

Abstract
There is great interest lately in the availability of analytical methods for quantification of 3-iodothyronamine from blood and tissues. To date, no validated method for determination of 3-iodothyronamine from biological matrices has been described. Detailed in this report is an LC-MS/MS method that permits accurate and reproducible quantification of pharmacological concentrations of 3-iodothyronamine from rat serum, with a 0.8 μM lower limit of quantification. Endogenous 3-iodothyronamine was observed from rodent and human serum (0.2 mL) at the method limit of detection. In summary, the LC-MS/MS method enables quantification of circulating 3-iodothyronamine to allow examination of a relationship with biological activity.
1. Introduction
3-Iodothyronamine (T1AM) is a recently discovered endogenous compound (Figure 1 Panel A) thought to arise from decarboxylation and deiodination of thyroxine (T4) [1]. T1AM is a potent agonist of the orphan G protein-coupled trace amine-associated receptor TAAR1 and is an inhibitor of catecholamine transport both at the plasma membrane and intracellular vesicles [2]. In vivo administration of T1AM induces a hypometabolic state characterized by behavioral inactivity, hypothermia, reduction in cardiac performance, and hyperglycemia [3]. In addition, T1AM administration induces a shift in fuel utilization away from carbohydrates and toward lipids in both mice and Siberian hamsters, a hibernating rodent species [4].
Fig. 1.

Endogenous 3-iodothyronamine (T1AM) is thought to arise from enzymatic decarboxylation and deiodination of thyroxine (T4). Chemical structures for T1AM and its proposed precursor T4 are shown in (A). The positive mode electrospray ionization (ESI) MS profile for T1AM exhibited abundant [M+H]+ m/z 356 ion (B) that fragmented with MS/MS to give dominant [M+H-NH3]+ m/z 339 and [M+H-NH3-I]+ m/z 212 products ions (C).
The availability of sensitive and selective analytical methods for in vivo quantification of T1AM would enable a greater understanding of the pharmacology and physiology of this agent. Little is known about the distribution or clearance of T1AM administered in animal models [1, 4–7] or naturally occurring concentrations of T1AM. To date, T1AM has been demonstrated to occur in vivo by detection from rodent brain, heart and liver tissues [1, 5] and as a circulating molecule in mice, guinea-pigs [1] and Siberian hamsters [4]. Serum concentrations of endogenous T1AM were estimated to reach up to 5 nM (1.8 μg/L) [1, 4]. This compares to human serum concentrations for the principle thyroid hormones, T4 and 3,3',5-triiodothyronine (T3), that range from 50–110 μg/L and 0.5–2.0 μg/L respectively [8, 9]. Measurements of serum T3 and T4 have routinely been performed using methods based upon immunoassays, an approach with high sensitivity but that is prone to method interference. This has led to the development of mass spectrometry (MS) methods for the measurement of thyroid hormones, particularly liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods [10–12]. LC-MS/MS demonstrates improved method selectivity with less method interference. Molecule identification is dependant on factors that include LC retention time and the presence of parent (precursor) and fragmented product ions. LC-MS/MS was previously utilized for the detection of T1AM from rodent blood and tissues [1]. Detection using LC-MS/MS also allows for isotope dilution MS using deuterated [2H4]3-iodothyronamine (T1AM-d4) internal standard to improve analyte quantification. Quantification is accomplished through measurement of the ratio of analyte signal to signal derived from stable isotope. Isotope dilution LC-MS/MS was recently used for T1AM quantification from assays examining the deiodinase-catalyzed deiodination of thyronamines [13]. Analyte detection was with LC-MS/MS experiments monitoring for a loss of ammonia product ion obtained from T1AM [M+H]+ precursor ion (MS and MS/MS profile for T1AM Figure 1 Panels B and C respectively).
It is worth noting that for determination of low-level endogenous molecules from biological matrices MS/MS may not always selectively differentiate between precursor ions of the same m/z that demonstrate common ion losses such as water and ammonia. Potential method interference must be carefully evaluated by examination of the peak shape, peak shoulder, and peak area ratio of two transitions acquired for each analyte [14]. LC-MS/MS methods can overcome interference by utilizing a more selective transition or with improved front-end chromatographic separation. Isocratic HPLC elution often enables optimal resolution of analyte from interferents. In this article we describe validation of a viable isocratic approach for determination of T1AM with LC-MS/MS. T1AM was detected as a symmetric peak from rat serum within 6 min.
2. Experimental
2.1. Chemicals and Reagents
T1AM and T1AM-d4 internal standard were synthesized as described previously [7, 15]. Stock solutions of T1AM and T1AM-d4 were prepared at 10 mM in dimethyl sulfoxide (DMSO). Working T1AM and T1AM-d4 stocks were diluted in water from 0.1–10 μM. For HPLC purposes methanol and water were purchased from Burdick and Jackson (Muskegon, MI, USA) and 99.999% ammonium acetate and HPLC-grade TFA and were obtained from Aldrich (Milwaukee, WI, USA). For sample work-up GR grade ammonium hydroxide was purchased from Merck (Darmstadt, Germany) and anhydrous methanol and acetone from Mallinckrodt Chemicals (Phillipsburg, NJ, USA). Hydrochloric acid and potassium phosphate were obtained from Fisher (Fairlawn, NJ, USA). Cation-exchange solid-phase extraction (SPE) cartridges (Bond Elut Certify, 130 mg/3mL) were purchased from Varian (Lake Forest, CA, USA). Volume 0.5 mL Ultrafree-MC centrifugal filters (0.45 μm) were from Millipore (Bedford, MA, USA). Auto-sampler 12×32 mm vials with 200 μL glass inserts were from Sun SRi (Rockwood, TN, USA). Centrifuges used were an Allegra 6R from Beckman (Fullerton, CA, USA) and a model 5402 from Eppendorf (Westbury, NY, USA). The Speedvac used was an SPD 121P Thermosavant model from Thermo Scientific (Waltham, MA, USA).
2.2. Animal studies
Animal experiments were performed in accordance with NIH approved guidelines for the use and care of animals. Male Sprague-Dawley rats (n=3) weighing 350 g were subjected to 20 mg/kg intravenous (IV) injection of T1AM. T1AM was dissolved in 60% DMSO and 40% physiological saline pH 7.4 for IV injections of 100 μL. The animals were bled 5 min prior to IV injection to obtain a baseline sample and further blood was collected at time points from 1 to 120 min post injection. Serum was generated by allowing the blood to clot at room temperature for 60 min. The supernatant was removed and spun at 3,000 RPM for 10 min to remove any remaining blood cells and insoluble material. The samples were stored at −80°C.
2.3. Preparation of calibrators and samples
Rat serum calibrator concentrations ranged from 0.8–40 μM T1AM (500 fmol-25 pmol on-column injection). Calibrators were made by spiking commercially available pooled rat serum (Aldrich) with T1AM. For method development studies we also used pooled mouse serum, as well as human serum from volunteers consented according to OHSU Institutional Review Board approved polices and procedures. For isolation of T1AM, serum samples (either 0.2 mL or a lesser volume made up to 0.2 mL with water) were thawed on ice. Samples were spiked with 6 pmol T1AM-d4 internal standard in 10 μL of water. Proteins were precipitated by addition of acetone acidified to pH 4 with concentrated hydrochloric acid (0.4 mL). The samples were vortexed for 30 seconds and centrifuged at 14,000 RPM for 5 min. The supernatant was removed in each case and evaporated to dryness using a speedvac at 35°C. Residues were dissolved in 1.5 mL pH 6 100 mM phosphate buffer for loading onto 130 mg/3mL cation-exchange SPE cartridges. The cartridges were preconditioned under positive pressure with argon using methanol (2 mL), de-ionized water (2 mL) and phosphate buffer (1.5 mL). Samples were loaded onto the column twice under gravity and were sequential washed with water (2 mL) and 100 mM HCl. T1AM was eluted with 2% (v/v) ammonium hydroxide in methanol (2 mL, then 0.5 mL twice). The solvent was removed in vacuo and the dried organic extract residues were reconstituted in 80 μL of equal parts methanol and 0.1 M HCl. The samples were centrifuged and filtered with centrifugal filters (0.45 μm) prior to T1AM determination with MS.
2.4. LC-MS/MS
The method was validated using a Thermo TSQ Quantum Discovery triple-quadrupole mass spectrometer (San Jose, CA, USA) equipped with an electrospray ionization (ESI) source. The ionization interface was operated in the positive mode using the following settings: spray voltage, 3.0 kV; sheath and aux nitrogen gas flow rates, 45 and 20 respectively; tube lens voltage, 150 V; capillary voltage, 35 V; and capillary temperature, 325°C. An instrument method was created to monitor for the T1AM transitions from m/z 356 precursor to m/z 212 and 339 product ions and T1AM-d4 transitions from m/z 360 to m/z 216 and 343 ions. The collision energy was 18 V and collision gas pressure at 1.0 mTorr. Scan event settings were scan width 1.2 m/z, scan time 0.25 s and peak width (FWHM) 0.7 and 0.6, for Q1 and Q3 respectively. The LC-MS system was composed of an in-line Thermo Surveyor auto-sampler and HPLC pump. T1AM was resolved from closely eluting endogenous interferents using an 200×2.1 (i.d.) mm, 5 μm Hypurity C18-HPLC column with guard from ThermoHypersil (Waltham, MA, USA). The HPLC column temperature was 30°C. The isocratic mobile phase was 5 μM ammonium formate methanol:water (45:55 v/v) at 0.01% TFA delivered at a flow rate of 0.3 mL/min. The relatively low concentration of TFA used had a minimal suppression effect on T1AM ionization but improved the LC peak shape obtained for T1AM. An 8 min column wash was included. The sample injection volume was 20 μL.
3. Results
3.1 HPLC-MS/MS
The mass spectrometry method was optimized using infusion experiments to examine T1AM ionization and fragmentation. A syringe pump was used to provide a constant analyte stream from 1–10 ng/min into the HPLC flow using a T-connection. Maximum T1AM signal intensity was achieved using ESI in the positive mode. Although in-source fragmentation is normally minimal with ESI, some occurrence of in-source CID was noted for T1AM with the ionization source and instrument settings we routinely use for ESI-MS. A low concentration of 5 μM ammonium formate in the mobile phase was found to increase signal intensity for T1AM. As can be seen in Figure 1 a full scan mass spectrum demonstrated abundant protonated T1AM [M+H]+ ion that readily fragmented with MS/MS to give dominant putative [M+H-NH3]+ and [M+H-NH3-I]+ product ions. The fragmentation of T1AM was also examined with a high-resolution (15,000) tandem mass spectrometer (MicrOTOF-Q, Bruker, Billerica, MA, USA) to confirm the loss of 17 product ion was from loss of NH3. Results for experiments examining fragmentation of T1AM precursor ion to product ions with the Q-q-TOF instrument (collision energy 10–20 eV) were similar to results for MS/MS experiments with the TSQ Quantum Q-q-Q instrument and are included in supplemental data available online. Use of the MicrOTOF-Q enabled an accurate mass measurement for T1AM [M+H]+ ion of m/z 356.0140 (0.5 ppm error). The calculated m/z for T1AM [M+H]+ ion is 356.0142. A measurement of m/z 338.9878 was obtained for putative [M+H-NH3]+ product ion. This confirms the product ion is formed from loss of NH3 (−0.6 ppm error) as opposed to loss of OH (69.7 ppm error).
T1AM spiked into serum could be detected with LC-MS/MS experiments monitoring for the transition from m/z 356 precursor to m/z 339 or 212 product ion. Despite the use of isocratic chromatography a number of closely eluting interferents were present when T1AM was detected with monitoring for the transition to [M+H-NH3]+ m/z 339 product ion. Monitoring for the transition to [M+H-NH3-I]+ m/z 212 product ion allowed for more selective detection of T1AM (extracted ion chromatograms for T1AM from human and rat serum Figure 2 Panel A). An on-column analyte injection of 500 fmol was readily detected from the lower limit of quantification (LLOQ) T1AM-spiked rat serum (2.5 μL) calibrant with a signal-to-noise ratio of >10:1, as well as from rat serum collected after administration of 20 mg/kg T1AM. Both human and rodent serum (0.2mL) demonstrated the presence of endogenous T1AM, although the concentrations were at the limit of detection (LOD) for this method, with signal-to-noise ratios ranging from 3–5 (extracted ion chromatograms Figure 2 Panel B). To our knowledge this is the first time that T1AM has been detected as a circulating molecule in humans.
Fig. 2.
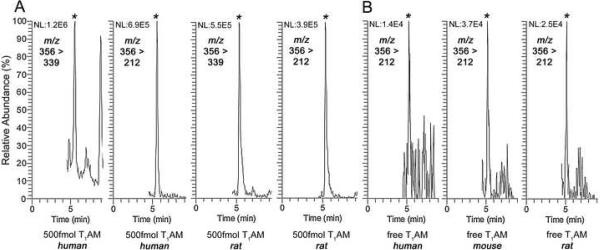
T1AM was detected from serum with LC-MS/MS experiments monitoring for the transition from m/z 356 precursor to either m/z 339 or 212 product ion (A). Monitoring for the transition to m/z 212 product ion resulted in extracted ion chromatograms demonstrating the detection of 500 fmol on-column T1AM injection from 0.2 mL of human and 2.5 mL rat serum (LLOQ calibrator). Detection of endogenous T1AM from 0.2 mL of human, mouse and rat serum was also observed (B). The retention time of T1AM (designated by *) was 5.5 min.
3.2. Method validation
The within- and between-run precision (RSD) for calculated T1AM was <20% for serum calibrators generated across the range 0.8–40 μM serum T1AM (500 fmol-25 pmol on-column injection). Between-run RSDs for calibrators analyzed over a month ranged from 7–20% (see Table 1). The LLOQ from serum was 500 fmol on-column injection with the between-run RSD determined as 20%. The LLOQ was based on monitoring for the m/z 212 product as quantifying ion and, to ensure selective detection, monitoring for the m/z 339 product as confirming ion. For detection of T1AM from all rat serum calibrants (n=31) an average area ratio between quantifying and confirming ion (m/z 212/339) was obtained with RSD of 8.9%. For the 0.8 μM LLOQ alone the RSD was 10.4%. A least-squares linear regression of peak area ratio (T1AM/T1AM-d4) versus concentration of T1AM (μM) was used for calibration. Serum calibrators were included with each sample set and monitored over one month. Calibration curves were reproducible with a typical linear regression equation of y = 0.4x − 0.007. Acceptable linearity was observed up to 40.0 μM with characteristic correlation coefficients (r2) >0.995. Experiments where T1AM was isolated from serum or plasma determined that recovery was the most consistent from serum (data not shown). T1AM was previously isolated from serum using liquid-liquid extraction with ethyl acetate [1, 5], methodology not suited to high-throughput sample processing. A solid-phase extraction (SPE) procedure using mixed-mode retention mechanisms of reversed-phase and cation-exchange was developed by Tai et al. to isolate T4 and T3 from serum [8, 9]. In our hands the Bond-Elut Certify SPE methodology proved to be efficient and reproducible for the high-throughput isolation of T1AM from up to 0.2 mL. All analysis described in this report was accomplished using the SPE methodology developed. To determine analyte recovery serum aliquots were spiked with T1AM and T1AM-d4 and the serum subjected to SPE. After reconstitution of dried residues in LC mobile phase, ion abundance detected for T1AM and T1AM -d4 was compared to ion abundance detected from the same quantity of T1AM and T1AM-d4 diluted into mobile phase. T1AM recovery averaged 63% across the range 1.2–4.0 μM (at 1.2 μM was 69%, at 2.0 μM was 62%, at 1.6 μM was 64%, and at 4.0 μM was 58%).. From the same experiment T1AM-d4 recovery averaged 63% across the calibrator range. T1AM spiked at 1.8 μM in serum was relatively stable after three freeze (−80°C)/thaw cycles with peak area ratios differing from a mean value by an average of 6.9% (−13.5, 8.8, −0.2, and 5.1%). T1AM was stable in serum for at least one year when samples were stored at −80ºC. Storage stability of reconstituted extracts in the auto-sampler at 4°C was evaluated by analysis at time zero and after 24 hours. T1AM and T1AM-d4 exhibited a loss in absolute signal but demonstrated <5% variation in isotope dilution ratios for up to 24 hours. Extracts in mobile phase were stable after storage at −80°C for one month. DMSO and aqueous dilutions of T1AM were stable at −80°C for at least one year.
Table 1.
Precision characteristics of LC-MS/MS method
| Serum T1AM (μM) | RSD, % |
|
|---|---|---|
| Between-run (n=5) | Within-run (n=3) | |
| 0.8 | 20.3 | 10.5 |
| 1.1 | 13.9 | 13.1 |
| 1.6 | 13.4 | 2.7 |
| 4.0 | 7.4a | 6.0 |
| 40 | 8.4a | 5.9 |
Three replicates
3.3 Applications of the LC-MS/MS method
Availability of a sensitive and selective LC-MS/MS method for quantification of T1AM from serum is of value for pharmacokinetic studies where sample is limited, as is often a factor in rodent studies. We set out to examine serum T1AM concentrations after administration in rodents, using the LC-MS/MS method to quantify T1AM from small volumes of serum. Serum was prepared from blood collected from male rats (n=3) after IV injection with a bolus of synthetic T1AM at 20 mg/kg. Maximal T1AM induction of hypothermia in rodents is achieved with 50 mg/kg i.p.; however, a lower dose of 20 mg/kg induces significant hypothermia in both mice and rats (Scanlan, T.S., unpublished results and [1]). After a 20 mg/kg IV injection the circulating concentration of T1AM could be determined from as little as 2.5 μL of rat serum. When the data for serum concentration versus time was plotted (semi-logarithmic plot Figure 3 Panel B) T1AM was seen to display multi-compartment characteristics following an IV dose. An initial rapid inter-compartmental distribution phase was evident, followed by the slower elimination of T1AM from circulation. To estimate the elimination rate constant (K) a regression analysis was performed on the elimination phase ln(concentration) data points versus time (Figure 3 Panel B). From the resultant slope K was determined as 0.0146 min−1, allowing calculation of a biological half-life value (t1/2) for T1AM of around 48 min.
Fig. 3.
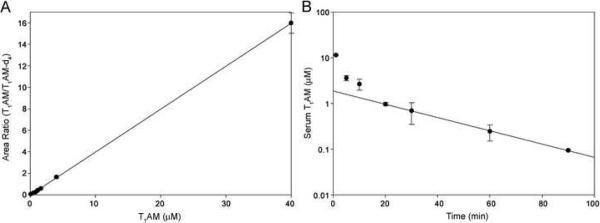
Calibration curves for quantifying T1AM were generated by a linear regression of the peak area ratios (T1AM/T1AM-d4) determined for rat serum calibrants versus specified T1AM concentration (standard deviation indicated, A). T1AM was quantified from rat serum (n=3) after IV injection with 20 mg/kg T1AM. A semi-logarithmic plot is shown for serum concentration versus time (standard deviation indicated, B). A linear regression for the elimination phase for T1AM resulted in an equation y = −0.0146x + 0.276 with a correlation coefficient (r2) = 0.999. From the slope K was determined as 0.0146 min−1, allowing calculation of a biological half-life (t1/2) value for T1AM of 48 min.
4. Discussion
To date, experiments performed to evaluate the in vivo biological effects of T1AM have been acute, single dose studies monitoring biological responses that occur within minutes to hours after dosing and which are attenuated after 6–9 h. For example, mice and Siberian hamsters given a single dose of T1AM display a peak drop in body temperature and metabolic rate within 1–2 h of dosing and these parameters return to normal 6–9 h later [4]. Use of the LC-MS/MS method for quantification from rat serum after administration of T1AM confirmed that circulating T1AM is rapidly cleared, with a half-life of 48 min. Based on the half-life obtained, T1AM is predicted to return to baseline levels after 6 h, a time point that correlates well with attenuation of the pharmacological effects for T1AM.
Availability of the LC-MS/MS method will also be useful for experiments studying metabolism of T1AM. It was recently determined that one mechanism for termination of T1AM activity in vivo may be by sulfotransferase-catalyzed sulfation in the liver [16]. After examination of a number of thyronamines, T1AM was found to be associated with the greatest sulfotransferase activity in a human liver preparation and in preparations of the expressed sulfotransferases 1A3 and 1E1. Another pathway for metabolism of T1AM is by deiodination to thyronamine (T0AM) [13]. Synthesis of endogenous T0AM was recently suggested to be possible from T1AM via deiodination by the type-III deiodinase Dio3 [13].
We have demonstrated that endogenous T1AM can be detected from human, mouse and rat serum with the LC-MS/MS method we describe. The endogenous concentrations of T1AM are at the LOD for the LC-MS/MS method. Previous estimates for the concentrations of T1AM detected from rodent serum are below 5 nM (1.8 μg/L) [1, 4]. The concentration of T1AM detected from human serum appears to be within the same range. The occurrence and extent of T1AM binding to proteins, and the effect of protein-binding on detection of T1AM is currently unknown. Although circulating concentrations of total T4 and T3 are in the μg/L range most circulating thyroid hormone is found bound to carrier proteins, such as thyroxine-binding globulin (TBG), transthyretin (TTR) and albumin [12]. In circulation, less than 0.3% of T4 and T3 is found in the free form not bound to carrier proteins [12]. In healthy adults with normal thyroid-stimulating hormone the reference intervals for concentration of free T4 and T3 were determined with LC-MS/MS to range from 8–22 ng/L and 0.9–6.8 ng/L respectively [10, 11]. Quantification of low-level T3 was achieved utilizing an API-5000 triple-quadrupole mass spectrometer equipped with a TurboSpray source (Applied Biosystems/MDS Sciex, Foster City, CA, USA) [10, 12]. Quantification of endogenous T1AM has proven to be technically challenging with a TSQ Quantum Discovery triple-quadrupole mass spectrometer. With the robust sample-work up and LC-MS/MS method developed, it remains to be seen if alternate mass spectrometers will enable quantification of endogenous T1AM to allow examination of a relationship with metabolic activity.
In summary, the LC-ESI-MS/MS method we have developed and validated demonstrates satisfactory precision and eliminates endogenous interference to ensure accurate determination of T1AM from serum. Sensitivity of the LC-MS/MS method allows for applications that include quantification of T1AM from limited sample for pharmacokinetic studies. We describe here quantification of T1AM from 2.5 μL rat serum across the range 500 fmol-25 pmol on-column analyte injection, with a 500 fmol on-column LLOQ.
Supplementary Material
Electrospray ionization of T1AM was also examined with a high-resolution (15 000) MicrOTOF-Q tandem mass spectrometry instrument. In-source fragmentation of T1AM was readily apparent under instrument conditions normally used to obtain full scan MS spectra with minimal fragmentation (in-source CID energy 0.0 eV, collision energy 6.0 eV; A). Use of the Q-q-TOF to examine fragmentation of T1AM precursor ion to product ions (collision energy 10 eV; B) allowed us to obtain an accurate mass measurement for T1AM [M+H]+ ion of m/z 356.0140 (0.5 ppm error). The calculated m/z for T1AM [M+H]+ ion is 356.0142. A measurement of m/z 338.9878 was obtained for putative [M+H-NH3]+ product ion. This confirms that the product ion is formed from loss of NH3 (−0.6 ppm error) as opposed to loss of OH (69.7 ppm error). Calibration was post-analysis with HyStar software utilizing injection of calibrant (1000 ppm aqueous sodium formate) along with T1AM. The ESI interface was operated in the positive mode using the following settings: end plate offset −500 V, capillary voltage −4500 V, nebulizer gas 1.6 bar, dry gas 4 L/min, dry temperature 200°C, funnel 1 RF 350 Vpp, funnel 2 RF 350 Vpp, hexapole RF 400 Vpp, collision energy 10 eV (for MS/MS normally ≥10 eV) and collision RF 300 Vpp.
Acknowledgments
This work was supported by a NIH grant (DK-52798 to T.S.S.) and was accomplished using instrumentation housed in the Department of Physiology and Pharmacology Bioanalytical Shared Resource and Department of Chemistry Mass Spectrometry Facility at Portland State University, Portland, OR. We thank Chris Toombs at Ikaria, Inc. for providing serum from T1AM treated rats.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- [1].Scanlan TS, Suchland KL, Hart ME, Chiellini G, Huang Y, Kruzich PJ, Frascarelli S, Crossley DA, Bunzow JR, Ronca-Testoni S, Lin ET, Hatton D, Zucchi R, Grandy DK. Nat. Med. 2004;10:638. doi: 10.1038/nm1051. [DOI] [PubMed] [Google Scholar]
- [2].Snead AN, Santos MS, Seal RP, Miyakawa M, Edwards RH, Scanlan TS. ACS Chem. Biol. 2007;2:390. doi: 10.1021/cb700057b. [DOI] [PubMed] [Google Scholar]
- [3].Regard JB, Kataoka H, Cano DA, Camerer E, Yin L, Zheng YW, Scanlan TS, Hebrok M, Coughlin SR. J.Clin.Invest. 2007;12:4034. doi: 10.1172/JCI32994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Braulke LJ, Klingenspor M, DeBarber A, Tobias SC, Grandy DK, Scanlan TS, Heldmaier G. J. Comp. Physiol. [B] 2008;178:167. doi: 10.1007/s00360-007-0208-x. [DOI] [PubMed] [Google Scholar]
- [5].Chiellini G, Frascarelli S, Ghelardoni S, Carnicelli V, Tobias SC, DeBarber A, Brogioni S, Ronca-Testoni S, Cerbai E, Grandy DK, Scanlan TS, Zucchi R. FASEB J. 2007;21:1597. doi: 10.1096/fj.06-7474com. [DOI] [PubMed] [Google Scholar]
- [6].Doyle KP, Suchland KL, Ciesielski TM, Lessov NS, Grandy DK, Scanlan TS, Stenzel-Poore MP. Stroke. 2007;38:2569. doi: 10.1161/STROKEAHA.106.480277. [DOI] [PubMed] [Google Scholar]
- [7].Hart ME, Suchland KL, Miyakawa M, Bunzow JR, Grandy DK, Scanlan TS. J. Med. Chem. 2006;49:1101. doi: 10.1021/jm0505718. [DOI] [PubMed] [Google Scholar]
- [8].Tai SS, Sniegoski LT, Welch MJ. Clin. Chem. 2002;48:637. [PubMed] [Google Scholar]
- [9].Tai SS, Bunk DM, White E, Welch MJ. Anal. Chem. 2004;76:5092. doi: 10.1021/ac049516h. [DOI] [PubMed] [Google Scholar]
- [10].Yue B, Rockwood AL, Sandrock T, La'ulu SL, Kushnir MM, Meikle AW. Clin. Chem. 2008;54:642. doi: 10.1373/clinchem.2007.098293. [DOI] [PubMed] [Google Scholar]
- [11].Gu J, Soldin OP, Soldin SJ. Clin. Biochem. 2007;40:1386. doi: 10.1016/j.clinbiochem.2007.08.007. [DOI] [PubMed] [Google Scholar]
- [12].Soldin SJ, Soukhova N, Janicic N, Jonklaas J, Soldin OP. Clin. Chim. Acta. 2005;358:113. doi: 10.1016/j.cccn.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Piehl S, Heberer T, Balizs G, Scanlan TS, Smits R, Koksch B, Kohrle J. Endocrinology. 2008;149:3037. doi: 10.1210/en.2007-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kushnir MM, Rockwood AL, Nelson GJ, Yue B, Urry FM. Clin. Biochem. 2005;38:319. doi: 10.1016/j.clinbiochem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- [15].Miyakawa M, Scanlan TS. Synth. Comm. 2006;36:891. [Google Scholar]
- [16].Pietsch CA, Scanlan TS, Anderson RJ. Endocrinology. 2007;148:1921. doi: 10.1210/en.2006-1172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Electrospray ionization of T1AM was also examined with a high-resolution (15 000) MicrOTOF-Q tandem mass spectrometry instrument. In-source fragmentation of T1AM was readily apparent under instrument conditions normally used to obtain full scan MS spectra with minimal fragmentation (in-source CID energy 0.0 eV, collision energy 6.0 eV; A). Use of the Q-q-TOF to examine fragmentation of T1AM precursor ion to product ions (collision energy 10 eV; B) allowed us to obtain an accurate mass measurement for T1AM [M+H]+ ion of m/z 356.0140 (0.5 ppm error). The calculated m/z for T1AM [M+H]+ ion is 356.0142. A measurement of m/z 338.9878 was obtained for putative [M+H-NH3]+ product ion. This confirms that the product ion is formed from loss of NH3 (−0.6 ppm error) as opposed to loss of OH (69.7 ppm error). Calibration was post-analysis with HyStar software utilizing injection of calibrant (1000 ppm aqueous sodium formate) along with T1AM. The ESI interface was operated in the positive mode using the following settings: end plate offset −500 V, capillary voltage −4500 V, nebulizer gas 1.6 bar, dry gas 4 L/min, dry temperature 200°C, funnel 1 RF 350 Vpp, funnel 2 RF 350 Vpp, hexapole RF 400 Vpp, collision energy 10 eV (for MS/MS normally ≥10 eV) and collision RF 300 Vpp.