors that target the active enzyme has proven to be a challenge (6,20,21). Early PKC inhibitors developed for experimental use, such as staurosporine and 1-(5-isoquinolinylsulfonyl)-2-methylpiperazine (H7), were only capable of blocking the activity of all PKCs rather than individual PKC isoforms (22). Staurosporine-like bisindolylmaleimide compounds, such as CGP41251, Go-6850, Ro-31–8220, and UCN-01, were subsequently developed, and act at the ATP-binding site of the enzyme (23). Notably, the macrocyclic bisindolylmaleimide compound LY333531 (Ruboxistaurin; Arxxant; Eli Lilly, Indianapolis, IN) was synthesized and previously was shown to be a competitive, reversible inhibitor of both PKC-β 1 and PKC-β2 (24,25).
King and colleagues (26) were the first to hypothesize that the PKC-β isoform primarily is responsible for the glucose-induced effects leading to diabetic microvascular complications. Further evidence for this hypothesis stems from reports that the aforementioned specific PKC-β isoform inhibitor ruboxistaurin ameliorates hyperglycemia-induced changes in the kidney (27–29). In rats with STZ-induced (type 1 diabetes model) and db/db mice (type 2 diabetes model), both characteristic pathological events of diabetic nephropathy, the development of albuminuria and mesangial expansion, could be prevented by the administration of ruboxistaurin (28,29). Kelly et al. (30) also demonstrated in STZ-induced diabetic and hypertensive (mRen-2) rats, a rodent model that is transgenic for the entire mouse renin gene (Ren-2), that in vivo inhibition of the PKC-β isoform with ruboxistaurin led to a reduction in renal TGF-β1 expression and structural injury of the kidney, as well as albuminuria.
The expected benefits from preclinical trials have not (so far) translated into meaningful outcome improvements in clinical trials. First, a multicenter, double-blind, placebo-controlled study suggested an additive effect of ruboxistaurin compared with placebo in 123 type 2 diabetic patients with proteinuria (mean albumin/creatinine ratio 764 mg/g) or near-normal serum creatinine who were already treated optimally with an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker and intensive glycemic control (31). The investigators reported that 32 mg ruboxistaurin daily for up to 1 year resulted in a reduction in urinary albumin/creatinine ratio in the ruboxistaurin group compared with the placebo group. Renal function, examined by estimated glomerular filtration rate, was maintained with ruboxistaurin treatment. In contrast, participants in the placebo group had a significant decline in estimated glomerular filtration rate that was within the range reported in recent clinical trials of angiotensin receptor blockers and angiotensin-converting enzyme inhibitors in patients with type 2 diabetes mellitus and overt nephropathy (31). It is important to note, however, that the reduction in albumin/creatinine ratio compared with the baseline level was not significant compared with the decrease achieved by placebo, according to conventional intergroup comparison. Another recent meta-analysis of the three major diabetic retinopathy trials (PKC-DRS, PKC-DMES, PKC-DRS2) demonstrated that kidney outcome rates after 3 years did not differ by treatment assignment, whereas serum creatinine doubling was 6.0%, progression to higher stages of diabetic nephropathy was 3.5%, and decline in glomerular filtration rate was ∼3–4 mL/min−1/year−1 (32).
Campochiaro et al. (33) also published an interesting article in 2004 reporting the results of a clinical trial using the PKC inhibitor CGP41251 (PKC412) in diabetic patients with proliferative retinopathy. This trial was a randomized, multicenter, double-masked, parallel-group study in which subjects received placebo (n = 34) or 50 mg/d CGP41251 (n = 32), 100 mg (n = 38), or 150 mg/d (n = 37) for up to 3 months. Subjects were 18 to 85 years of age and had retinal thickening. Treatment with high doses of CGP41251 caused a significant decrease in thickening in the region of greatest thickening and in the fovea, a decrease of the retinal volume, and a small improvement in visual acuity at 3 months compared with baseline in the 100 mg/d CGP41251 group. We therefore tested, in the current study, whether this inhibitor also could prevent the development of diabetic microvascular complications such as experimental diabetic nephropathy. We found that the occurrence of albuminuria was prevented and that already existing albuminuria was reduced by the treatment, but it had only little effects on renal hypertrophy and, at higher doses, an increased mortality. Because we were surprised by this observation, we then tested again the specificity of this so-called classical PKC inhibitor and found that, contrary to published data, CGP41251 is a rather unspecific pan-PKC inhibitor (15). Interestingly, we previously have demonstrated that deletion of PKC-ɛ in specific KO mice causes development of glomerulosclerosis and albuminuria, even in the nondiabetic state (13). We therefore concluded that upregulation of this novel PKC isoform in diabetes mellitus may represent a protective response to injury. (13) Thus, it is tempting to speculate that we would have been able to demonstrate even clearer results in the current study if CGP41251 were a truly specific PKC-α/β inhibitor that does not interfere with the rather protective PKCɛ signaling pathway in experimental diabetic nephropathy (13). The three-dimensional structures of distinct PKC isoforms have been identified, which certainly will provide valuable information for more selective pharmacological drug design (6,34).
In conclusion, we believe that PKC isoform selectivity does occur in vivo and exerts distinct pivotal functions in the development of murine experimental diabetic nephropathy. The results from PKC-α/β double KO mice and using the (pan)-PKC inhibitor CGP41251 presented within this study suggest that isolated inhibition of the two classical PKC isoforms, α and β, is beneficial and that such a dual pharmacological approach should be considered in the treatment of diabetic microvascular complications such as diabetic nephropathy.
ACKNOWLEDGMENTS
This work was supported by a grant-in-aid from the European Foundation for the Study of Diabetes (EFSD)/SERVIER grant for vascular complications of type 2 diabetes mellitus to M.M. and by the German Research Council (DFG) to J.M. (ME3143/1-1).
No potential conflicts of interest relevant to this article were reported.
J.M. and M.M. wrote the manuscript. J.M., N.S., and M.M. designed experiments. J.M., N.S., J.B., Y.K., R.L., J.-K.P., and M.M. performed experiments. N.S., J.B., H.H., and J.-K.P. reviewed the manuscript. N.S., H.H., and J.-K.P. edited the manuscript. J.M. and M.M. are the guarantors of this work and, as such, had full access to all the data and take responsibility for the integrity of data and the accuracy of data analysis.
The authors thank Petra Berkefeld (Medical School Hannover, Hannover, Germany) for excellent technical assistance. The authors thank Dr. Leitges (Medical School Hannover, Hannover, Germany) for providing them with the PKC-α and PKC-β knockout mice.
Footnotes
See accompanying commentary, p. 1010.
REFERENCES
- 1.Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: Diagnosis, prevention, and treatment. Diabetes Care 2005;28:164–176 [DOI] [PubMed] [Google Scholar]
- 2.Cooper ME. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia 2001;44:1957–1972 [DOI] [PubMed] [Google Scholar]
- 3.Lee T-S, Saltsman KA, Ohashi H, King GL. Activation of protein kinase C by elevation of glucose concentration: proposal for a mechanism in the development of diabetic vascular complications. Proc Natl Acad Sci USA 1989;86:5141–5145 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 4.Craven PA, DeRubertis FR. Protein kinase C is activated in glomeruli from streptozotocin diabetic rats. Possible mediation by glucose. J Clin Invest 1989;83:1667–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dempsey EC, Newton AC, Mochly-Rosen D, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol 2000;279:L429–L438 [DOI] [PubMed] [Google Scholar]
- 6.Gould CM, Newton AC. The life and death of protein kinase C. Curr Drug Targets 2008;9:614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menne J, Park J-K, Boehne M, et al. Diminished loss of proteoglycans and lack of albuminuria in protein kinase C-alpha-deficient diabetic mice. Diabetes 2004;53:2101–2109 [DOI] [PubMed] [Google Scholar]
- 8.Menne J, Meier M, Park JK, et al. Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase C alpha signaling in-vivo. Kidney Int 2006;70:1456–1462 [DOI] [PubMed] [Google Scholar]
- 9.Meier M, Park JK, Overheu D, et al. Deletion of protein kinase C-β isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes 2007;56:346–354 [DOI] [PubMed] [Google Scholar]
- 10.Meier M, Menne J, Park J-K, Haller H. Nailing down PKC isoform specificity in diabetic nephropathy two's company, three's a crowd. Nephrol Dial Transplant 2007;22:2421–2425 [DOI] [PubMed] [Google Scholar]
- 11.Meier M, Menne J, Haller H. Targeting the protein kinase C family in the diabetic kidney: lessons from analysis of mutant mice. Diabetologia 2009;52:765–775 [DOI] [PubMed] [Google Scholar]
- 12.Menne J, Meier M, Park JK, Haller H. Inhibition of protein kinase C in diabetic nephropathy—where do we stand? Nephrol Dial Transplant 2009;24:2021–2023 [DOI] [PubMed] [Google Scholar]
- 13.Meier M, Menne J, Park JK, et al. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol 2007;18:1190–1198 [DOI] [PubMed] [Google Scholar]
- 14.Ohshiro Y, Ma RC, Yasuda Y, et al. Reduction of diabetes-induced oxidative stress, fibrotic cytokine expression, and renal dysfunction in protein kinase Cbeta-null mice. Diabetes 2006;55:3112–3120 [DOI] [PubMed] [Google Scholar]
- 15.Seo MS, Kwak N, Ozaki H, et al. Dramatic inhibition of retinal and choroidal neovascularization by oral administration of a kinase inhibitor. Am J Pathol 1999;154:1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meier M, Menne J, Park J-K, et al. Deletion of protein kinase C-{varepsilon} signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol 2007;18:1190–1198 [DOI] [PubMed] [Google Scholar]
- 17.Tossidou I, Teng B, Menne J, et al. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS ONE 2010;5:e10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thallas-Bonke V, Thorpe SR, Coughlan MT, et al. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes 2008;57:460–469 [DOI] [PubMed] [Google Scholar]
- 19.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 2003;111:707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen GX. Selective protein kinase C inhibitors and their applications. Curr Drug Targets Cardiovasc Haematol Disord 2003;3:301–307 [DOI] [PubMed] [Google Scholar]
- 21.Swannie HC, Kaye SB. Protein kinase C inhibitors. Curr Oncol Rep 2002;4:37–46 [DOI] [PubMed] [Google Scholar]
- 22.Way KJ, Chou E, King GL. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci 2000;21:181–187 [DOI] [PubMed] [Google Scholar]
- 23.Teicher BA. Protein kinase C as a therapeutic target. Clin Cancer Res 2006;12:5336–5345 [DOI] [PubMed] [Google Scholar]
- 24.Jirousek MR, Gillig JR, Gonzalez CM, et al. (S)-13-[(dimethylamino)methyl]-10,11,14,15-tetrahydro-4,9:16, 21-dimetheno-1H, 13H-dibenzo[e,k]pyrrolo[3,4-h][1,4,13]oxadiazacyclohexadecene-1,3(2H)-d ione (LY333531) and related analogues: isozyme selective inhibitors of protein kinase C beta. J Med Chem 1996;39:2664–2671 [DOI] [PubMed] [Google Scholar]
- 25.Ruboxistaurin: LY 333531. Drugs R D 2007;8:193–199 [DOI] [PubMed] [Google Scholar]
- 26.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci USA 1992;89:11059–11063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science 1996;272:728–731 [DOI] [PubMed] [Google Scholar]
- 28.Koya D, Jirousek MR, Lin Y-W, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest 1997;100:115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koya D, Haneda M, Nakagawa H, et al. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J 2000;14:439–447 [DOI] [PubMed] [Google Scholar]
- 30.Kelly DJ, Zhang Y, Hepper C, et al. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes 2003;52:512–518 [DOI] [PubMed] [Google Scholar]
- 31.Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care 2005;28:2686–2690 [DOI] [PubMed] [Google Scholar]
- 32.Tuttle KR, McGill JB, Haney DJ, Lin TE, Anderson PW. for the PKC-DRS P-D, and PKC-DRS 2 Study Groups. Kidney outcomes in long-term studies of ruboxistaurin for diabetic eye disease. Clin J Am Soc Nephrol 2007;2:631–636 [DOI] [PubMed] [Google Scholar]
- 33.Campochiaro PA. C99-PKC412-003 Study Group. Reduction of diabetic macular edema by oral administration of the kinase inhibitor PKC412. Invest Ophthalmol Vis Sci 2004;45:922–931 [DOI] [PubMed] [Google Scholar]
- 34.Tang S, Xiao V, Wei L, Whiteside CI, Kotra LP. Protein kinase C isozymes and their selectivity towards ruboxistaurin. Proteins 2008;72:447–460 [DOI] [PubMed] [Google Scholar]