

Abstract
DC licensed by the interaction between pathogen products and pattern recognition receptors can activate naïve T cells to undergo antigen-dependent proliferation and cytokine production. In contrast, DC induced to mature by trans-acting inflammatory stimuli are believed to only be capable of supporting antigen-dependent proliferative responses. Here we show that uninfected DC matured as a consequence of Leishmania-induced inflammation induce CD8+ T cells to proliferate in the absence of their cognate antigen. We separated splenic DC from Leishmania donovani-infected mice into those that contained parasites and had been activated to induce IL-12p40, from those that had received only partial maturation, measured by increased CD86 expression in the absence of IL-12p40 induction. We then showed that these partially matured DC could induce exogenous peptide-independent proliferation of OT-I and F5 CD8+ TCR transgenic T cells, as well as polyclonal CD8+ T cells. Proliferation of OT-I cells was significantly inhibited in vitro and in vivo by anti-CD86 mAb but not by anti-CD80 mAb and could also be inhibited by cyclosporine A. Proliferating OT-I cells did not produce IFNγ, even when re-exposed to mature DC. However, these primed OT-I cells subsequently produced effector cytokines, not just on exposure to their cognate peptide but, more importantly, to weak exogenous TCR agonists that otherwise failed to induce IFNγ. We further showed that OT-I cells undergoing locally driven proliferation to another pathogen, Streptococcus pneumoniae, rapidly seeded other lymphoid tissues, suggesting that CD8+ T cells primed in this way may play a role in rapidly countering pathogen dissemination.
Introduction
Dendritic cells (DC) are highly potent antigen-presenting cells crucial for the innate and adaptive immune response and for maintaining immune tolerance towards self-antigens (1, 2). To maximise the efficiency of naive T cell activation, antigen-bearing DC undergo a complex process of maturation, guided by signals transmitted through a broad range of pattern recognition receptors (PRR), including Toll like receptors (3, 4), C-type lectin receptors (5) and NOD-like receptors (6) that interact with cognate ligands derived from pathogens. Maturation mediated through PRRs has also been shown, at least for CD4+ T cells, to assist in the process of selecting peptides for MHCII-dependent antigen presentation (7). However, an increasing body of literature suggest that DC can also be indirectly stimulated to mature by endogenous danger signals such as uric acid (8), kinins (9, 10), and pro-inflammatory cytokines and chemokines (11-14). Although these signals may induce levels of DC maturation sufficient to induce antigen-dependent CD4+ T cell proliferation, it has been argued that in the absence of ‘licensing’ by cognate engagement of pattern recognition receptors, mature DC are unable to induce full CD4+ effector T cell differentiation (12). Recent evidence suggests that extension of the half life of MHC-peptide complexes, allowing increased serial triggering of TCRs, may provide a molecular basis for ‘licensing’ by PRRs (15).
Although DC maturation and the multiplicity of signals required for optimal naïve T cell activation combine to promote specificity, a large body of evidence indicates that that under certain conditions antigen-independent activation of T lymphocytes can occur after infection. For example, early studies by Ehl and colleagues demonstrated that a low frequency of LCMV-specific TCR transgenic CD8+ T cells became activated after vaccinia virus infection (16), and a number of subsequent reports have shown proliferation and cytokine production by ‘bystander’ CD4+ and CD8+ T cells in mice with ongoing infection, for example with Mycobacterium avium (17), Burkholderia pseudomallei (18) and Leishmania donovani (19). In most of these systems, bystander activation for cytokine production, as opposed to bystander proliferation, has been attributed to high levels of infection-associated pro-inflammatory cytokine production by mononuclear phagocytes, notably of IL-12 and IL-18 in the case of IFNγ production by CD8+ T cells (18). In contrast, the contribution of DC maturation to the induction of bystander proliferation has not been examined in detail.
Here, we have re-examined the issue of bystander CD8+ T cell activation, focusing on whether DC maturation independent of pathogen uptake and largely resulting from inflammation is sufficient to drive this process. Using Leishmania donovani infection as a model to induce systemic inflammation, we have shown that inflammation-induced maturation of DC infection is sufficient to confer on DC the capacity to induce proliferation of OT-I cells in the absence of their cognate antigen. Both in vitro and in vivo, this process was largely attributable to enhanced CD86-dependent costimulation. Although OT-I cells proliferating in this way did not produce IFNγ, they were nevertheless primed to do so upon exposure to otherwise ineffective weak TCR agonist peptides. Using Streptococcus pneumoniae as a model infection where DC maturation can be restricted to distinct lymphoid organs, we also showed that OT-I cells primed in the absence of their cognate antigen seeded sites distal from that of their initial activation. Collectively, our data suggest that CD86-dependent but cognate peptide -independent proliferation of CD8+ T cells induced by mature DC may be a common mechanism to increase the efficiency of immune surveillance against systemic pathogen spread.
Materials and Methods
Mice and infection
C57BL6 (Charles River, UK), OT-I RAG1−/− (a gift from Dr B Seddon, NIMR, London) and F5 RAG1−/− mice were used. All mice were housed under specific pathogen free conditions and used at 6-8 weeks of age. Amastigotes of Leishmania donovani (LV9) were isolated (22), labelled (5μM CFSE; 37°C for 10 minutes) and injected (5 × 107 i.v.) into mice. In some experiments LPS was adsorbed onto fluorescent microspheres (2um) for 24h (100μg/ml/109 microspheres) and then washed extensively in PBS before injection (5×107 i.v). All animal care and procedures were in accord with U.K. Home Office requirements and performed with local ethical approval.
DC isolation and enrichment
Conventional DC (CD11chi MHCIIhi) from naïve and infected mice were enriched by digesting the spleens or lymph nodes in RPMI supplemented with 0.25 mg/ml collagenase (5ml per spleen) for 25 minutes at RT. All subsequent steps were done between 0-4°C. Following collagenase digestion, 5ml of 50mM EDTA/PBS solution was added and the digest was passed through a 100μM strainer to make a single cell suspension. To enrich DC, dead cells and highly phagocytic cells were first depleted prior to CD11c enrichment by magnetic cell sorting using basic microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s protocol. Briefly, digested splenocytes were incubated with basic magnetic microbeads for 10 mins on ice. After washing, cells that had non-specifically bound or phagocytosed microbeads were trapped on a separation column. CD11c+ cells were then enriched by incubating anti-CD11c microbeads for 30mins on ice and then passed twice over a separation column. CD11chi cells (85-98% pure) were stained with CD11c-PE, MHCII-APC and isolated (99% purity) using a MoFlo (Beckman-Coulter). Cytospins were Giemsa-stained and visualised by light microscopy. For immunofluorescence, cytospins were fixed (2% paraformaldehyde, 30 min, RT), mounted in antifade agent (Prolong Gold; Molecular Probes) and examined using a Zeiss Axioplan LSM 510 confocal microscope.
Flow cytometry
Cells were incubated with 10 μg/ml 2.4G2 anti-Fc receptor mAb (ATCC, Rockville, MD) followed by staining with directly-conjugated monoclonal antibodies, including: FITC-conjugated anti-CD8α (53-6.7), anti-CD86 (GL1); phycoerythrin (PE)-conjugated anti-MHCII (M5/114.15.2), anti-CD11c (HL3), anti-CD62L (MEL-14); PE-Cy7-conjugated, CD11c (N418); APC-conjugated, anti-CD4 (RM4-5), anti-CD44 (IM7), anti-CD86 (GL1); APC-Cy7-conjugated, anti-CD25 (PC61.5), anti-CD8α(53-6.7) (All from eBioscience). Minimal background staining was observed using appropriate fluorochrome-labeled isotype controls. Flow cytometric analysis was performed with a Cyan ADP (Beckman-Coulter) and analysed using Summit software (Beckman-Coulter). Absolute cell numbers were quantitated by spiking samples with beads of known amount. DAPI (0.5μg/ml) was used to discriminate live/dead cells.
Real-time RT-PCR
RNA was isolated from sort purified DC using an RNeasy kit according to manufacturer’s instructions (Qiagen). RNA was then reverse transcribed into cDNA using the first strand cDNA synthesis kit according to the manufacturer’s instructions (Invitrogen). Oligonuclotides (5′-3′) used for amplification of IL-12/23p40 and HPRT were as described (30). Real-time quantitative PCR was performed with the SYBR Green PCR kit in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems). Expression of target genes were normalized to HPRT and expressed as either absolute copy number (target molecules/1000 HPRT molecule) or relative expression using the change in cycle threshold (ΔΔCT) analysis method.
Proliferation of CD8 T cells in vitro
Naïve CD8+CD62Lhi CD44lo T cells from WT, OT-1.RAG−/− or F5.RAG−/− mice were sort-purified, CFSE-labelled cells and cultured (1 × 105) with sort purified splenic DC subsets (2×104), with or without the appropriate cognate peptide (0.1μg/ml). sIL-15r (10μg/ml) or anti-CD86 (GL1) (10μg/ml) as well as isotype controls were added as required. At d5, proliferation was assessed by flow cytometry. In some experiments DCnaive or DCmat loaded with or without OVA-peptide (0.1μg/ml) were fixed with 1% paraformaldehyde for 15mins. Fixed DC were then washed and incubated for 20 mins with 10uM glycine/PBS. Fixed and washed DC (2×104 cells/well) were then cultured with CFSE labelled naïve sorted OT-1 T cells. Some samples were supplemented (20% v/v) with 24h culture supernatants derived from DCmat. In some in vitro experiments LPS was added at 100ng/ml at the start of DC and T cell co-cultures. After 5 days in culture CFSE profiles were assessed using flow cytometry.
Priming of OT-I cells in vitro
Naïve OT-1 (2×105 cells/well) primed with 5×104 DCmat for 3 days or naïve OT-1 cells were cultured with the indicated DC in the presence or absence of SIINFEKL, EIINFEKL (E1) or SIIGFEKL (G4). After 72h, surviving OT-I cells were stimulated with 106 peptide-pulsed CD45.1 spleen cells for 6h. BFA was added during the last 4h. Cells were washed in ice-cold PBS, then surface-stained with monoclonal antibodies followed by fixation with 2% paraformaldehyde for 10 mins. Samples were resuspended in PBS containing 2% FCS and 5mM EDTA and left overnight in the fridge before intracellular staining.
Intracellular cytokine staining
Fixed cell samples were resuspended in PBS/EDTA/FCS containing 0.5% bovine serum albumin (BSA) and 0.5% saponin (saponin buffer). Cells were then stained with pacific blue-conjugated anti-IFNγ (XMG 1.2) or APC-conjugated anti-IL-2 (JES6-5H4) for 45 mins on ice. Isotype controls were used to set gates. Samples were washed twice in saponin buffer and once in PBS without saponin. Flow cytometric analysis was performed using a CyanADP (Beckman-Coulter). 50,000 cells of interest were collected and analysed using Summit software (Beckman-Coulter). Absolute numbers of cells were quantitated by spiking samples with beads of known amount and gating on cells of interest.
Adoptive transfer
Naïve CFSE-labelled CD45.2 WT polyclonal or transgenic monoclonal OT-I cells were injected (2×106) i.v. into CD45.1 mice 1d prior to infection with L. donovani (i.v) or S. pneumoniae (i.v or i.n ~1×107 CFU). After 5 days, proliferation of OT-1 cells in spleens and mediastinal LNs was assessed. Blocking mAbs against CD80 (16-10A1) and CD86 (GL1) or control mAbs were administered (200ug) 12h prior to infection as required.
Statistics
Statistical analysis was performed using a paired Student t test. p<0.05 was considered significant. All experiments were performed independently at least twice and with 3 technical replicates performed for each sample.
Results
Bystander maturation of DC
To obtain DC that had been activated by inflammation, we infected mice with CFSE-labelled L. donovani and sorted splenic DC into parasitized vs. non-parasitized populations (Fig. 1). As anticipated from other studies (20), only a minor population of DC (here referred to as DCinf) contained parasites (3 ± 1.41%, following injection of 5×107 parasites; Fig. 1A). By light and confocal microscopy, sorted CFSEhi DCinf contained Leishmania whereas neither intact parasites not CFSE-labelled cellular debris was associated with CFSE− DC (Fig. 1A). Within the CFSEhi and CFSE− DC, the major subsets of splenic DC (CD4+, CD8+ and double negative) were equally represented (data not shown), indicating that each sub-population of splenic DC was equally susceptible to infection with L. donovani in vivo.
Figure 1. Bystander activation of DC in response to L donovani infection occurs in vivo.

(A) CFSE-labelled parasites were injected i.v. into B6 mice and 5h p.i, enriched CD11c+ cells were sorted for CD11c+CFSE− and CD11c+CFSE+ cells. Sorted DC were examined by light or confocal microscopy. (B) CD11chi MHCII+ DC from naïve and infected mice were examined for CD86 expression. MFI of CD86 is annotated within histograms as mean ± SD from replicate mice (n=3) p<0.001 for naïve vs. DCmat and DCinf. (C) DC from naïve mice (DCnaive) as well as DCmat or DCinf from 5h infected mice were sorted and the accumulation IL-12/23p40 mRNA was determined by quantitative RT-PCR. Experiments A and B were performed 5 times; C was performed twice.
We then compared CFSE− and CFSEhi DC for their expression of costimulatory molecules associated with DC maturation. As anticipated, compared to DC in naïve mice (referred to as DCnaive), DCinf expressed high levels of CD86 (increased >20-fold and >4-fold for CD8− and CD8+ DC respectively compared to naïve mice).
Nevertheless, compared to DCnaive, CFSE− DC from infected mice also had significantly elevated levels of expression of CD86. On CD8− DC, CD86 increased ~18-fold and expression of CD86 on CD8+ DC increased by 4-fold (Fig. 1B). Herein, we refer to these mature but uninfected DC that we obtain from infected mice as ‘DCmat’. Although we cannot rule out the possibility that DCmat may have encountered trace amounts of soluble leishmanial antigen, only DCinf contained demonstrable mRNA for IL-12p40 (Fig. 1C) and indicator of maturation mediated by engagement of PRRs (12). Thus, DCmat represent a population of DC that have been partially matured in trans by ‘inflammatory’ signals.
DC matured by inflammation stimulate naïve OT-I T cell proliferation
To assess the capacity of DCmat to stimulate sorted naïve CD8+ T cells (Supplementary Fig. S1), we used OVA-specific CD8+ OT-1 cells. As expected, DCnaive and DCmat induced a robust OT-I response in the presence of SIINFEKL, as measured by CFSE dilution analysis of viable (DAPI−) cells (Fig. 2A) and by quantifying the total viable T cell recovery after 5 days (Fig. 2B). DCmat (and to the same extent, DCinf) also stimulated OT-I proliferation in the absence of SIINFEKL, though the total recoverable cell yield was reduced by ~10-fold compared to that seen in the presence of this peptide. Examination of the CFSE content of DAPI+ (i.e. dead) cells within these cultures (Fig. 2A)) indicated that, similar to OT-I cells cultured in the absence of DC, most dead cells in the cultures stimulated with DCmat had undiluted CSFE content. These observation suggests that death by neglect rather than reduced survival after proliferation was responsible for the low cell recovery (Fig. 2B) compared to that seen in the presence of antigen. Collectively, therefore, these data indicate that only limited numbers of OT-I cells are activated by DCmat in the absence of an exogenous source of their cognate antigen, but once activated, these cells progress effectively through multiple divisions.
Figure 2. DC from infected mice drive the proliferation of antigen-specific CD8+ T cells.

DCnaive, DCmat and DCinf were cultured with CFSE-labelled naïve OT-I cells with or without SIINFEKL (100ng/ml). (A) CFSE dilution of viable (DAPI−) and non-viable (DAPI+) OT-1 cells, and (B) the number of recovered viable cells in the absence (open bars) and presence (black bars) of peptide, was determined at d5. Numbers in lower gate in (A) indicate mean % viable cells ± SD. Experiments A-C were performed 5 times. Representative data are shown from one experiment with 3 replicate cultures. ** p<0.01 compared to DCnaive in (B) and for DCmat and DCinf vs.DCnaïve in the absence of peptide in (A).
To address if CD8+ T cells from a different transgenic system behaved similarly, we used influenza nucleoprotein specific (F5) CD8+ T cells. F5 CD8+ T cells also proliferated when stimulated with DCmat in the absence of their cognate peptide (31 ± 4% dividing at least once; Supplementary Fig. S2). We next tested whether polyclonal naïve CD8+ T cells from WT mice exhibited a similar response. Compared to DCnaive, DCmat caused significant proliferation of naïve polyclonal CD8+ T cells (Fig. 3A), with approximately 10-fold greater cell recovery (Fig. 3B). Each of the three major DCmat subsets had similar potential to induce proliferation of polyclonal CD8+ T cells (Fig. 3C). In addition, the activity of DC was augmented in the presence of LPS, even when DCnaive were used as stimulators (Fig. 3D), suggesting that inflammation-induced activation and TLR-induced activation may have additive effects. Finally, to replicate inflammation-induced DC maturation in vivo but in the absence of an antigenic stimulus, we injected mice with LPS-coated fluorescent microspheres and purified DCmat that did not contain beads. DCmat in this system were again more efficient at inducing proliferation of naïve polyclonal CD8+ T cells than naïve DC (Fig. 3E).
Figure 3. DC from infected mice drive the proliferation of WT polyclonal CD8+ T cells.

(A,B) Sorted DCnaive and DCmat from naive or L. donovani infected mice were cultured with CFSE-labelled naïve polyclonal CD8+ T cells and CFSE dilution (A) and number (B) of viable polyclonal cells T cells was determined at d5. (C) Numbers of viable CD8+ T cells at d5 of co-culture with DCnaive and DCmat separated into subsets on the basis of CD4 and CD8 expression. (D) DCnaive and DCmat were co-cultured with CD8+ T cells in the presence or absence of LPS (100ng/ml). CFSE dilution is shown, with percentage of divided cells indicated in each plot. (E) Sorted DCnaive and DCmat from naive mice or mice injected with LPS-coated fluorescent microspheres (5×107) were cultured with CFSE-labelled CD8+ T cells, and CFSE dilution of viable cells was determined at d5. These experiments were performed twice. Data in (B) represents mean ± SD of triplicate cultures. * p<0.05, ** p<0.01 compared to DCnaive in (B) and (C), p<0.01 for DCnaive vs. DCmat in the presence or absence of LPS in (A) and (D) and p<0.001 for (E).
Proliferation of OT-I cells is CD86-dependent
To dissect the mechanism by which DCmat are able to activate OT-I, we first tested the stimulatory capacity of fixed DCmat. SIINFEKL-independent proliferation of OT-I cells induced by DCmat was unaffected by fixation (Fig. 4A). Conversely, the ability of DCnaive to induce SIINFEKL -independent proliferation of OT-I was not markedly improved by addition of supernatant collected from DCmat. Not surprisingly, this supernatant also did not enhance the ability of fixed DCmat to induce OT-I proliferation (Fig. 4B). As CD86 expression was significantly increased on DCmat (Fig. 1) and this costimulatory pathway has been shown to be fixation-resistant (21), we blocked CD86-CD28 signalling. As shown in Fig. 4C, CD86 mAb significantly reduced OT-I proliferation induced by DCmat. The frequency of recoverable OT-I cells was also reduced by 58 ± 1 % (p=0.002; Fig. 4D).
Figure 4. OT-1 proliferation is largely mediated through CD86 costimulation.

(A) DCnaive and DCmat were pulsed with SIINFEKL (100ng/ml) and fixed in 1% PFA. Fixed DC were then cultured with naïve CFSE-labelled OT-I cells and proliferation determined at d5. (B) Fixed DC and OT-I cells were supplemented with supernatant (20%) derived from 24h culture of DCmat. (C, D) DCnaive or DCmat were cultured with CFSE-labelled naïve OT-I cells without peptide but in the presence of either a control rat IgG2a or anti-CD86 (GL1) mAb. After 5 days, CFSE dilution of OT-I cells was analysed by flow cytometry. Data are shown as representative dot plots (C) and mean ± SD of percentage divided OT-I cells from triplicate cultures (D). (E, F) 2×106 CFSE-labelled OT-1I cells were adoptively transferred into CD45.1 mice 1 day prior to infection with L. donovani. Blocking CD80 and CD86 mAb or control mAb was administered i.p 12h prior to infection. After 5 days, CFSE dilution of splenic donor OT-I cells was analysed by flow cytometry (E) and the frequency of divided viable OT-I cells was enumerated (F). (G) DCmat were cultured with CFSE-labelled naïve OT-I cells with or without peptide as well as in the presence or absence or cyclosprorin A (1uM). After 5 days, CFSE dilution of OT-I cells was analysed by flow cytometry. Figures in plots represent % viable cells having gone through more than one round of division. Experiments A-G were performed twice with a representative data set shown. Individual data represent mean ± SD of triplicate cultures. p<0.01 for DCmat with vs. without peptide in (A) and p<0.001 for anti-CD86 and antiCD86+CD80 vs. isotype in (E), and p<0.001 for with and without peptide in (G).
To determine whether similar CD86-dependent proliferation also occurred in vivo, we transferred CFSE-labelled naïve OT-I cells into mice and subsequently infected these mice with L. donovani. OT-I cells recovered from the spleen 5 days after infection had significantly diluted CSFE, indicating proliferation in vivo, and this response was significantly blocked by prior administration of CD86 mAb (Fig. 4E and F). Similar results were noted 5 days post transfer of polyclonal CD8+ T cells into L. donovani infected mice (Supplementary Fig. S3A and B). In contrast, blockade of CD80, which we have previously shown is not up-regulated on DC from infected mice (22), had no effect on OTI-proliferation (Fig. 4E and F). Furthermore, in vitro proliferation of OT-I cells by DCmat in the absence of SIINFEKLwas blocked by addition of cyclosporine A (Fig. 4G) suggesting signals mediated via the TCR were - also required.
IL-7 and IL-15 are both known to play a role in T cell proliferation and homeostasis and at least for IL-15, a membrane bound form has been observed on a variety of APC (23). To determine whether these cytokines played a role in the capacity of DCmat to induce proliferation of OT-I cells, we first measured mRNA accumulation for IL-7 and IL-15 in DCnaive and DCmat. Our data indicated an increase (~8 fold) in IL-15 mRNA accumulation in DCmat whereas IL-7 mRNA accumulation was unchanged (Supplementary Fig. 4A). However, using soluble IL-15R, we found no evidence to support a role of IL-15 in the proliferation of OT-I cells in response to DCmat (Supplementary Fig. 4B). We also did not find evidence of IL-2 production by DCmat during co-culture with OT-I T cells (Supplementary Figure 4C).
Several reports have demonstrated that co-stimulation lowers the threshold of T cell activation (24, 25) and may allow recognition of weak TCR agonist peptides (26, 27). We therefore compared the capacity of DCnaive and DCmat to present variant OVA peptides to OT-I cells. As measured by OT-I recovery at 72h., SIINFEKL induced a maximal response in OT-I cells at all concentrations tested (from 1-1000pg/ml), irrespective of whether DCnaive or DCmat were used as APC (Fig. 5A). G4 was at least three orders of magnitude less effective than SIINFEKL at stimulating OT-I when presented by DCnaive. In contrast, stimulation by G4 was significantly enhanced by presentation on DCmat, with similar numbers of OT-I cells recovered as seen with SIINFEKL presented by DCnaive (Fig. 5B). E1 (26) was poorly presented by DCnaive, but near maximal numbers of OT-I cells were recovered when this peptide was presented by DCmat (Fig. 5C). Collectively our data indicate that DCmat have a heightened capacity to induce proliferation of OT-I cells through enhanced CD86-dependent recognition of weak peptide agonists.
Figure 5. Presentation of weak affinity peptides is enhanced by DCmat.

OT-I cells were cultured with DCnaive ( ) or DCmat (
) or DCmat ( ) in the presence or absence of SIINFEKL (A), E1 (B) or G4 (C) and total number of OT-I cells was determined at d5. The experiment was performed three times. Representative data set is shown. Data represent mean ± SD of triplicate cultures. p<0.05, ** p<0.01 *** p<0.001.
) in the presence or absence of SIINFEKL (A), E1 (B) or G4 (C) and total number of OT-I cells was determined at d5. The experiment was performed three times. Representative data set is shown. Data represent mean ± SD of triplicate cultures. p<0.05, ** p<0.01 *** p<0.001.
Characterisation of proliferating CD8+ T cells
Next, we characterised OT-I cells that had been stimulated by DCmat. Compared to naïve cells, OT-I cells activated by DCmat up-regulated CD44, and down-regulated CD62L and CCR7 (Supplementary Figure 5). Autocrine signals through the heterotrimeric IL-2R are known to promote naïve T cell proliferation and survival. Therefore, we examined the requirements for IL-2R mediated signals to promote the proliferation of naïve CD8+ T cells by DCmat. In the absence of antigen, OT-I cells stimulated by either DCmat or DCnaive produced little detectable IL-2, compared to cells stimulated for 24h by these DC populations in the presence of SIINFEKL (Fig. 6A). Whereas addition of exogenous rIL-2 to OT-I cells expanded by DCmat in the presence of SIINFEKL enhanced OT-I proliferation, this response was more muted in OT-I cells expanded using DCmat in the absence of antigen (Fig. 6B). This reduced response correlated with low expression of CD25 on OT-I cells stimulated by DCmat in the absence compared to in the presence of SIINFEKL (MFI for CD25 expression of 15.5 ± 0.2 vs. 455 ± 13, respectively, p=0.005; Fig. 6C). To assess whether this relative lack of IL-2 production and CD25 expression affected the functional differentiation of OT-I cells, we cultured naïve OT-I cells with DCmat for 3days, followed by a further 2 days in the presence or absence of exogenous rIL-2. At day 5, surviving OT-I cells were restimulated with SIINFEKL and assessed for IFNγ production. As shown in Fig. 6D, OT-I cells activated to proliferate in the absence of antigen required additional exogenous rIL-2 to develop into IFNγ-secreting effector cells.
Figure 6. OT-I proliferation induced by DCmat does not lead to functional commitment.
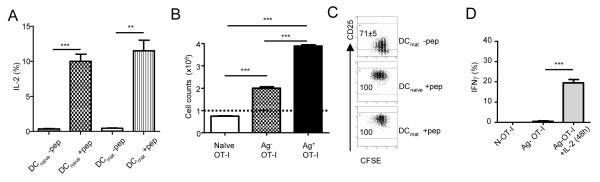
(A) DCnaive and DCmat with (black bars) or without (open bars) SIINFEKL were cultured with naïve OT-I cells for 24h. BFA was added during the final 6h of culture and intracellular IL-2 accumulation was measured by flow cytometry. Data represent % IL2+ cells ± SD. (B) OT-I cells (1×105) primed in the presence (Ag+; black bar) or absence (Ag−; grey bar) of SIINFEKL were cultured in IL-2 (100U/ml) and their expansion was analysed after 48h compared to naïve OT-I cells (open bar). (C) CD25 expression on OT-I cells primed for 5 days by either DCnaive or DCmat in the presence or absence of SIINFEKL. In the absence of SIINFEKL, insufficient OT-I cell s were recovered from cultures with DCnaive for accurate phenotyping. (D) OT-I cells were primed in the absence of peptide using DCmat for 3 days (Ag− OT-I), and then supplemented with (black bar) or without (grey bar) IL-2 for a further 48h. IFNγ responses were examined by restimulating samples with SIINFEKL-pulsed APC for 7h. BFA was added to cultures during the last 4h and intracellular IFNγ accumulation was measured by ICS. Naïve OT-1 T cells (open bar) were used as controls. The experiments were performed twice. Data represent mean ± SD of triplicate cultures. * p<0.05, ** p<0.01 *** p<0.001.
To determine whether proliferation induced by DCmat impacted on subsequent functional competence, we generated a population of OT-I cells that had been primed for 3d with DCmat and then compared their response with that of naïve OT-I cells to restimulation with SIINFEKL, G4 and E1. As shown in Fig. 7A, the frequency of IFNγ secreting OT-I cells was comparable when SIINFEKL was presented by either DCmat or DCnaive, and irrespective of whether the OT-I cells had been primed using DCmat. Importantly, no IFNγ was produced by cells primed and subsequently restimulated with DCmat alone. Priming, therefore, neither led to functional commitment in the absence of exogenous antigen nor to loss of function upon subsequent antigen exposure. Stimulation of naïve OT-I cells using G4 mirrored that observed by proliferation (Fig. 5B), with DCmat inducing a greater response than DCnaive. In addition, a significant effect of priming OT-I cells was observed at low antigen doses (Fig. 7B). With DCnaive, E1 failed to induce IFNγ production and only a weak response was generated using DCmat. However, priming of OT-I using DCmat significantly enhanced the IFNγ response to this otherwise ineffectual weak TCR agonist (Fig. 7C).
Figure 7. Pre-primed OT-1 T cells are more sensitive to stimulation by weak affinity peptides on DC.

(A-C) Naïve OT-I (solid line) or OT-I cells primed by DCmat in the absence of exogenous peptide (Ag− OT-I; dashed line) were cultured with either DCnaive ( ) or DCmat (
) or DCmat ( ) in the presence or absence of SIINFEKL (A), E1 (B) or G4 (C). After 3 days in culture, samples were restimulated with splenic APC pulsed with the appropriate peptide for 7h. BFA was added for the last 4h and intracellular IFNγ accumulation was measured by flow cytometry. No IFNγ was detected by T cells in the absence of antigen in all conditions tested, including after priming and restimulation with DCmat. The experiment was performed twice with triplicate cultures. * p<0.05, ** p<0.01 Ag− OT-1 compared to naïve OT-1.
) in the presence or absence of SIINFEKL (A), E1 (B) or G4 (C). After 3 days in culture, samples were restimulated with splenic APC pulsed with the appropriate peptide for 7h. BFA was added for the last 4h and intracellular IFNγ accumulation was measured by flow cytometry. No IFNγ was detected by T cells in the absence of antigen in all conditions tested, including after priming and restimulation with DCmat. The experiment was performed twice with triplicate cultures. * p<0.05, ** p<0.01 Ag− OT-1 compared to naïve OT-1.
OT-I cells redistribute amongst lymphoid tissues
The results described above demonstrate that splenic DC activated by inflammation during L. donovani infection can stimulate OT-I cells to proliferate independently of exposure to their cognate antigen. To explore the generality of this response, we therefore examined the in vivo response of OT-I cells following infection with Streptococcus pneumoniae. Mice were infected with S. pneumoniae via the intravenous route (allowing comparison with responses generated with L. donovani) or via the intranasal route, to examine the response in the lung-draining LN (28). 48h after intravenous infection with S. pneumoniae, splenic DC and to a lesser extent lung-draining LN DC had responded by an increase in expression of CD86. In contrast, following intranasal infection, DC maturation was restricted to the lung-draining LN (Fig. 8A). We next adoptively transferred naïve OT-I cells into B6.CD45.1 mice prior to intranasal or intravenous administration of 107 S. pneumoniae. 5d later, OT-I cells were recovered and CFSE dilution assessed. OT-I cells were found in both spleen and lung-draining LN after both i.v. and intranasal infection (Fig. 8B). The increased numbers of recoverable cells in dLN from i.n vs. i.v. infected mice (21374 ± 5853 vs. 549 ± 67, respectively) was likely a reflection of the lymph node enlargement that accompanies this route of infection (data not shown). More importantly, however, OT-I cells with identical profiles of CFSE dilution were isolated from both organs, irrespective of the route of administration and therefore independently of whether local DC maturation had occurred (Fig. 8C).
Figure 8. Ag− T cells re-distribute to distal sites lacking DC maturation.

(A) B6 mice were infected either intravenously (i.v.) or intranasally (i.n.) with Steptococcus pneumoniae (107). After 48h, CD86 expression on DC (gated on CD11chiMHCIIhi) from the spleen or lung dLN was examined. (B,C) Donor CFSE-labelled OT-I cells (2×106) were adoptively transferred into host CD45.1 mice prior to either i.n or i.v. infection with S. pneumoniae. After 5 days, the number of OT-I cells (B) and the extent of CFSE dilution (C) was determined in the spleen and lung dLN. This experiment was performed twice each time with 3 biological replicates. * p<0.05, ** p<0.01.
Discussion
We have shown that CD8+ T cells are stimulated to proliferate without functional commitment when exposed in vitro or in vivo to DC that have undergone maturation in the presence of inflammatory signals. This CD8+ T cell response is costimulation-dependent and requires signals through the TCR, suggesting that these CD8+ T cells are recognising endogenous self-ligands that behave as weak peptide agonists when presented by matured DC. CD8+ T cells stimulated to proliferate in this manner were not retained at the site of priming and appeared to circulate to other lymphoid organs, suggesting that self recognition on DCmat may increase the number of CD8+ T cells available for pathogen recognition at distal sites.
Earlier work by Reis e Sousa and colleagues working with CD4+ T cells (12) suggested that DC matured by inflammatory signals evoked by LPS injection were only capable of inducing T cell proliferation and not commitment to cytokine production. In contrast, DC that had been licensed for IL-12p40 production through MyD88-dependent signalling were able to present cognate antigen and induce functional commitment. In these studies, however, neither the ability of DC to induce proliferation of CD4+ T cells in the absence of antigen, nor their ability to stimulate CD8+ T cells was reported. Here, we injected mice with CFSE-labelled L. donovani parasites to generate an in vivo inflammatory microenvironment and then examined the functional ability of DC directly ex vivo. Using IL-12p40 expression as a means of distinguishing DC activated by cognate TLR signalling, we found that IL-12p40 was restricted to cells containing CFSE-labelled parasites or parasite fragments. We then showed that these DCmat could effectively induce proliferation in three independent target populations; OT-I and F5 TCR transgenic T cells and polyclonal CD8+ T cells. The capacity of DCmat to induce proliferation in a seemingly antigen-independent manner was unlikely to merely reflect presentation of Leishmania antigens, not detectable by CFSE labelling and that served as weak agonists for the OT-I and F5 TCRs. First, we used transgenic T cells on a RAG−/− background, to rule out the possibility that recombination had generated new Leishmania-specific TCRs. Second, if OT-I cells recognised Leishmania antigens, it would have been expected that DCinf would stimulate a greater proliferative response than DCmat, yet this was not observed. Third, proliferation in CD8+ T cells was also induced by ‘bead-negative’ DC isolated from mice injected with LPS-coated fluorescent latex beads. Thus, inflammation alone appears sufficient to endow DC with the ability to induce naïve CD8+ T cell proliferation. We did not set out to identify the factors responsible for inflammation-induced maturation of DC. However, these trans-acting inflammatory signals may include cytokines produced by DC themselves (22), cytokines produced by other infected cells such as macrophages and neutrophils (29), uric acid (8), or kinins and othr products of the coagulation pathway (9, 10).
The role of CD28-dependent costimulation in regulating the activation threshold signal for antigen-specific T cell proliferation is well known (30). The data presented here indicate that CD86 but not CD80 are involved in the proliferation of CD8+ T cells induced by DCmat. This finding is in keeping with the minimal regulation of CD80 on DC following infection with visceralising species of Leishmania observed by us and others (20). Lack of complete inhibition of the response using CD86 mAb likely reflects the additional participation of other pathways not investigated in this study. Costimulation-dependence, cyclosporine A-dependence and the ability to drive proliferation with DC that either do not contain or have not been exposed to exogenous antigen all collectively argue that the CD8+ T cell proliferation observed here is due to self-antigen recognition. We believe that the most likely explanation for the limited frequency of OT-I cells that respond to DCmat in this manner is that although DC are known to express a variety of self antigens containing peptide analogues of SIINFEKL (31), such expression may not be uniform. In this regard, it would be of future interest to determine whether interference with negative costimulatory receptor-ligand interactions, such as that between PD-1 and PD-L1 (32), might further enhance the magnitude of this response. Our data are also consistent with the notion that OT-I proliferation induced by DCmat is tempered by limiting availability of, and responsiveness to, IL-2, a feature of T cell activation also known to be dependent on strength of signal (33). Although we have not found a role for IL-15 and IL-7 in this response, and our data using fixed DCmat argue against an obligatory requirement for the release of soluble mediators, we do not rule out the possibility that other cytokines contribute to early OT-I proliferation, as suggested by others (34).
Our study bears some similarities yet has important differences to one recently report by Bevan and colleagues. Zehn et. al used the elegant approach of expressing different OVA peptide agonists as transgenes in Listeria monocytogenes (35), whereas we have utilised Leishmania with no detectable cross-reactivity with the OT-I TCR and have measured the response to OT-I agonist peptides selected from the endogenous self peptide repertoire. Nevertheless, both studies report expansion of OT-I cells by low affinity ligands, with similar observable division rates, low survival and impaired CD25 expression. Our data, like that of Zehn et. al., also indicates that CD8+ T cells activated by weak agonists have differential migration patterns. Here, we showed that OT-I cells induced to proliferate by DCmat in the lung draining LN were found in the spleen 5 days after transfer, having presumably trafficked there via the efferent lymphatics and subsequently via the bloodstream. These kinetics are very similar to those obtained by Zehn et al. who showed i) that OT-I cells stimulated with weak agonist peptides Q4 and V4 appeared in blood at day 4.5 post infection and ii) that the avidity of endogenous polyclonal CD8+ T cells in the blood was lower at day 4.5 than at day 7.5 post infection (35). Our data extends the scope of their conclusions, however, by including self peptide recognition as a means for expanding the pool of pathogen-specific T cell clones and broadening the avidity of the response. We further suggest that expansion of potentially pathogen-responsive CD8+ T cells, though self-peptide recognition promoted by augmented costimulation, provides a limited window of opportunity for enhancing peripheral responsiveness to microbial insult. Thus, pathogen-specific T cells primed in a foreign antigen-independent manner become more readily able to be functionally committed even by activation with weak agonists, and this priming therefore serves to effectively enhance the precursor frequency of pathogen-specific T cells available for recruitment into the systemic immune response. Our model also predicts that T cell responses would mature with time, partly as the relative advantage of bystander proliferation is lost due to poor survival and partly due to the numerical gain associated with activated and proliferation mediated by DC that acquire direct pathogen induced maturation signals.
In conclusion, we have shown that upon activation by inflammatory signals, mature DC can stimulate proliferation of naïve CD8+ T cells in the absence of their cognate antigen. Whilst self recognition due to enhanced costimulation is likely to drive this proliferation, such activation remains below the threshold required for IFNγ production. Nevertheless, proliferation induced in this way primes CD8+ T cells for more robust cytokine production on subsequent encounter with weak TCR agonist peptides. The continued requirement for exogenous antigen should help limit, though not necessarily exclude, any autoimmune consequence of such proliferation, but by reducing the threshold for activation by exogenous peptides, this pathway may provide for more effective surveillance against systemic pathogen spread.
Supplementary Material
Acknowledgements
The authors thank Drs B. Seddon and D. Kioussis for providing OT-I.RAG1−/− and F5 mice, Prof. F.Y. Liew for the sIL-15Rα and M4 proteins, and the staff of the University of York Biological Services Facility and Technology Facility for their assistance.
Non-standard Abbreviations
- DC
conventional DC
- p.i.
post infection
Footnotes
This work was supported by grants from the Wellcome Trust and the British Medical Research Council
References
- 1.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 2.Ueno H, Klechevsky E, Morita R, Aspord C, Cao T, Matsui T, Di Pucchio T, Connolly J, Fay JW, Pascual V, Palucka AK, Banchereau J. Dendritic cell subsets in health and disease. Immunol Rev. 2007;219:118–142. doi: 10.1111/j.1600-065X.2007.00551.x. [DOI] [PubMed] [Google Scholar]
- 3.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R, Janeway CA., Jr. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 5.Geijtenbeek TB, van Vliet SJ, Engering A, t Hart BA, van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- 6.Ting JP, Davis BK. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu Rev Immunol. 2005;23:387–414. doi: 10.1146/annurev.immunol.23.021704.115616. [DOI] [PubMed] [Google Scholar]
- 7.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 8.Kool M, Soullie T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205:869–882. doi: 10.1084/jem.20071087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aliberti J, Viola JP, Vieira-de-Abreu A, Bozza PT, Sher A, Scharfstein J. Cutting edge: bradykinin induces IL-12 production by dendritic cells: a danger signal that drives Th1 polarization. J Immunol. 2003;170:5349–5353. doi: 10.4049/jimmunol.170.11.5349. [DOI] [PubMed] [Google Scholar]
- 10.Svensjo E, Batista PR, Brodskyn CI, Silva R, Lima AP, Schmitz V, Saraiva E, Pesquero JB, Mori MA, Muller-Esterl W, Scharfstein J. Interplay between parasite cysteine proteases and the host kinin system modulates microvascular leakage and macrophage infection by promastigotes of the Leishmania donovani complex. Microbes Infect. 20068:206–220. doi: 10.1016/j.micinf.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Munz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med. 2005;202:203–207. doi: 10.1084/jem.20050810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6:163–170. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- 13.Tam MA, Sundquist M, Wick MJ. MyD88 and IFN-alphabeta differentially control maturation of bystander but not Salmonella-associated dendritic cells or CD11cintCD11b+ cells during infection. Cell Microbiol. 2008;10:1517–1529. doi: 10.1111/j.1462-5822.2008.01144.x. [DOI] [PubMed] [Google Scholar]
- 14.Yadav R, Zammit DJ, Lefrancois L, Vella AT. Effects of LPS-mediated bystander activation in the innate immune system. J Leukoc Biol. 2006;80:1251–1261. doi: 10.1189/jlb.0406253. [DOI] [PubMed] [Google Scholar]
- 15.Rudd BD, Brien JD, Davenport MP, Nikolich-Zugich J. Cutting edge: TLR ligands increase TCR triggering by slowing peptide-MHC class I decay rates. J Immunol. 2008;181:5199–5203. doi: 10.4049/jimmunol.181.8.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehl S, Hombach J, Aichele P, Hengartner H, Zinkernagel RM. Bystander activation of cytotoxic T cells: studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J Exp Med. 1997;185:1241–1251. doi: 10.1084/jem.185.7.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilbertson B, Germano S, Steele P, Turner S, Fazekas de St Groth B, Cheers C. Bystander activation of CD8+ T lymphocytes during experimental mycobacterial infection. Infect Immun. 2004;72:6884–6891. doi: 10.1128/IAI.72.12.6884-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lertmemongkolchai G, Cai G, Hunter CA, Bancroft GJ. Bystander activation of CD8+ T cells contributes to the rapid production of IFN-gamma in response to bacterial pathogens. J Immunol. 2001;166:1097–1105. doi: 10.4049/jimmunol.166.2.1097. [DOI] [PubMed] [Google Scholar]
- 19.Polley R, Sanos SL, Prickett S, Haque A, Kaye PM. Chronic Leishmania donovani infection promotes bystander CD8+-T-cell expansion and heterologous immunity. Infect Immun. 2005;73:7996–8001. doi: 10.1128/IAI.73.12.7996-8001.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Trez C, Brait M, Leo O, Aebischer T, Torrentera FA, Carlier Y, Muraille E. Myd88-dependent in vivo maturation of splenic dendritic cells induced by Leishmania donovani and other Leishmania species. Infect Immun. 2004;72:824–832. doi: 10.1128/IAI.72.2.824-832.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel P, Gribben JG, Freeman GJ, Zhou LJ, Nozawa Y, Abe M, Nadler LM, Wakasa H, Tedder TF. The B7-2 (B70) costimulatory molecule expressed by monocytes and activated B lymphocytes is the CD86 differentiation antigen. Blood. 1994;84:1402–1407. [PubMed] [Google Scholar]
- 22.Maroof A, Kaye PM. Temporal regulation of interleukin-12p70 (IL-12p70) and IL-12-related cytokines in splenic dendritic cell subsets during Leishmania donovani infection. Infect Immun. 2008;76:239–249. doi: 10.1128/IAI.00643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity. 2002;17:537–547. doi: 10.1016/s1074-7613(02)00429-6. [DOI] [PubMed] [Google Scholar]
- 24.Anderson DE, Ausubel LJ, Krieger J, Hollsberg P, Freeman GJ, Hafler DA. Weak peptide agonists reveal functional differences in B7-1 and B7-2 costimulation of human T cell clones. J Immunol. 1997;159:1669–1675. [PubMed] [Google Scholar]
- 25.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 26.Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005;3:e356. doi: 10.1371/journal.pbio.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ, Gascoigne NR, Palmer E, Jameson SC. The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity. 2001;15:59–70. doi: 10.1016/s1074-7613(01)00173-x. [DOI] [PubMed] [Google Scholar]
- 28.Kirby AC, Raynes JG, Kaye PM. CD11b regulates recruitment of alveolar macrophages but not pulmonary dendritic cells after pneumococcal challenge. J Infect Dis. 2006;193:205–213. doi: 10.1086/498874. [DOI] [PubMed] [Google Scholar]
- 29.Kaye PM, Svensson M, Ato M, Maroof A, Polley R, Stager S, Zubairi S, Engwerda CR. The immunopathology of experimental visceral leishmaniasis. Immunol Rev. 2004;201:239–253. doi: 10.1111/j.0105-2896.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- 30.Chambers CA. The expanding world of co-stimulation: the two-signal model revisited. Trends Immunol. 2001;22:217–223. doi: 10.1016/s1471-4906(01)01868-3. [DOI] [PubMed] [Google Scholar]
- 31.Santori FR, Kieper WC, Brown SM, Lu Y, Neubert TA, Johnson KL, Naylor S, Vukmanovic S, Hogquist KA, Jameson SC. Rare, structurally homologous self-peptides promote thymocyte positive selection. Immunity. 2002;17:131–142. doi: 10.1016/s1074-7613(02)00361-8. [DOI] [PubMed] [Google Scholar]
- 32.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, Schacker T, Roederer M, Douek DC, Koup RA. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Stipdonk MJ, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, Schoenberger SP. Dynamic programming of CD8+ T lymphocyte responses. Nat Immunol. 2003;4:361–365. doi: 10.1038/ni912. [DOI] [PubMed] [Google Scholar]
- 34.D’Souza WN, Lefrancois L. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J Immunol. 2003;171:5727–5735. doi: 10.4049/jimmunol.171.11.5727. [DOI] [PubMed] [Google Scholar]
- 35.Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.