

Abstract
The thiolato complex [Pt(II)(bipyridine)(N,S-aminoethanethiolate)]+ Cl- (1) undergoes sequential reactions with singlet oxygen to initially form the corresponding sulfenato complex [Pt(II)(bipyridine)(N,S(=O)-aminoethansulfenate)]+ (2) followed by a much slower reaction to the corresponding sulfinato complex. In contrast with many Pt dithiolato complexes, 1 does not produce any singlet oxygen, but its rate constant for singlet oxygen removal (kT) is quite large (3.2 × 107 M-1sec-1) and chemical reaction accounts for ca. 25 % of the value of kT). The behavior of 1 is strikingly different from the complex Pt(II)(bipyridine)(1,2-benzenditholate) (4). The latter complex reacts with 1O2 (either from an external sensitizer or via a self-sensitized pathway) to form a sulfinato complex. These two very different reactivity pathways imply different mechanistic pathways: The reaction of 1 with 1O2 must involve O-O bond cleavage and intermolecular oxygen atom transfer, while the reactive intermediate in complex 4 collapses intramolecularly to the sulfinato moiety.
The chemistry of platinum diimine thiolato complexes has been extensively studied during the past three decades. When dithiolato ligands are employed, such complexes exhibit strong luminescence due to a charge-transfer complex formed from donation from the dithiolate to the diimine ligands.1,2 Many of these complexes are also sensitizers for the production of singlet oxygen, although the actual quantum yields for these processes are not known.2-4 Some of these Pt-dithiolato complexes undergo self-sensitized oxidation reactions, with products ranging from monosulfinates and dinsulfinates4 to dithiolenes.3 Interestingly, products with singly oxidized sulfur sites, i.e. platinum sulfenates, have not been reported for these complexes. The factors that determine which product is formed and the nature of the intermediates involved are poorly understood. Platinum diimine complexes bearing one or several alkylthiolato complexes are also of interest as small model complexes for Pt-bound cysteine. Such complexes are formed during the interaction of cis-platinum drugs with the sulfur-rich sites of metallothionein, leading to inactivation of this enzyme and the well-known nephrotoxicity of these drugs.5 The chemistry and photophysics of Pt(diimine) complexes with just one alkyl thiolato ligand (as a model for one metal-bound cysteine site) has received much less attention than that of the dithiolate complexes. We now report a detailed study of the photooxidation of a Pt(diimine) monothiolato complexes, namely [Pt(II)(bipyridine)(N,S-aminoethanethiolate)]+ Cl- (1).6 In striking contrast with the dithiolate complexes which form sulfinato products upon photooxidation, complex 1 undergoes sequential oxidation reactions with singlet molecular oxygen, initially forming only the sulfenato adduct followed by slow formation of a sulfinato complex.
Unlike the Pt(II)(bipyridine)(dithiolato) complexes,2-4 [Pt(II)(bipyridine)(N,S-aminoethanethiolate)]+ (1) does not sensitize the production of singlet oxygen. However, upon reaction with 1O2 produced by an external photosensitzer (in water or methanol/water, sens=Methylene Blue, tungsten-halogen lamp, cut-off filter at 493 nm to prevent excitation of 1) complex 1 is cleanly converted to the corresponding sulfenato complex 2. We have followed the reaction by IR, UV/vis, 1H and 195Pt NMR, and liquid chromatography/mass spectroscopy (LC-MS). No Pt(IV) intermediates or products other than 2 were observed until conversion of 1 to 2 was complete.7 When the reaction was carried out in D2O, no deuterium incorporation at the methylene site of 1 adjacent to the sulfur atom was observed by mass spectroscopy. Spectroscopic data of the complexes are summarized in Table S1. Sulfenato complex 2 can also be obtained upon reaction of 1 with one equiv. of hydrogen peroxide. The assignment of 2 as a Pt(II) sulfenato complex was confirmed by an X-ray molecular structure.
The bond distance of the S=O bond of complex 2 (1.520 Å) is within the expected range of metal sulfenato complexes (1.5-1.6 Å).8 It is longer than that of the sulfinato complex 5 (1.45 Å)4, consistent with the fact that the S=O bond of a sulfenate is more polar than that of a sulfinate.8 Continuous exposure of the sulfenato complex 2 to singlet oxygen leads to slow conversion to the corresponding sulfinato complex 3. This complex was also characterized by IR, UV/vis and 1H and 195Pt NMR. While we were unable to obtain an X-ray molecular structure of 3, its identity as an S-bound sulfinate was confirmed by the characteristic IR stretches for the Pt-S(=O)2 moiety at 1210 cm-1 and 1070 cm-1.5 An 18O labeling experiment showed a change of these peaks to 1162 cm-1 and 1024 cm-1, consistent with their assignment as S-bound sulfinato bands.9
Kinetic analyses of the photooxidation of complexes 1 and 2 is consistent with formation of the sulfenate 2 as the only initial product. The total rate constant (kT) of singlet oxygen removal by 1 is about an order of magnitude higher than that of complex 2, i.e. 3.2 × 107 M-1sec-1 for 1 and 2.5 × 106 M-1sec-1 for the sulfenato complex 2 (Figure S3). It appears that the singlet oxygen removal is largely due to interaction of the metal-thiolato or metal-sulfenato moiety, as the sulfinato complex 3, where the metal-sulfur bond is completely oxidized, has a 1O2 removal rate constant that is quite small, i.e. kT = 9.8 × 105 M-1sec-1.
Chemical reaction with singlet oxygen constitutes a significant fraction of the kT value for complex 1: We carried out competition experiments [in CD3OD/D2O (2:1)] with the known singlet oxygen acceptor, 3-(10-(2-carboxy-ethyl)- anthracen-9-yl)-propionic acid [kT = 1.8 × 107 M-1sec-1 in CD3OD/D2O (2:1, we used this mixture as the solubility of the anthracene derivative in neat water is very poor)] and the thiolato complex 1.10 The rate constant for chemical reaction (kr) between 1 and singlet oxygen is 1.0 × 107 M-1sec-1. We re-measured the total rate of singlet oxygen removal kT in the 2:1 CD3OD/D2O mixture and found it to be 4.1 × 107 M-1sec-1, very similar to the value in D2O. Thus chemical reaction accounts for ca. 25 % of the total rate of singlet oxygen removal of complex 1 in methanol/water (2:1), and about four times the total rate of singlet oxygen removal (kT) by sulfenato complex 2. These kinetic data are consistent with the surprising observation that no sulfinato product 3 is observed prior to complete conversion of the starting complex 1 to the sulfenato product 2.
The reaction of 1 with singlet oxygen is rather different from the self-sensitized photooxidations of Pt dithiolate complexes studied by Schanze and co-workers3 and especially the reaction of singlet oxygen with the complex Pt(II)(bipyridine)(1,2-benzenditholate) (4) reported by Connick and Gray.4 Complex 4 undergoes a self-sensitized photooxidation leading to formation of only mono and bis-sulfinato products while complex 1 reacts with singlet oxygen to produce the sulfenato complex 2. Even though the dithiolato complex 4 has a second thiolato group that could act as an intramolecular trap for a peroxidic intermediate formed from attack of 1O2 on the thiolate ligand, no sulfenate or bis-sulfenate formation was observed by Gray et al. during the photooxidation of 4.4 This implies that intramolecular collapse of the peroxidic intermediate during the photooxidation of 4 must be preferred over oxygen atom transfer whereas during the photooxidation of 1, intermolecular oxygen atom transfer from a peroxidic intermediate must be the sole reaction channel leading to oxygenated products. The difference in reactivity is not due to protic vs. aprotic conditions (reactions for complex 4 have been carried out in CH2Cl2 and CH3CN), as addition of water to an acetonitrile solution of 4 did not change the outcome of the reaction.
Given this striking and unexpected contrast between complexes 1 and 4, we decided to reinvestigate some aspects of the photochemistry of complex 4. Singlet oxygen luminescence measurements (external reference sensitizer: C60, ΦΔ = 1.0) confirmed that unlike complexes 1-3, complex 4 is indeed a singlet oxygen sensitizer with a quantum yield of 0.50 (Table 1). Time-resolved singlet oxygen luminescence quenching experiments with 1O2 generated by an external photosensitizer (methylene blue, exited at 532 nm) demonstrated that 1O2 interacts with complex 4, with a total rate constant of singlet oxygen removal of 4.8 × 107 M-1sec-1, in reasonable agreement with the value of 1 × 108 M-1sec-1 obtained by Gray et al. via transient absorption spectroscopy during the self-sensitized photooxidation of 4.4 Photooxidation of complex 4 using externally generated singlet oxygen without exciting 4 (sensitizer = Methylene Blue, cut-off filter at 645 nm) leads to the same sulfinate product (5) that was reported by Gray et al. when complex 4 is irradiated under aerobic conditions without an external sensitizer confirming that singlet oxygen indeed oxidizes 4 to the sulfinate (Scheme 1b). No sulfenate or bis-sulfenate was obtained in the presence of externally generated singlet oxygen.
Table 1.
Rate Constants for Singlet Oxygen Removal by Complexes 1-4, and Singlet Oxygen Quantum yields
| Rate constant of 1O2 removal (kT) × 107 M-1s-1 | 1O2 quantum yieldc (ΦΔ) | |
|---|---|---|
| Pt(II)(bpy)(N,S-aminoethanethiolate)a1 | 3.2 ± 0.3 | 0.00 |
| Pt(II)(bpy)(N, S(=O)-aminoethanesulfenate)a2 | 0.25 ± 0.03 | 0.00 |
| Pt(II)(bpy)(N,S(=O)2 - aminoethanesulfinate)a3 | 0.098 ± 0.008 | 0.00 |
| Pt(II)(bpy)(1,2-benzenditholate)b4 | 4.8 ± 0.4 | 0.50 ± 0.05 |
In D2O, average of 3-5 runs, error is one standard deviation
In CD2Cl2, average of 3-5 runs, error is one standard deviation
Determined at λexc. = 532 nm from the near-infrared 1O2 emission.
Scheme 1.

(a) Sequential Reaction of Complex 1 with singlet oxygen (b) Direct conversion of Pt dithiolato complex 4 sulfinato complex 5.
It is possible that the sulfinato product 5 is the result of an intramolecular isomerization of a bis-sulfenato complex which could be the initial product; a similar hypothesis was raised by Darensbourg et al. for the formation of a Ni(II) sulfinato product during the oxidation of Ni(II) dithiolato complexes.11 To investigate this possibility, we carried out the photooxidation of 4 at low temperature (–42 °C) in an NMR tube placed into a transparent Dewar flask. If a bis-sulfenato product were initially formed, its isomerization should be sufficiently slow at –42 °C so that it could be observed by low-temperature NMR. However, no intermediate during the photooxidation of 4 was detected even at –42°C. While the lack of observation of a low-temperature intermediate does not conclusively rule out a transient bis-sulfenato complex, the sulfinato product 5 may well be the primary product of the photooxidation of 4.
A key step during the photooxidation of organic sulfides is the abstraction of a proton on the α-carbon by the primary peroxidic intermediate (most likely a persulfoxide) leading to a hydroperoxy sulfonium ylide which undergoes intramolecular oxygen atom transfer reaction leading to sulfoxide products.12 It is not known whether or not the reaction of singlet oxygen with metal thiolates proceeds via an analogous mechanism.13 It is striking that complex 1 (which undergoes intramolecular O-atom transfer) does posses two hydrogen atoms on the α carbon atom while complex 4 does not. However, no H/D exchange was observed at the methylene site adjacent to the sulfur atom during the photooxidation of 1 in D2O, in contrast with the photooxidation of organic sulfides which does proceed via a hydroperoxysulfonium ylide and concomitant H/D exchange at the methylene group adjacent to the sulfur atom.14 Thus, alternatively, the intermediate responsible for intermolecular oxygen atom transfer may possibly be a thiadioxirane-type moiety. Both of these possible secondary intermediates are depicted in Scheme 2 below.
Scheme 2.

Possible secondary intermediates formed during photooxidation of 1 and intermolecular oxygen atom transfer.
Formation of the thiadioxiranes has been ruled out for photooxidation of organic sulfides, as the sulfur lone pair would have to rotate away from the second oxygen-sulfur bond formed from the rearrangement of the initial persulfoxide.12 On the other hand, for a metallo-thiolate, the in-plane 3p orbital of the sulfur interacts with the metal,15 perhaps thereby making formation of the thiadioxirane easier. If this were the case, interaction between the π system of the benzene ring of complex 4 and the thiolate ligand would inhibit the ring-closing step required to form a thiadioxirane. In conclusion, we have demonstrated that the photooxidation of a Pt monothiolato complex proceeds via sequential oxidation reactions. Unlike for Pt benzenedithiolato complexes, a peroxidic intermediate in this process is capable of rapid intermolecular oxygen atom transfer. The nature of the reactive intermediates in these reactions appears to be different from those involved in the photooxidation of organic sulfides.
Supplementary Material
Figure 1.
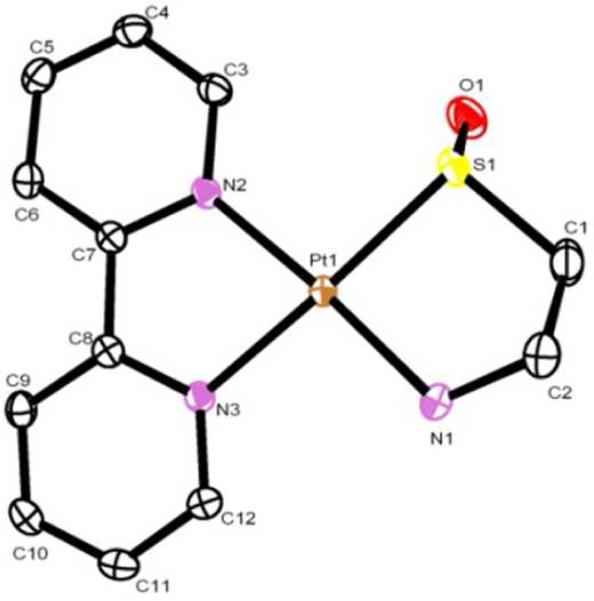
X-ray molecular structure of Pt(II)(bipyridine)-(N,S(=O)-aminoethansulfenate) (2), solvent molecules, hydrogen atoms and Cl- counterion have been omitted for clarity.
ACKNOWLEDGMENT
We thank Dr. A. Ali Jabalameli (CSULA) for assistance with the 195Pt NMR experiments. This work was supported by the NIHNIGMS (5SC1GM084776). L.T. acknowledges support from NIH-NIGMS GM61331. We also acknowledge partial support by the NSF-CREST program (NSF HRD-0932421).
Footnotes
Supporting Information. Experimental details, and X-ray crystallographic data in CIF format for 2. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.a Cummings SD, Eisenberg R. J. Am. Chem. Soc. 1996;118:1949. [Google Scholar]; b Zuleta JA, Eisenberg R. Coord. Chem. Rev. 1990;97:47. and references therein. [Google Scholar]
- 2.a Anbalagan V, Srivastava TS. J. Photochem. Photobiol, A: Chem. 1995;89:113. [Google Scholar]; b Anbalagan V, Srivastava TS. J. Photo-chem. Photobiol., A: Chem. 1994;77:141. [Google Scholar]; c Anbalagan V, Srivastava TS. Polyhedron. 1994;13:291. [Google Scholar]; d Anbalagan V, Srivastava TS. J. Photochem. Photobiol., A: Chem. 1992;66:345. [Google Scholar]; e Kamath SS, Srivastava TS. J. Photochem. Photobiol., A: Chem. 1990;52:83. [Google Scholar]; f Shukla S, Kamath SS, Srivastava TS. J. Photo-chem. Photobiol., A: Chem. 1988;44:143. [Google Scholar]
- 3.Zhang Y, Ley KD, Schanze KS. Inorg. Chem. 1996;35:7102. doi: 10.1021/ic960685x. [DOI] [PubMed] [Google Scholar]
- 4.Connick WB, Gray HB. J. Am. Chem. Soc. 1997;119:11620. [Google Scholar]
- 5.a Pattanaik A, Bachowski G, Laib J, Lemkuil D, Shaw CF, IIIrd, Petering DH, Hitchcock A, Saryan L. J. Biol. Chem. 1992;267:16121. [PubMed] [Google Scholar]; b Karotki AV, Vasak M. Biochemistry. 2008;47:10961. doi: 10.1021/bi801253x. [DOI] [PubMed] [Google Scholar]; c Karotki AV, Vasak M. J. Biol. Inorg. Chem. 2009;14:1129. doi: 10.1007/s00775-009-0557-x. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell KA, Jensen CM. Inorg. Chem. 1995;34:4441. [Google Scholar]
- 7.This is in contrast with a methylplatinum(II) tridentate amino-substituted terpyridine complex which undergoes a self-sensitized photooxidation to a methylperoxo complex via a Pt(IV) peroxo intermediate. Taylor RA, Law DJ, Sunley GJ, White AJP, Britovsek GJP. Angew. Chem. Int. Ed. 2009;48:5900. doi: 10.1002/anie.200806187.
- 8.a Masitas CA, Mashuta MS, Grapperhaus CA. Inorg. Chem. 2010;49:5344. doi: 10.1021/ic100414c. [DOI] [PubMed] [Google Scholar]; b Adzamli IK, Libson K, Lydon JD, Elder RC, Deutsch E. Inorg. Chem. 1979;18:303. [Google Scholar]
- 9.S-bound sulfinates typically show IR stretches at 1250-1150 and 1100-1020 cm-1. See, for example, Farmer PJ, Soluki T, Mills DK, Soma T, Russel DH, Reibenspies JH, Darensbourg MY. J. Am. Chem. Soc. 1992;114:4601.
- 10.a Hernandez B, Wang Y, Zhang D, Selke M. Chem. Commun. 2006:997. doi: 10.1039/b514601a. [DOI] [PubMed] [Google Scholar]; b Higgins R, Foote CS, Cheng H. Adv. Chem. Ser. 1968;77:102. [Google Scholar]
- 11.Grapperhaus CA, Maguire MJ, Tuntulani T, Darensbourg MY. Inorg. Chem. 1997;36:1860. doi: 10.1021/ic970050d. [DOI] [PubMed] [Google Scholar]
- 12.Clennan EL. Acc. Chem. Res. 2001;34:875. doi: 10.1021/ar0100879. [DOI] [PubMed] [Google Scholar]; b Jensen F, Greer A, Clennan EL. J. Am. Chem. Soc. 1998;120:4439. [Google Scholar]
- 13.Zhang D, Hernandez B, Selke M. J. Sulf. Chem. 2008;299:377. doi: 10.1080/17415990802146980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishiguro K, Hayashi M, Sawaki Y. J. Am. Chem. Soc. 1996;118:7265. [Google Scholar]
- 15.Solomon EI, Gorelsky SI, Dey A. J. Comput. Chem. 2006;27:1415–1428. doi: 10.1002/jcc.20451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.