

Abstract
3-Iodothyronamine (T1AM) is an endogenous derivative of thyroxine. Recently there have been numerous reports of analytical methods to quantify endogenous T1AM levels, but substantial discrepancies in concentration depending on the method of analysis (LC-MS/MS or immunoassay) suggest endogenous T1AM may be covalently modified in vivo. Using information dependent acquisition methods to perform unbiased scans for T1AM metabolites following a single IP injection in mice, we have identified O-sulfonate-T1AM, N-acetyl-T1AM and T1AM-glucuronide as conjugates occurring in vivo, as well as the oxidatively deaminated 3-iodothyroacetic acid and non-iodinated thyroacetic acid. 3-iodothyroacetic acid, O-sulfonate-T1AM and T1AM-glucuronide are present in serum at greater concentrations that unmodified T1AM and all metabolites are extensively distributed to tissues. These results suggest covalent modifications of T1AM may play a critical role in regulating distribution and biological activity of T1AM, and analytical methods to quantify endogenous T1AM should be able to account for these metabolites as well.
Keywords: 3-iodothyronamine, information dependent acquisition, LC-MS/MS
1. Introduction
3-Iodothyronamine (T1AM) is an endogenous derivative of thyroxine (T4), the main thyroid hormone produced by the thyroid gland. T1AM is found in circulation and in tissues of rat, mice, guinea pig, hamster and human [1–5]. T1AM is a decarboxylated and deiodinated derivative of T4 (Figure 1) and has a unique pharmacological profile [1]. In rodents, T1AM acutely induces hypothermia, decreases cardiac function and shifts the respiratory quotient (RQ) from predominantly carbohydrate to predominantly lipid utilization [1–3], suggesting a role for T1AM in regulating metabolic homeostasis.
Figure 1.

Structures of T4, T1AM, and T1AM metabolites TA1, T0AM, S-T1AM, TA0, Ac-T1AM and T1AM-glucuronide.
Due to the unique pharmacological effects of T1AM, there has been much interest in measuring endogenous levels of T1AM. Multiple analytical assays have been developed to quantify endogenous T1AM in circulation and in tissues of multiple species. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods have been validated using multiple reaction monitoring (MRM) to increase selectivity for T1AM by monitoring multiple m/z transitions [6–8]. In addition, an immunoassay has also been developed to quantify T1AM in human serum [4], and unexpectedly shows serum T1AM to be 100 fold higher than when measured by LC-MS/MS [4,8]. This discrepancy led us to hypothesize that endogenous T1AM is covalently modified in vivo, resulting in pools of endogenous modified T1AM recognizable by the T1AM antibody but not by an MRM LC-MS/MS method. To date, the only in vivo study on the metabolism of T1AM shows oxidative deamination in rat to form 3-iodothyroacetic acid (TA1) (Figure 1), another thyroid hormone derivative [9]. In vitro data show T1AM is a substrate for the type 3 inner ring deiodinase enzyme and human liver sulfotransferases, resulting in the non-iodinated thyronamine, T0AM [10] and O-sulfonate-T1AM (S-T1AM) [11], respectively (Figure 1). To date, there are no data on covalent modifications of T1AM occurring in vivo.
LC-MS/MS methods are valuable analytical tools due to the selectivity and sensitivity that can be routinely achieved. In the current situation, however, the selectivity of existing LC-MS/MS methods may be limiting studies of endogenous T1AM since the MRM methods monitor only for unmodified T1AM [6–8] and are unable to monitor for potential covalent modifications. Unknown modifications of T1AM can be studied using LC-MS/MS when information dependent acquisition (IDA) methods are used. IDA methods utilize unbiased survey scans to identify possible compounds that are structurally related to the parent compound, T1AM, and generate MS/MS fragmentation spectra that provide additional structural information. In this study we 1) use IDA methods to identify in vivo metabolites of T1AM, 2) develop and validate an MRM LC-MS/MS method to quantify metabolites of T1AM in mouse serum following a single injection of T1AM, and 3) provide a relative assessment for the tissue distribution of T1AM metabolites.
2. Materials and Methods
2.1 Chemicals and Reagents
T1AM, T0AM, d4-T1AM, TA1 and d4-TA1 were synthesized as described previously [9,12,13]. N-Acetyl-T1AM (Ac-T1AM) was synthesized in one step from T1AM with acetic anhydride and triethylamine in THF, and O-sulfonate-T1AM (S-T1AM) was synthesized in one step from Boc-protected T1AM using thionyl chloride, followed by hydrolysis of the Boc-protecting group. Mouse serum was purchased from Invitrogen (Carlsbad, CA) and mouse liver microsomes were purchased from BD Biosciences (San Jose, CA). β-glucuronidase from bovine liver was purchased from Sigma-Aldrich (St. Louis, MO). HPLC grade solvents were purchased from Burdick and Jackson (Muskegon, MI) or Fisher (Pittsburg, PA).
TA0 and d4-TA0 were synthesized using similar protocols to those described for TA1 and d4-TA1 [9]. Briefly, (4-hydroxy-phenyl)-acetic acid tert-butyl ester was coupled to a TIPS-boronic acid, and the protecting groups were cleaved to produce TA0. Similarly, (4-hydroxy-phenyl)-acetic acid tert-butyl ester was coupled to p-triisopropylsilanyloxy-d4-phenyl boronic acid (from [d6]-phenol starting material), and cleavage of the protecting groups produced d4-TA0. Briefly, d4-T0AM was synthesized by coupling BOC-tyramine to p-triisopropylsilanyloxy-d4-phenyl boronic acid, and cleavage of the protecting groups yielded d4-T0AM-chloride.
T1AM-glucuronide was synthesized enzymatically using a modified protocol for T4 glucuronidation [14]. Briefly, 150 μl reaction buffer (75 mM Tris-HCl pH 7.8, 7.5 mM MgCl2, 30 mM UDP-glucuronic acid, 1 μM T1AM, 0.1 μM iopanoic acid, 0.1 μM iproniazid) was incubated with 50 μL of a mouse liver microsome dilution (4 μg/μL in reaction buffer) for 60 minutes at 37 °C. The reaction was stopped by adding 200 μL ice cold methanol and samples were left on ice for 20 minutes. Samples were centrifuged at 12,000 × g for 45 minutes at 5 °C and supernatants were filtered with 0.22 μm centrifugal filters prior to injection for LC-MS/MS analysis.
2.2 Animal Studies
Experimental protocols were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Oregon Health & Science University Institutional Animal Care & Use Committee. Wild type male C57Bl/6 mice, aged 8–10 weeks, were housed in a climate controlled room with a 12 hour light-dark cycle with ad libitum access to food and water. T1AM was dissolved in 1:1 DMSO:saline (0.9% NaCl). Mice were injected intraperitoneally (IP) with 25 mg/kg T1AM. Following euthanasia of four mice at each timepoint (10, 20, 45 and 90 minutes, 3 and 6 hours post-injection, and vehicle injected, t = 0), blood was removed and allowed to clot on ice for a minimum of 30 minutes prior to centrifugation at 7500 × g. Sera was removed and stored at −80 °C until further use. Tissues were frozen immediately on dry ice and stored at −80 °C until further use. Samples were used within one month of collection.
2.3 Sample preparation
2.3.1 Calibration standards and serum samples
Commercial mouse serum was used to prepare samples for use in calibration curves. A mix of standards (T1AM, T0AM, S-T1AM, Ac-T1AM, TA1 and TA0) was added to commercial mouse serum to produce the following final concentrations: 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, 10 and 40 μM, and stored at −80 °C until analysis. Four serum samples were analyzed at each time point following T1AM injection, and four vehicle injected (t = 0) samples were analyzed. Each calibrant or serum sample was prepared by adding 10 μL of internal standard mix (10 μM each of d4-T1AM, d4-T0AM, d4-TA1 and d4-TA0) to 40 μL of serum standard. Samples were vortexed and 100 μL of sodium acetate (100 mM, pH 5.0) was added. To each calibrant, 50 μL of β-glucuronidase (1 mg/100 μL 0.2% NaCl) was added. Each experimental serum sample was processed in the absence and presence of β-glucuronidase by adding 50 μL 0.2% NaCl (negative β-glucuronidase) or 50 μL β-glucuronidase (0.1 mg/mL in 0.2% NaCl). All samples were incubated for 60 minutes at 37 °C. Reactions were stopped with the addition of ice-cold methanol (200 μL) and samples were left on ice for 20 minutes. Samples were centrifuged at 12000 × g for 10 minutes at 5 °C and supernatants were filtered with 0.22 μm centrifugal filters prior to injection for LC-MS/MS analysis.
Mouse serum spiked with calibrants of known concentrations were aliquoted and stored at −80 °C to be analyzed for determination of precision, accuracy, lower limits of detection (LLOD) and lower limits of quantitation (LLOQ). LLOD was determined using a signal to noise (S/N) ratio of 3:1, and LLOQ was determined using a S/N ratio of 10:1 along with precision and accuracy within 20%. Calibration curves were analyzed with a 1/× weighted regression analysis. To determine intra-assay precision and accuracy, six samples were analyzed at the LLOQ for each compound and at 40 μM. To determine inter-assay precision and accuracy, calibrants at the LLOQ and at 40 μM were processed and analyzed on at least four separate days over the course of one month. Precision is calculated as relative standard deviation (RSD), using the equation: RSD = 100*(σ/x̄), where σ is the standard deviation and x̄ is average concentration. Accuracy is calculated as % bias, using the following equation: % bias = 100*(x−μ)/μ where x is the measured concentration and μ is the nominal concentration. Precision of the analyte retention times are demonstrated as the standard deviation of at least seven samples, composed of both the LLOQ and 40 μM concentrations, run on seven different days.
2.3.2 Tissue sample preparation
All tissues were analyzed using samples from three mice. Liver, kidney, heart, brain brown adipose tissue (BAT) and white adipose tissue (WAT) were weighed and sodium acetate (100 mM, pH 5.0) was added in a ratio of 1 mL per 1 gram of tissue, except for BAT samples, which each received 150 μL. Tissues were homogenized using 20 passes in a motorized dounce homogenizer. Samples were centrifuged at 12000 × g for 10 minutes, and the volume of supernatant was recorded. For each sample, 10 μL of internal standard mix was added to 50 μL of homogenized tissue supernatant. Samples were vortexed and 50 μL ice-cold methanol was added. Samples were vortexed again and incubated on ice for 20 minutes to allow for complete protein precipitation. Samples were centrifuged at 12000 × g for 10 minutes and supernatants were filtered with 0.22 μm centrifugal filters prior to injection for LC-MS/MS analysis.
2.4 LC-MS/MS
2.4.1 General
Instrumention was housed in the Bioanalytical Shared Resource/Pharmacokinetics Core at Oregon Health & Science University. A Shimadzu Prominence HPLC (CANBY OR) was coupled to a 4000 QTRAP triple quadrupole/linear ion trap mass spectrometer (AB Sciex, Foster City, CA) using electrospray ionization (ESI). For all samples, 10 μL was injected onto a Poroshell120 2.1 × 100 mm C18 column (Agilent Technologies, Santa Clara, CA). Solvent A was water with 0.05% acetic acid and Solvent B was acetonitrile with 0.05% acetic acid. The flow rate was 0.3 mL/min and the gradient conditions were as follows: 10% B to 45% B from 0 to 14 min; 45% B to 95% B from 14 to 15 min; 95% B from 15 to 18 min; 95% B to 10% B from 18 to 19 min; 10% B from 19 to 22 min. The autosampler was kept at 4 °C and the oven temperature was 30 °C.
2.4.2 IDA Methods
IDA methods were used to screen for potential metabolites of T1AM. Four IDA methods were created using the LightSight software and used for metabolite screening. IDA methods included a precursor survey scan method using the m/z 212 fragment of T1AM and a predicted MRM (pMRM) survey scan method using Q1/Q3 ion pairs generated using LightSight software (AB Sciex) based on predicted biotransformations of T1AM. Since TA1 is the only known in vivo metabolite of T1AM, previously observed in rat [9], additional IDA methods were generated using TA1 as the parent compound to look for potential covalent modifications. The TA1 based IDA methods included a precursor survey scan method using the m/z 127 fragment and a pMRM survey scan method using Q1/Q3 ion pairs generated in LightSight based on predicted biotransformation of TA1. T1AM based IDA methods were operated in positive ion mode while TA1 IDA methods were operated in negative ion mode. Data were analyzed manually and LightSight was used to identify possible metabolites of T1AM.
For all methods, the IDA experiment was an enhanced product ionization (EPI) scan. For the precursor ion survey scan methods, the detection of the product ion of interest (m/z 212 or 127) triggered an EPI scan of the precursor ion which was producing the product ion of interest (m/z 212 or 127). For the pMRM methods, and EPI scan was triggered upon the detection of a predicted Q1/Q3 ion pair. EPI scans were triggered when the intensity exceeded 500 cps for the MRM survey scans or 1000 cps for the precursor ion survey scans. For all EPI scans, the scan rate was 4000 Da/s over the mass ranges 80–179.2 and 174–609.2 (precursor 127), 174–596 (precursor 212), 174–692 (T1AM MRM) or 174–681.2 (TA1 MRM). The following parameters were used for all IDA methods: curtain gas (CUR), 20; collision gas (CAD), high; temperature, 550 °C; ion source gas 1, 50; ion source gas 2, 50. For the precursor 212 and T1AM MRM methods, the ionspray voltage (IS) was 5200 V and for the precursor 127 and TA1 MRM methods the IS was −4500 V. The precursor 212 and precursor 127 methods used the following parameters, respectively: declustering potential (DP), 40 V and −5 V; collision energy (CE), 27 eV and −12 eV; and collision exit potential (CXP), 14 V and −17 V.
2.4.3 MRM Methods
Two separate MRM methods were validated to quantify T1AM metabolites in serum. Method 1 was operated in the positive ionization mode to analyze T0AM, d4-T0AM, T1AM-glucuronide, S-T1AM, T1AM, d4-T1AM, and Ac-T1AM, with the following parameters: CUR, 30; CAD, medium; IS, 4500 V; temperature, 500 °C; ion source gas 1, 50; ion source gas 2, 40; scan rate, 15 msec. DP, CE and CXP values for Method 1 MRM transitions are given in Table 1 Method 2 was operated in the negative ionization mode to analyze TA0, d4-TA0, TA1, and d4-TA1, with the following parameters: CUR, 50; CAD, medium; IS, −4000; temperature, 500 °C; ion source gas 1, 30; ion source gas 2, 40; scan rate, 75 msec. DP, CE and CXP values for Method 2 MRM transitions are given in Table 2.
Table 1.
MRM Method 1 instrument parameters.
| Analyte | Q1 (m/z ) | Q3 (m/z ) | DP (V) | CE (eV) | CXP (V) |
|---|---|---|---|---|---|
| T1AM | 356 | 339* | 81 | 10 | 17 |
| 212 | 81 | 10 | 27 | ||
| d4-T1AM | 360 | 343* | 96 | 17 | 10 |
| 216 | 96 | 27 | 22 | ||
| T0AM | 230 | 213* | 66 | 27 | 14 |
| 109 | 66 | 35 | 20 | ||
| d4-T0AM | 234 | 217* | 71 | 17 | 16 |
| 113 | 76 | 43 | 6 | ||
| T1AM-glucuronide | 532 | 515 | 81 | 10 | 11 |
| 356 | 81 | 10 | 11 | ||
| 339 | 81 | 10 | 11 | ||
| 212 | 81 | 10 | 11 | ||
| S-T1AM | 436 | 419* | 81 | 10 | 12 |
| 339 | 81 | 10 | 10 | ||
| 212 | 81 | 10 | 14 | ||
| Ac-T1AM | 398 | 356 | 66 | 27 | 14 |
| 339 | 66 | 27 | 14 | ||
| 271 | 66 | 27 | 14 | ||
| 212* | 66 | 27 | 14 |
, m/z fragment used for quantification.
Table 2.
MRM Method 2 instrument parameters.
| Analyte | Q1 (m/z ) | Q3 (m/z ) | DP (V) | CE (eV) | CXP (V) |
|---|---|---|---|---|---|
| TA1 | 369 | 325 | −5 | −8 | −13 |
| 127* | −5 | −12 | −17 | ||
| d4-TA1 | 373 | 329 | −50 | −6 | −9 |
| 127* | −50 | −26 | −11 | ||
| TA0 | 243 | 199 | −5 | −14 | −39 |
| 106* | −5 | −26 | −19 | ||
| d4-TA0 | 247 | 203 | −30 | −14 | −17 |
| 106* | −30 | −26 | −15 | ||
| 97 | −30 | −31 | −12 |
, m/z fragment used for quantification.
2.5 Statistics
The serum T1AM metabolite time-course was analyzed using two-way ANOVA to determine statistical significance.
3. Results
3.1 IDA Screen
Mice received a single IP injection of T1AM (25 mg/kg) to increase the concentration of potential metabolites. An initial screen with IDA methods using serum collected 10 minutes post-T1AM injection identified potential metabolites corresponding to m/z shifts of +176, +80, +42 and −112 using the positive ion mode methods, and a metabolite corresponding to unmodified TA1 using the negative ion mode methods. Mass shifts of +176, +80 and +42 correspond to the addition of glucuronide, sulfonate and acetyl groups, respectively, while the mass shift of −112 would correspond to the predicted transformation of combined deiodination and oxidative deamination to produce thyroacetic acid, TA0.
TA1, TA0, and S-T1AM have all been previously synthesized and were used as authentic standards to confirm the identity of the IDA identified metabolites. To confirm the identity of metabolites corresponding to m/z shifts of +42 and +176, N-acetyl-T1AM (Ac-T1AM) was synthesized chemically and T1AM-glucuronide was synthesized enzymatically using mouse liver microsomes (Figure 1). MS/MS product ion spectra were compared to the EPI spectra generated in the IDA methods for the +42 and +176 compounds. As shown in Figure 2, spectra for synthetic Ac-T1AM and the +42 compound both show ions with an m/z of 398, 356, 339, 271 and 212 (Figure 2A), and spectra for T1AM-glucuronide and the +176 compound both show ions with an m/z of 532, 515, 356, 339 and 212 (Figure 2B). Since Ac-T1AM was synthesized chemically, it was produced in a great enough quantity to be used in the generation of a calibration curve to quantify serum Ac-T1AM; enzymatically generated T1AM-glucuronide was not produced in a great enough quantity to purify and generate a calibration curve for quantification. Cleavage of T1AM-glucuronide resulted in undetectable T1AM-glucuronide and an increase in unmodified T1AM (Figure 3), allowing for indirect quantification of serum T1AM-glucuronide. In the MRM method, T1AM-glucuronide was monitored using Q1/Q3 ion pairs based on fragments identified in the MS/MS spectra (Figure 2B).
Figure 2.

Identification of Ac-T1AM (A) and T1AM-glucuronide in mouse serum 10 minutes post-T1AM injection. A) The EPI spectrum of the +42 metabolite identified in serum (top panel) and the MS/MS spectrum of synthetic N-acetyl-T1AM (bottom panel). B) The EPI spectrum of the +176 metabolite identified in serum (top panel) and the EPI spectrum generated for enzymatically synthesized T1AM-glucuronide (bottom panel) with the chemical structure inset.
Figure 3.
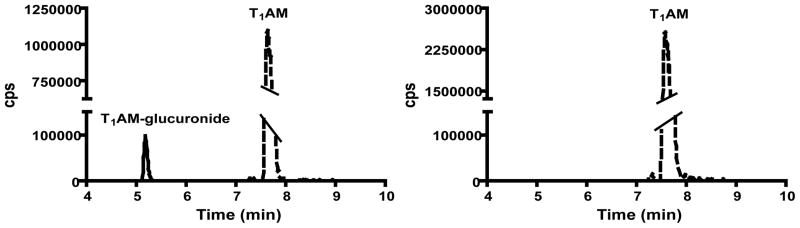
Chromatographic traces for T1AM-glucuronide (RT = 5.2 min) and T1AM (RT = 7.6 min) from a serum sample 20 minutes post-T1AM injection, in the absence (left) or presence (right) of β-glucuronidase. Dashed line corresponds to T1AM-glucuronide and solid line corresponds to T1AM. There is no detectable peak corresponding to T1AM-glucuronide following treatment with β-glucuronidase.
3.2 LC-MS/MS MRM Method Validation
Two separate LC-MS/MS MRM methods were developed and validated to quantify T1AM metabolites in mouse serum. Method 1 was operated in the positive mode to quantify T1AM, T0AM, Ac-T1AM, S-T1AM and T1AM-glucuronide. D4-T0AM was used as the internal standard for T0AM, and d4-T1AM was used as the internal standard for T1AM, S-T1AM and Ac-T1AM (Table 1). Method 2 was operated in the negative mode to quantify TA1 and TA0 using d4-TA1 and d4-TA0, respectively, as internal standards (Table 2). Although T0AM was not identified in any IDA methods, it was included in the MRM method since T1AM was previously shown to be an in vitro substrate for deiodination [10]. Calibration curves were constructed in commercially available mouse serum since the experimental samples being quantified were also mouse serum. Calibrants covered the range of 0.05 μM to 40 μM. Intra- and inter-assay precision (RSD) and accuracy were within 20% for all compounds at the LLOQ and within 15% for all compounds at 40 μM (Table 3). Typical slopes for linear regression of calibration curves, as well as retention times, LLOQ and LLOD for each compound are listed in Table 4.
Table 3.
Precision and accuracy for Method 1 and Method 2.
| Intra-assay | |||||||
|---|---|---|---|---|---|---|---|
| 0.1 uM | 0.25 uM | ||||||
| Avg (μM) | Bias (%) | RSD (%) | Avg (μM) | Bias (%) | RSD (%) | Avg (μM) | |
| T1AM | 0.11 | 9 | 13 | 40.6 | |||
| T0AM | 0.10 | −2 | 13 | 39.3 | |||
| Ac-T1AM | 0.10 | −3 | 15 | 44.3 | |||
| S-T1AM | 0.22 | −12 | 7 | 41.7 | |||
| TA1 | 0.24 | −4 | 13 | 38.4 | |||
| TA0 | 0.24 | −2 | 13 | 40.2 | |||
| Inter-assay | |||||||
|---|---|---|---|---|---|---|---|
| 0.1 uM | 0.25 uM | ||||||
| Avg (μM) | Bias (%) | RSD (%) | Avg (μM) | Bias (%) | RSD (%) | Avg (μM) | |
| T1AM | 0.10 | 2 | 19 | 40.8 | |||
| T0AM | 0.11 | 9 | 17 | 41.3 | |||
| Ac-T1AM | 0.12 | 15 | 12 | 42.5 | |||
| S-T1AM | 0.22 | −12 | 16 | 38.9 | |||
| TA1 | 0.26 | 4 | 10 | 39.9 | |||
| TA0 | 0.26 | 3 | 11 | 42.1 | |||
n = 4–7 per analyte
Table 4.
Typical regression equations for calibration curves, retention times, LLOD and LLOQ.
| Analyte | Slope | Regression Intercept | r2 | RT (minutes) | LLOD (μM) | LLOQ (μM) |
|---|---|---|---|---|---|---|
| T1AM | 0.31 | 0.006 | 0.998 | 7.63 ± 0.05 | 0.05 | 0.1 |
| T0AM | 0.496 | −0.004 | 0.998 | 4.95 ± 0.03 | 0.05 | 0.1 |
| S-T1AM | 0.0204 | 0.0003 | 0.989 | 7.28 ± 0.04 | 0.1 | 0.25 |
| Ac-T1AM | 0.305 | −0.019 | 0.964 | 12.86 ± 0.05 | 0.05 | 0.1 |
| TA1 | 0.407 | 0.016 | 0.994 | 13.84 ± 0.11 | 0.05 | 0.25 |
| TA0 | 0.351 | 0.001 | 0.999 | 10.71 ± 0.07 | 0.05 | 0.25 |
RT, retention time, +/− standard deviation.
3.3 T1AM Metabolites in Serum
The validated LC-MS/MS MRM methods were used to quantify T1AM metabolites in serum following a single IP injection of T1AM. Sera were analyzed between 0 (vehicle injected) and 6 hours post T1AM injection. Serum samples were processed in the presence or absence of β-glucuronidase in order to quantify T1AM-glucuronide. At 10 minutes post T1AM injection, all metabolites are detectable (Figure 4). Each metabolite was quantified at all time points collected to determine the metabolite profile of T1AM in serum (Figure 5A). At 10 minutes post-injection, the serum concentration of TA1 is 17.7 ± 4.6 μM, which is equal to the concentration of unmodified T1AM, 16.6 ± 8.5 μM. At 20 minutes post-injection, the concentrations of TA1 and T1AM are 23.5 ± 13.2 μM and 5.6 ± 3.1 μM, respectively, suggesting oxidative deamination is a major and rapid pathway of T1AM metabolism. Between 20 minutes and 6 hours, the concentrations of TA1, S-T1AM and T1AM-glucuronide are all greater than or equal to the concentrations of unmodified T1AM. The maximum serum concentrations for TA1, S-T1AM and T1AM-glucuronide all occur at 20 minutes post T1AM injection, and are 23.5 ± 13.2 μM, 10.2 ± 3.3 μM and 6.74 ± 3.01 μM, respectively. Ac-T1AM is quantifiable in at least 3 out of 4 samples between 0 and 90 minutes, reaching a maximum serum concentration of 0.30 ± 0.11 μM at 20 minutes, and decreases to remain measurable at the LLOQ in only one sample at 3 hours. T1AM concentration is significantly greater than T1AM-glucuronide, S-T1AM, Ac-T1AM and TA0 at 10 minutes post-injection (p < 0.001 for all), and concentration of TA1 is significantly greater than T1AM at 20 (p < 0.001), 45 (p < 0.001), and 90 (p < 0.05) minutes post-injection.
Figure 4.

Representative chromatographic traces for T1AM metabolites from mouse serum at 10 minutes post-T1AM injection. Traces correspond to the following m/z transitions: T0AM, 230-213; d4-T0AM, 234-217; T1AM-glucuronide, 532-515; S-T1AM, 436-418; T1AM, 356-339; d4-T1AM, 360-343; TA0, 243-106; d4-TA0, 247-106; Ac-T1AM, 398-212; TA1, 369-127; d4-TA1, 373-127.
Figure 5.

T1AM metabolites in mouse serum. A) Concentrations of T1AM, T1AM-glucuronide, S-T1AM, Ac-T1AM, TA1 and TA0 between 10 minutes and 6 hours after a single IP injection of T1AM. Inset shows data from 0 to 2 hours; n = 3–4 per point, except for the following: T1AM and T1AM-glucuronide, 6 hours, n = 2; Ac-T1AM, 3 hours, n = 1; TA0, 10 and 45 minutes, n = 2; TA0, 20 minutes and 3 hours, n = 1. B) Representative chromatographic traces of T0AM in serum at 20 minutes post-T1AM injection in the absence (left) or presence (right) of β-glucuronidase.
TA0 and T0AM, the deiodinated thyroacetic acid and thyronamine, respectively, were not observed in high quantities in serum. TA0 is quantifiable in two out of four samples at 10 and 45 minutes, and one out of four samples at 20 minutes and 3 hours. The greatest concentration of TA0 occurs is at 6 hours post-injection (0.77 ± 0.16 μM). T0AM is occasionally detectable, but always below the LLOQ. Interestingly, the T0AM peak intensity increases at some time points following incubation with β-glucuronidase (Figure 5B), which suggests T0AM-glucuronide may be a minor metabolite. T0AM remains below the LLOQ even after incubation with β-glucuronidase. Measured concentrations for all metabolites at all time points are reported in Supplemental Table 1.
3.5 Tissue Distribution of T1AM Metabolites
The method reported here is validated to quantify metabolites from mouse serum. To determine if the calibration curves constructed in mouse serum could be used to quantify metabolites in tissue homogenates, untreated mouse liver, kidney and brain homogenates were spiked with 1 μM calibrants and analyzed using the mouse serum calibration curves. For all metabolites in each tissue matrix, the calculated concentrations were outside the acceptable precision and accuracy established during validation of serum quantification (data not shown). Since no information is currently known on the tissue distribution of T1AM metabolites, a preliminary investigation into tissue distribution of metabolites was conducted in order to determine if tissue specific calibration curves should be generated in the future to quantify tissue levels of T1AM metabolites. To determine the relative tissue distribution of metabolites, the area ratio (peak area of metabolite to deuterated internal standard) for each metabolite was normalized to the mass of tissue analyzed. Figure 6 illustrates the relative tissue distributions of metabolites following T1AM injection. Tissues analyzed include liver, kidney, heart, brain, BAT and WAT. T1AM is distributed in all six tissues at 6 hours, the final time point analyzed. T0AM is detectable at 6 hours in liver and kidney, but only intermittently at earlier time points in brain, heart and WAT, and not detectable in BAT at any time point. T1AM-glucuronide remains detectable in both liver and kidney at 6 hours, suggesting these tissues may be sites of T1AM-glucuronide accumulation. S-T1AM is transiently present in BAT (at 90 minutes), but remains in liver, kidney, heart and WAT at 6 hours, while brain is not a major site of distribution. Ac-T1AM is distributed to all tissues analyzed, and remains in kidney, heart and brain at 90 minutes, and in liver, BAT and WAT at 6 hours. TA1 is also distributed to all tissues analyzed, but only minimally to brain (to 90 minutes) while remaining present in liver, kidney, heart, BAT and WAT at 6 hours. TA0 was detectable in liver and kidney between 0 and 6 hours, but was only detectable in heart at 6 hours and was not detectable at any time point in brain, BAT or WAT.
Figure 6.

Relative tissue distribution of T1AM metabolites to liver, kidney, heart, brain, BAT and WAT. Peak areas were integrated with respect to the deuterated internal standard (d4-T1AM used for T1AM-glucuronide, S-T1AM and Ac-T1AM) and the area ratio was normalized to the amount of tissue analyzed; n = 1–3 per point. T0AM was not detectable in BAT. TA0 was not detected in brain, BAT or WAT, and was only detected in heart at 6 hours (n = 2).
4. Discussion
IDA methods offer a means to perform an unbiased screen for unknown metabolites of small molecules by LC-MS/MS. Here we report the use of IDA methods to identify novel in vivo metabolites of T1AM. This study provides the first report of thyronamine metabolites in mouse, including TA1, which was previously shown to be a metabolite in rat [9], and the first report of S-T1AM occurring in vivo, which was previously suggested as a metabolite in an in vitro enzymatic assay [11]. The use of IDA methods also identified two novel compounds that occur as T1AM metabolites, Ac-T1AM and T1AM-glucuronide.
Since little was known about T1AM metabolites in serum prior to this study, the compounds identified using IDA methods were used to develop a validated MRM method to quantify this panel of metabolites in mouse serum. In order to determine which compounds may represent major pathways of metabolism, mouse serum was analyzed following a single IP injection of T1AM. At 20 minutes post-injection, three metabolites, TA1, S-T1AM and T1AM-glucuronide, are present in serum at greater concentrations than unmodified T1AM. This suggests that oxidative deamination, sulfation and glucuronidation represent the major pathways of T1AM metabolism in mice. Ac-T1AM and TA0 were also quantified in serum, although in lesser concentrations than the other metabolites, indicating these may be minor metabolic pathways. Since T1AM is known to be a substrate for deiodination to form T0AM [10], it was interesting that T0AM was never present above the LLOQ for the method, suggesting this is a minor pathway in vivo. Although it remained below the LLOQ, the increased peak intensity for T0AM following treatment with β-glucuronidase suggests that T0AM-glucuronide may be also be a minor T1AM metabolite. Another reason for low T0AM levels could be that deiodination to T0AM results in rapid elimination of the compound from the body, preventing an accumulation of T0AM in serum.
While absolute values were not quantified, we also investigated the relative tissue distribution of T1AM metabolites. Similar to the serum results, T0AM and TA0 appear to be minor metabolites and minimally distributed to tissues. TA0 does appear to accumulate in liver and kidney, and was detectable in brain at 6 hours post-injection. This is a similar pattern of accumulation that was seen in serum, in which the highest concentration of TA0 (0.77 ± 0.16 μM) was observed at 6 hours post-injection. T0AM also appears to accumulate in liver and kidney at 6 hours post-injection, which contrasts with serum, where T0AM was not quantifiable. Since it appears that T0AM-glucuronide is present to some extent in serum (Figure 5B), tissue distribution of T0AM-glucuronide may be interesting to investigate in the future.
TA1 and S-T1AM are extensively distributed and remain in most tissues at 6 hours post-injection. This is similar to the elevated levels of TA1 and S-T1AM measured in serum. TA1 is distributed to all tissues analyzed, although is not detectable in brain longer than 90 minutes post-T1AM injection. S-T1AM does not appear to be distributed to brain, and is not detectable in BAT longer than 90 minutes post-injection. This suggests that tissue specific distribution of these metabolites may be important for regulating their action. Ac-T1AM is measurable in liver, kidney and WAT at 6 hours (Figure 6), whereas it is not quantifiable in serum 3 hours post-injection (Figure 5A). This apparent accumulation of Ac-T1AM in liver and WAT likely reflects specific tissue binding of this metabolite since fat is more poorly perfused than liver. Likewise, T1AM-glucuronide appears to accumulate in liver and kidney, which is evidenced by detection of this metabolite in these tissues at 6 hours; unlike Ac-T1AM, T1AM-glucuronide is still detectable in serum 6 hours post-injection. This accumulation of T1AM-glucuronide in liver and kidney is similar to a recent report of T4-glucuronide present in liver and kidney [15], suggesting these tissues may serve as storage for glucuronide-conjugates.
We have reported the occurrence of novel in vivo metabolites of T1AM, relative tissue distribution of metabolites and a validated LC-MS/MS MRM method to quantify these metabolites from mouse serum. The number of T1AM metabolites (Figure 1) is similar to the extensive metabolism observed for T4 [16] and rich, diverse metabolism such as this is not generally seen with synthetic drugs or xenobiotics. This suggests that covalent modifications may be critical for regulating endogenous T1AM exposure and biological activity. As interest in measuring T1AM remains high and new analytical methods continue to be developed [4,8], this method represents a valuable analytical tool to study endogenous T1AM metabolites. Since several metabolites, notably S-T1AM, Ac-T1AM and T1AM-glucuronide remain detectable in tissues at 6 hours, investigating endogenous tissue distribution of these metabolites is essential for improving our understanding of T1AM action and regulation.
Supplementary Material
Highlights.
T1AM metabolites were identified using information dependent acquisition methods.
T1AM is extensively metabolized in mouse serum following a single IP injection.
MRM LC-MS/MS methods were validated to quantify T1AM metabolites in serum.
T1AM-glucuronide and Ac-T1AM were identified as novel compounds.
Acknowledgments
This work was supported by a grant from the NIH (DK-52798, T.S.S). This work used instrumentation maintained by the Bioanalytical Shared Resource/Pharmacokinetics Core at Oregon Health & Science University. We are grateful to Warren Wood, Aaron Nilsen and Federico Espinosa for preparing synthetic standards.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sarah A. Hackenmueller, Email: hackenmu@ohsu.edu.
Thomas S. Scanlan, Email: scanlant@ohsu.edu.
References
- 1.Scanlan TS, Suchland KL, Hart ME, Chiellini G, Huang Y, Kruzich PJ, Frascarelli S, Crossley DA, Bunzow JR, Ronca-Testoni S, Lin ET, Hatton D, Zucchi R, Grandy DK. Nat Med. 2004;10:638. doi: 10.1038/nm1051. [DOI] [PubMed] [Google Scholar]
- 2.Chiellini G, Frascarelli S, Ghelardoni S, Carnicelli V, Tobias SC, DeBarber A, Brogioni S, Ronca-Testoni S, Cerbai E, Grandy DK, Scanlan TS, Zucchi R. Faseb J. 2007;21:1597. doi: 10.1096/fj.06-7474com. [DOI] [PubMed] [Google Scholar]
- 3.Braulke LJ, Klingenspor M, DeBarber A, Tobias SC, Grandy DK, Scanlan TS, Heldmaier G. J Comp Physiol [B] 2008;178:167. doi: 10.1007/s00360-007-0208-x. [DOI] [PubMed] [Google Scholar]
- 4.Hoefig CS, Kohrle J, Brabant G, Dixit K, Yap B, Strasburger CJ, Wu Z. J Clin Endocrinol Metab. 2011;96:1864. doi: 10.1210/jc.2010-2680. [DOI] [PubMed] [Google Scholar]
- 5.Galli E, Marchini M, Saba A, Berti S, Tonacchera M, Vitti P, Scanlan TS, Iervasi G, Zucchi R. J Clin Endocrinol Metab. 2012;97:E69. doi: 10.1210/jc.2011-1115. [DOI] [PubMed] [Google Scholar]
- 6.DeBarber AE, Geraci T, Colasurdo VP, Hackenmueller SA, Scanlan TS. J Chromatogr A. 2008;1210:55. doi: 10.1016/j.chroma.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piehl S, Heberer T, Balizs G, Scanlan TS, Kohrle J. Rapid Commun Mass Spectrom. 2008;22:3286. doi: 10.1002/rcm.3732. [DOI] [PubMed] [Google Scholar]
- 8.Saba A, Chiellini G, Frascarelli S, Marchini M, Ghelardoni S, Raffaelli A, Tonacchera M, Vitti P, Scanlan TS, Zucchi R. Endocrinology. 2010;151:5063. doi: 10.1210/en.2010-0491. [DOI] [PubMed] [Google Scholar]
- 9.Wood WJ, Geraci T, Nilsen A, Debarber AE, Scanlan TS. Chembiochem. 2009;10:361. doi: 10.1002/cbic.200800607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piehl S, Heberer T, Balizs G, Scanlan TS, Smits R, Koksch B, Kohrle J. Endocrinology. 2008;149:3037. doi: 10.1210/en.2007-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pietsch CA, Scanlan TS, Anderson RJ. Endocrinology. 2007;148:1921. doi: 10.1210/en.2006-1172. [DOI] [PubMed] [Google Scholar]
- 12.Miyakawa M, Scanlan TS. Synthetic Communications. 2006;36:891. [Google Scholar]
- 13.Hart ME, Suchland KL, Miyakawa M, Bunzow JR, Grandy DK, Scanlan TS. J Med Chem. 2006;49:1101. doi: 10.1021/jm0505718. [DOI] [PubMed] [Google Scholar]
- 14.Hood A, Klaassen CD. Toxicol Sci. 2000;55:78. doi: 10.1093/toxsci/55.1.78. [DOI] [PubMed] [Google Scholar]
- 15.Buitendijk M, Galton VA. Thyroid. 2012;22:187. doi: 10.1089/thy.2011.0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu SY, Green WL, Huang WS, Hays MT, Chopra IJ. Thyroid. 2005;15:943. doi: 10.1089/thy.2005.15.943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.