

Abstract
Amyloid-β (Aβ) producing enzymes are key targets for disease-modifying Alzheimer’s disease (AD) therapies since Aβ trafficking is at the core of AD pathogenesis. Development of such drugs might benefit from the identification of markers indicating in vivo drug effects in the central nervous system. We have previously shown that Aβ1-15 is produced by concerted β- and α-secretase cleavage of amyloid-β protein precursor (AβPP). Here, we test the hypothesis that this pathway is more engaged upon γ-secretase inhibition in humans and cerebrospinal fluid (CSF) levels of Aβ1-15/16 represent a biomarker for this effect. Twenty healthy men were treated with placebo (n=5) or the γ-secretase inhibitor semagacestat (100 mg [n=5], 140 mg [n=5], or 280 mg [n=5]). CSF samples were collected hourly over 36 hours and 10 time points were analyzed by immunoassay for Aβ1-15/16, Aβx-38, Aβx-40, Aβx-42, sAβPPα and sAβPPβ. The CSF concentration of Aβ1-15/16 showed a dose-dependent response over 36 hours. In the 280 mg treatment group, a transient increase was seen with a maximum of 180% relative to baseline at 9 hours post administration of semagacestat. The concentrations of Aβx-38, Aβx-40 and Aβx-42 decreased the first 9 hours followed by increased concentrations after 36 hours relative to baseline. No significant changes were detected for CSF sAβPPα and sAβPPβ.Our data shows that CSF levels of Aβ1-15/16 increase during treatment with semagacestat supporting its feasibility as a pharmacodynamic biomarker for drug candidates aimed at inhibiting γ-secretase-mediated AβPP-processing.
Keywords: Alzheimer’s, Aβ, Amyloid, γ-secretase
Introduction
Alzheimer’s disease (AD) is a slowly progressive brain disease which impairs episodic memory and ultimately the ability to carry out simple tasks such as decision-making and orientation. It is the main cause of dementia in the ageing population, accounting for 50–60% of approximately 35 million dementia cases around the world [1]. Neuropathologically, AD is characterized by accumulation of intraneuronal neurofibrillary tangles and extracellular plaques consisting mainly of N-terminally truncated amyloid-β (Aβ) species [2, 3].
Among the main focus of AD-modifying therapies are drugs targeting Aβ production in the brain by inhibiting Aβ generating enzymes, or increasing Aβ clearance from the brain using approaches such as immunotherapy. Even though AD modifying treatments have positive effects in animal models [4, 5], direct biochemical biomarker evidence of target engagement in trials on human subjects has been difficult as decreasing Aβ concentrations with secretase inhibition has been difficult to quantitate [6]. Thus, there is a need for indirect, more easily measured biomarkers to more fully characterize and monitor the biochemical effects of candidate disease-modifying drugs in AD clinical trials.
γ-Secretase is a membrane-bound protease complex which is, directly or indirectly, involved in generating Aβ consisting of 42 amino acids (Aβ1-42) and C-terminally truncated Aβ species ranging from Aβ1-40 down to Aβ1-17 [7, 8]. Aβ in CSF is mainly expressed as C-terminally truncated peptides ranging from Aβ1-13 and up to Aβ1-42; Aβ1-40 is the most abundant isoform [9, 10].
γ-Secretase is recognized as one of the potential targets for disease-modifying therapy against AD. The γ-secretase inhibitor semagacestat lowers the concentration of Aβ1-40 and Aβ1-42 in human plasma. Lowering of Aβ concentrations due to inhibition of amyloid-β protein precursor (AβPP) processing in the central nervous system initially could not be demonstrated by directly measuring the concentrations of Aβ1-40 and Aβ1-42 in CSF [11]. The initial failure to detect an acute pharmacodynamic (PD) effect of semagacestat on CSF Aβ levels was presumed to be due to a modest transient PD response that could not be readily detected owing to poor timing of single time point sample collection by lumbar puncture and marked variability in CSF Aβ levels repeatedly collected via an indwelling thecal sac cathether. However, using a recently developed stable-isotopic labeling kinetics (SILK) assay of Aβ metabolism, semagacestat was shown to decrease newly synthesized Aβ for at least 12 hours without affecting clearance in the central nervous system [12].
AβPP can undergo a catabolic processing pathway by concerted β- and α-secretase cleavages, releasing several short Aβ species such as Aβ1-15 and Aβ1-16 [7, 13]. This pathway may become particularly prevalent when C99 levels accumulate subsequent to γ-secretase inhibition. Using mass spectrometry together with immunoprecipitation we have previously shown that this pathway is enhanced by semagacestat chronic treatment resulting in increased relative levels of Aβ1-15 and Aβ1-16 [13–15]. Here, we test the hypothesis that increased CSF concentrations of Aβ1-15/16 measured by a high-throughput immunoassay can be used as a biomarker for γ-secretase inhibition in humans with acute administration of semagacestat, a γ-secretase inhibitor.
Material and Methods
Study participants and sample collection
The study cohort and clinical procedures have been described before in detail [16]. Briefly, a randomized, double-blind, placebo-controlled study was performed to determine the central nervous system effect of single oral doses of the γ-secretase inhibitor semagacestat. The first two participants were single-blinded and given a single oral dose of 140 mg semagacestat, after which all participants were given study drug in a double-blinded fashion. Healthy 21- to 50-year-old male volunteers were invited to participate, screened, and enrolled. Each participant received either placebo or a single oral dose of 100, 140, or 280 mg of semagacestat 1 hour before the start of the CSF collection. A lumbar catheter was placed at the L3-4 interspace, intravenous catheters placed in both arms, and CSF sampled hourly.
The study was approved by the Washington University Human Studies Committee, and all participants gave written consent.
Immunoassay measurements
CSF was analyzed for Aβ1-15/16 using the AlphaLISA® Amyloid Aβ1-15/16 immunoassay research kit according to the manufacturer’s protocol. Briefly, the AlphaLISA format is a proven technology with a wide range of applications which provides a chemiluminescent, no-wash, high-throughput immunoassay technique based on acceptor and donor beads together with specific antibodies against target analyte [17]. One antibody is specific to the β-secretase cleavage site of Aβ at the N-terminus (82E1) while the second antibody is directed against the C-terminus of human sAβPPα (2B3). The assay plates were read on an EnVision® multilabel plate reader equipped with the AlphaScreen® module (PerkinElmer Inc.). The assay has a lower detection limit of 8.1 pg/mL, the intra-assay precision and inter-assay precision had coefficients of variations < 6% and <8%, respectively and the cross-reactivity (specificity) were 5% and 0.8% for Aβ1-14 and Aβ1-17, respectively. The Aβ1-15/16 immunoassay research kit contains Aβ1-16 as standard. However, we have detected an enzymatic activity corresponding to a carboxypeptidase(s) present in fetal bovine serum, a component of the buffers used, that degrades Aβ1-16 to Aβ1-15, which is then detected by the immunoassay (manuscript in preparation). Thus, the AlphaLISA® Amyloid Aβ1-15/16 immunoassay research kit measures Aβ1-15 and a significant fraction of Aβ1-16 through its conversion into Aβ1-15.
CSF levels of Aβx-42, Aβx-40 and Aβx-38 were determined using the Aβ Triplex assay (Human Aβ peptide Ultra-Sensitive Kits) provided and developed by Meso Scale Discovery (MSD, Gaithersburg, Maryland, USA) as described elsewhere [18]. Briefly, this assay uses C terminus specific antibodies to capture the different Aβ peptides and a SULFO-TAG labeled anti-Aβ antibody (4G8) for detection with electrochemiluminescence. A drug-independent curvature in the placebo groups was, for each analyte and dose, eliminated by fitting the empirical function to placebo data (t: time, a, b, c: fitting parameters) and then subtracting the curvature that is represented by the first term in the function.
CSF concentrations of α-secretase cleaved soluble AβPP (sβAPPα) and β-secretase cleaved soluble APP (sAβPPβ) were determined using the MSD sAβPPα/sAβPPβ multiplex assay as described by the manufacturer (MSD, Gaithersburg, Maryland, USA). This assay employs the 6E10 antibody to capture sAβPPα and a neoepitope-specific antibody to capture sAβPPβ. Both isoforms are detected by the SULFO-TAG labelled anti-APP antibody p2-1.
Immunoprecipitation on CSF using the monoclonal 2B3 antibody (epitope in the C-terminal part of human sAβPPα, IBL Hamburg Germany) and mass spectrometry was performed as described previously [9].
Statistical analysis
The time series for each treatment were analysed using Friedman’s test (SAS, Cary, NC, USA). Due to Bonferroni correction, the treatment was considered significant if p<0.0083.
Results
To test the specificity of the 2B3 antibody used in the AlphaLISA Aβ1-15/16 immunoassay research kit, a CSF sample was analyzed by immunoprecipitation in combination with MALDI-TOF/TOF mass spectrometry. The only peak detected in the mass spectrum was Aβ1-15 (Fig. 1).
Figure 1.

Representative MALDI-TOF mass spectrum displaying Aβ1-15. The 2B3 antibody selectively immunoprecipitated Aβ1-15 without other species.
The CSF concentration of Aβ1-15/16 showed a time-dependent increase in the 140 mg and 280 mg treatment groups (p=0.016 and p=0.024, respectively) with a maximum increase of 66% and 180% 9 hours after administration of semagacestat compared to baseline (0 hours) (Fig. 2). The CSF concentration of Aβ1-15/16 for placebo and 100 mg doses demonstrated 11% and 35% change 9 hours after placebo or drug administration respectively. The increase was followed by a decrease returning to baseline concentrations 36 hours after treatment.
Figure 2.

The CSF concentrations, relative baseline, of Aβ1-15/16 as a function of time for placebos and after administration of 100 mg, 140 mg and 280 mg of the γ-secretase inhibitor semagacestat. Friedman’s test was used for calculating p-values. The error bars represent standard errors of the mean. N=4 in the 100 mg group due to a missing base line (0 hours) value.
The placebo subjects displayed increased concentrations of Aβx-38/40/42 ending at higher levels 36 hours after administration compared to baseline with a mean increase of 85, 52 and 73% for Aβx-38, Aβx-40 and Aβx-42, respectively (p=0.00038, 0.012 and 0.025 for Aβx-38, Aβx-40 and Aβx-42, respectively) (Fig. 3). This drug-independent effect was subtracted for each analyte and dose revealing a significant time-dependent response in the 280 mg treatment group with decreases 9 hours post treatment for Aβx-38/40/42 (Fig. 3).
Figure 3.

The CSF concentrations of Aβx-38, Aβx-40 and Aβx-42 (black boxes) as a function of time for placebos and after administration of 100 mg, 140 mg and 280 mg of the γ-secretase inhibitor semagacestat. A drug-independent curvature in the placebo groups was, for each analyte and dose, eliminated by fitting the empirical function to placebo data (t: time, a, b, c: fitting parameters) and then subtracting the curvature (open boxes) that is represented by the first term in the function. Friedman’s test was used for calculating p-values. The error bars represent standard errors of the mean.
Finally, the ratio of Aβ1-15/16:Aβx-40 was tested with the result that both 140 mg and 280 mg dosages showed a significant time-dependent increase (p=0.0036 and 0.0037 for 140 mg and 280 mg, respectively) while no significant change was observed in the placebo treated subjects (Fig. 4). No significant changes were detected for sAβPPα and sAβPPβ (Fig. 5).
Figure 4.

The CSF Aβ1-15/16:Aβx-40 ratio as a function of time for placebos and after administration of 100 mg, 140 mg and 280 mg of the γ-secretase inhibitor semagacestat. Friedman’s test was used for calculating p-values. The error bars represent standard errors of the mean.
Figure 5.
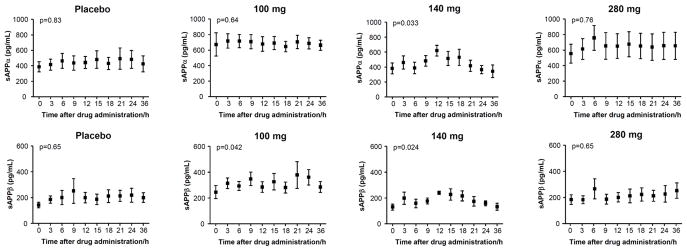
The CSF concentrations of sAβPPα and sAβPPβ as a function of time for placebos and after administration of 100 mg, 140 mg and 280 mg of the γ-secretase inhibitor semagacestat. Friedman’s test was used for calculating p-values. The error bars represent standard errors of the mean.
Discussion
Treatment with the γ-secretase inhibitor semagacestat clearly increased the CSF concentration of Aβ1-15/16 up to 9 hours post treatment followed by a decrease towards baseline concentrations after 36 hours. This is in line with previous findings that the relative CSF signals of Aβ1-15 from dogs treated with the same inhibitor increased up to 8 hours after administration followed by a decrease towards baseline levels 24 hours after treatment [15]. Recently, we showed that the relative levels of CSF Aβ1-15 significantly increased in AD patients treated with semagacestat for 14 weeks [14]. However, the method used in the two studies was based on non-absolute quantitative mass spectrometry in combination with immunoprecipitation and only the relative levels were reported. The results presented here support the use of Aβ1-15 as an acute pharmacodynamic marker in clinical trials with the aim of inhibiting γ-secretase using a high-throughput immunoassay technique enabling absolute quantification, which may be useful to identify treatment responders, facilitate dosing and ensure stable treatment effects over time.
The expected effect of semagacestat treatment on Aβ1-40 and Aβ1-42 in the central nervous system had not been demonstrated until only recently [11]. However, direct measurement of Aβ production and clearance by measurements in CSF, demonstrated that semagacestat significantly inhibits Aβ production in the central nervous system [12]. Further, a trend towards decreased levels 9-12 hours post treatment can be observed for Aβ1-X, Aβ1-40 and Aβ1-42 of which the area under the curve for Aβ1-42 showed a significant change at the high dosage (280 mg). In addition to this drug treatment effect, CSF Aβ levels gradually increased over the 36 hour sampling interval in all treatment cohorts, placebo-treated patients included. The steady increase in CSF Aβ levels did not return to baseline and thus, is inconsistent with a diurnal process. The most plausible cause of this effect is the existence of a cephalic to caudal gradient for the longer Aβ peptides which is not readily evident with lumbar puncture but apparent when larger total volumes of CSF are collected over 36 hours. Here, we replicate this finding on Aβx-38/40/42 with decreased concentrations 9 hours post treatment and increased levels over 36 hours.
There were significant changes in the levels of Aβx-38/40/42 in placebo treated subjects and in the treatment groups from 0 to 36 hours with a placebo induced increase of Aβ over time. Such variability has previously been described and has been attributed in part to dynamic changes in the production or clearance rate of Aβ in the central nervous system [12]. However, Aβ1-15/16 did not show the same time-dependent drift over 36 hours since the concentrations measured returned to baseline levels after the increase 9 hours post treatment.
A comparison of the results from CSF obtained after 12 weeks treatment with semagacestat [14] with those from a single dose as reported here are of interest. After 12 weeks of treatment with 100 mg semagacestat daily, a significant 20–30% increase in Aβ1-15/16 was found in CSF obtained 6 hours after the final dosing. In this single-dose study, a 100 mg dose of semagacestat did not cause a significant increase in Aβ1-15/16. Although the plasma half-life of semagacestat is only approximately 2.5 hours [19], the accumulation of Aβ1-15/16 in CSF after multiple doses suggests that PD effects in the central compartment may be not predicted accurately by peripheral pharmacokinetic properties of a drug. This conclusion is supported by the observations in this study that Aβ1-15/16 concentrations did not return to baseline until 24–36 hours after a single dose, and that Aβx-38/40/42 concentrations remained reduced for 12–15 hours.
By using the Aβ1-15/16:Aβx-40 ratio, significant time-dependent responses for the 140 mg and 280 mg dosages was achieved while no significant change was observed for the placebo groups. Thus, the ratio of the two isoforms seems promising in clinical trials with a γ-secretase inhibitor as a potential disease modifying drug.
The primary goal of γ-secretase inhibitors is to decrease the longer forms of Aβ species, especially Aβ42 and Aβ40. Although Aβ1-15/16 does not directly measure these products, increasing Aβ15/16 has been correlated with decreasing Aβ40 and Aβ42. This study further supports a relationship between decreased Aβ40 and Aβ42 production and increased concentrations of Aβ15/16 in human CSF.
In conclusion, Aβ1-15/16 seems to be a sensitive acute pharmacodynamic biomarker for γ-secretase inhibition and may have several advantages compared to measuring longer CSF Aβ species. For example, Aβ1-15/16 is less affected by the time of CSF sampling as it does not demonstrate the same rise compared to longer forms. The most sensitive measures for Aβ1-15/16 are when the ratio of Aβ1-15 and longer Aβ species (e.g. Aβ1-40) are used. The increase in Aβ1-15/16 combined with the decrease in Aβ1-40 increase the power of this biomarker to detect a difference. Moreover, Aβ1-15/16 seems to be a direct measure of the effects of semagacestat on APP-processing in humans.
Acknowledgments
This work was supported by grants from the Swedish Research Council (projects 2006-6227, 2006-2740, 2006-3505 and 14002), the Alzheimer’s Association (NIRG-08-90356), cNEUPRO, the Söderberg Foundation, the Royal Swedish Academy of Sciences, the Sahlgrenska University Hospital, Swedish Brain Power, Stiftelsen Gamla Tjänarinnor and Alzheimer Foundation, Sweden. This work was supported by an investigator initiated research grant from Eli Lilly (H6L-MC-LFAM), K23 AG030946 NIH R01 NS065667, and the Knight Initiative for Alzheimer Research to R. Bateman, and the NIH CARS 1UL1 RR024992.
References
- 1.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.Caselli RJ, Beach TG, Yaari R, Reiman EM. Alzheimer’s disease a century later. J Clin Psychiatry. 2006;67:1784–1800. doi: 10.4088/jcp.v67n1118. [DOI] [PubMed] [Google Scholar]
- 3.Portelius E, Bogdanovic N, Gustavsson MK, Volkmann I, Brinkmalm G, Zetterberg H, Winblad B, Blennow K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010;120:185–193. doi: 10.1007/s00401-010-0690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Best JD, Smith DW, Reilly MA, O’Donnell R, Lewis HD, Ellis S, Wilkie N, Rosahl TW, Laroque PA, Boussiquet-Leroux C, Churcher I, Atack JR, Harrison T, Shearman MS. The novel gamma secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1, 1-trifluoromethanesulfonamide (MRK-560) reduces amyloid plaque deposition without evidence of notch-related pathology in the Tg2576 mouse. J Pharmacol Exp Ther. 2007;320:552–558. doi: 10.1124/jpet.106.114330. [DOI] [PubMed] [Google Scholar]
- 5.Imbimbo BP, Del Giudice E, Colavito D, D’Arrigo A, Dalle Carbonare M, Villetti G, Facchinetti F, Volta R, Pietrini V, Baroc MF, Serneels L, De Strooper B, Leon A. 1-(3′,4′-Dichloro-2-fluoro[1,1′-biphenyl]-4-yl)-cyclopropanecarboxylic acid (CHF5074), a novel gamma-secretase modulator, reduces brain beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease without causing peripheral toxicity. J Pharmacol Exp Ther. 2007;323:822–830. doi: 10.1124/jpet.107.129007. [DOI] [PubMed] [Google Scholar]
- 6.Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 7.Portelius E, Price E, Brinkmalm G, Stiteler M, Olsson M, Persson R, Westman-Brinkmalm A, Zetterberg H, Simon AJ, Blennow K. A novel pathway for amyloid precursor protein processing. Neurobiol Aging. 2011;32:1090–1098. doi: 10.1016/j.neurobiolaging.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 8.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 9.Portelius E, Tran AJ, Andreasson U, Persson R, Brinkmalm G, Zetterberg H, Blennow K, Westman-Brinkmalm A. Characterization of amyloid beta peptides in cerebrospinal fluid by an automated immunoprecipitation procedure followed by mass spectrometry. J Proteome Res. 2007;6:4433–4439. doi: 10.1021/pr0703627. [DOI] [PubMed] [Google Scholar]
- 10.Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of beta-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- 11.Fleisher AS, Raman R, Siemers ER, Becerra L, Clark CM, Dean RA, Farlow MR, Galvin JE, Peskind ER, Quinn JF, Sherzai A, Sowell BB, Aisen PS, Thal LJ. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol. 2008;65:1031–1038. doi: 10.1001/archneur.65.8.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bateman RJ, Siemers ER, Mawuenyega KG, Wen G, Browning KR, Sigurdson WC, Yarasheski KE, Friedrich SW, Demattos RB, May PC, Paul SM, Holtzman DM. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann Neurol. 2009;66:48–54. doi: 10.1002/ana.21623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook JJ, Wildsmith KR, Gilberto DB, Holahan MA, Kinney GG, Mathers PD, Michener MS, Price EA, Shearman MS, Simon AJ, Wang JX, Wu G, Yarasheski KE, Bateman RJ. Acute gamma-secretase inhibition of nonhuman primate CNS shifts amyloid precursor protein (APP) metabolism from amyloid-beta production to alternative APP fragments without amyloid-beta rebound. J Neurosci. 2010;30:6743–6750. doi: 10.1523/JNEUROSCI.1381-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Portelius E, Dean RA, Gustavsson MK, Andreasson U, Zetterberg H, Siemers E, Blennow K. A novel Abeta isoform pattern in CSF reflects gamma-secretase inhibition in Alzheimer disease. Alzheimers Res Ther. 2010;2:7. doi: 10.1186/alzrt30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Portelius E, Van Broeck B, Andreasson U, Gustavsson MK, Mercken M, Zetterberg H, Borghys H, Blennow K. Acute effect on the Abeta isoform pattern in CSF in response to gamma-secretase modulator and inhibitor treatment in dogs. J Alzheimers Dis. 2010;21:1005–1012. doi: 10.3233/JAD-2010-100573. [DOI] [PubMed] [Google Scholar]
- 16.Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–861. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bielefeld-Sevigny M. AlphaLISA immunoassay platform- the “no-wash” high-throughput alternative to ELISA. Assay Drug Dev Technol. 2009;7:90–92. doi: 10.1089/adt.2009.9996. [DOI] [PubMed] [Google Scholar]
- 18.Mattsson N, Zetterberg H, Bianconi S, Yanjanin NM, Fu R, Mansson JE, Porter FD, Blennow K. Gamma-secretase-dependent amyloid-beta is increased in Niemann-Pick type C: a cross-sectional study. Neurology. 2011;76:366–372. doi: 10.1212/WNL.0b013e318208f4ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siemers ER, Dean RA, Friedrich S, Ferguson-Sells L, Gonzales C, Farlow MR, May PC. Safety, tolerability, and effects on plasma and cerebrospinal fluid amyloid-beta after inhibition of gamma-secretase. Clin Neuropharmacol. 2007;30:317–325. doi: 10.1097/WNF.0b013e31805b7660. [DOI] [PubMed] [Google Scholar]